
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEarly detection of bacteria using SPR imaging and event counting: experiments with Listeria monocytogenes and Listeria innocua†
Marine Boulade ab,
Alexandra Morlayac,
Felix Piatc,
Yoann Roupioza,
Thierry Livachead,
Paul G. Charetteb,
Michael Canvab and
Loïc Leroy*a
ab,
Alexandra Morlayac,
Felix Piatc,
Yoann Roupioza,
Thierry Livachead,
Paul G. Charetteb,
Michael Canvab and
Loïc Leroy*a
aINAC-SyMMES, Univ. Grenoble Alpes, CEA, CNRS, 38000 Grenoble, France. E-mail: loic.leroy@univ-grenoble-alpes.fr
bLaboratoire Nanotechnologies Nanosystèmes (LN2), CNRS UMI-3463, Université de Sherbrooke, UGA, 3000 Boulevard Université, J1K OA5, Québec, Canada
cPrestodiag, 1 Mail du Professeur Georges Mathé, F-94800 Villejuif, France
dAryballe Technologies, 17 Avenue des Martyrs, 38000 Grenoble, France
First published on 17th May 2019
Abstract
Foodborne pathogens are of significant concern in the agrifood industry and the development of associated rapid detection and identification methods are of major importance. This paper describes the novel use of resolution-optimized prism-based surface plasmon resonance imaging (RO-SPRI) and data processing for the detection of the foodborne pathogens Listeria monocytogenes and Listeria innocua. With an imaging spatial resolution on the order of individual bacteria (2.7 ± 0.5 μm × 7.9 ± 0.6 μm) over a field of view 1.5 mm2, the RO-SPRI system enabled accurate counting of individual bacteria on the sensor surface. Using this system, we demonstrate the detection of two species of Listeria at an initial concentration of 2 × 102 CFU mL−1 in less than 7 hours. The surface density of bacteria at the point of positive detection was 15 ± 4 bacteria per mm2. Our approach offers great potential for the development of fast specific detection systems based on affinity monitoring.
1 Introduction
Rapid microbial detection and identification is of great concern for consumer safety, in particular in the agri-food industry where perishable goods must be screened for pathogen contamination.1–3 Listeriosis for example, a bacterial infection by Listeria monocytogenes, one of the main foodborne pathogens, causes a variety of severe illnesses with fatality rates as high as 30%.4 However, all current testing methods for these bacteria require an incubation period lasting from 24 h to several days.5 As a result, significant work in academia and industry has focused on the development of rapid and affordable pathogen detection tools6,7 such as dedicated biosensors.8,9Various types of surface-based biosensors have proven effective for the detection of pathogens such as surface acoustic wave (SAW) sensors,10 impedance sensors,11–13 the quartz crystal microbalance (QCM),14,15 and surface plasmon resonance (SPR).16,17 Such sensors are compact, versatile, respond in real time, can be fabricated at low cost, and do not require molecular labeling.18 Using modern microfabrication methods, these sensors can be configured in a matrix layout, thus enabling parallel detection to screen for multiple species or concentrations simultaneously. With SPR, the entire sensor surface can be imaged (termed “SPRI”), with the surface partitioned into arbitrary sized “spots” with location-specific surface functionalization for parallel detection applications.19
In the case of biosensors that rely on surface detection, improvement in performance rely on three main strategies: improving the probability of contact of the target molecules with the sensing surface (efficacy and dynamics of mass transport towards the surface), increasing the efficiency of the surface probes (specificity and efficacy of capture of the targets), and enhancing the sensitivity of the transducer to improve the lower limit of detection. The first strategy is used, for instance, in biosensors that employ microfluidic mixing,20,21 electrophoresis22 or immunoseparation.23 The second strategy has spawned much research in optimizing the nature of the probes24 and immobilization strategies.25–28 In the third strategy, efforts have focused on increasing the performance of the transducer,29 labelling the targets with non-intrusive secondary molecules,30 and on chemically amplifying the response.31
SPRI systems are easily automated and compatible with real food samples.9,19,32,33 Optical coupling can be achieved with gratings,34 microscope objectives35,36 or prisms,19,32,37,38 with prism-based SPRI being the most frequently used configuration for the detection of bacteria. Prism-based SPRI systems are often designed with a relatively wide field of view (around 1 cm2) for parallel measurements on a matrix of spots (spotted microarray) with detection levels as low as 100 pM in the case of DNA–DNA hybridization.38,39 Though prism-based SPRI systems are limited in spatial resolution to tens of microns or more by geometrical aberrations caused by the prism,40 this is not a problem for spotted microarray applications since the spots are sufficiently large (100 microns in diameter or more) and spatially well separated. In true imaging applications of biological objects such as bacteria, however, where the morphology and/or dynamical behaviour of individual objects is the basis for the information sought, the low spatial resolution results in loss of important information.
In this article, we propose a new strategy to improve the performance of bacterial detection using resolution optimized (RO) surface plasmon resonance imaging (RO-SPRI). The strategy is based on extending the high spatial resolution of SPRI, typically restricted to a narrow band in the middle of the image and falling off towards the edges, to the entire field of view, thereby significantly improving the accuracy of the surface kinetics analysis.40 We demonstrate faster positive signature identification based on single bacteria observation compared to SPRI analysis based on aggregate spots responses. Our observations are confirmed with standard differential interference coherence (DIC) microscopy.41 To our knowledge, this is the first time that SPRI is used for bacterial detection based on counting and trajectory analysis of individual bacteria.
2 Experimental section
2.1 Microbiology culture
Two Listeria species were used in this study: Listeria monocytogenes and Listeria innocua from naturally contaminated food products provided by the Institut Scientifique d'Hygiène et d'Analyse (ISHA, Massy, France). The bacterial species, from stock suspension conserved at −80 °C, were streaked on Tryptone-Soy Agar (TSA, Biomérieux, France) plates and incubated overnight at 37 ± 1 °C. Prior to experiments, one colony of the strain of interest was inoculated in 10 mL of Tryptic Soy Broth (TSB, Sigma-Aldrich, Missouri, USA) at 37 ± 1 °C, 150 rpm for 18 h ± 2 h. The bacterial concentration was then adjusted to 1 ± 0.2 MacFarland (McF) with a densitometer (Biomérieux, France) by adding Buffer Peptone Water (BPW, Biomérieux, France).For each experiment, six serial dilutions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) were carried out with TSB. 200 μL of one of the six dilutions corresponding to the targeted concentration was injected into the RO-SPRI fluidic chamber and processed on the biochips. Other samples of 200 μL were also done in parallel and kept at the same temperature as the experimental chamber (37 °C). The bacterial concentration was then controlled every hour for four hours (plus initial time) by spreading 100 μL of the sixth dilution on two TSA plates. The colony counting was performed after overnight incubation at 37 ± 1 °C.
:10) were carried out with TSB. 200 μL of one of the six dilutions corresponding to the targeted concentration was injected into the RO-SPRI fluidic chamber and processed on the biochips. Other samples of 200 μL were also done in parallel and kept at the same temperature as the experimental chamber (37 °C). The bacterial concentration was then controlled every hour for four hours (plus initial time) by spreading 100 μL of the sixth dilution on two TSA plates. The colony counting was performed after overnight incubation at 37 ± 1 °C.
2.2 Biochip preparation
N-SF66 glass prisms with an apex angle of 83° and a width of 12 mm (ref. 40) (Schott, Switzerland) formed the base of the RO-SPRI biochips. Thin films of 2 nm of chromium and 53 nm of gold were deposited on the prism base (the sensor surface) by evaporation. The prism bases were then plasma cleaned (40 W, 0.6 mbar, 75% oxygen, 25% argon) for 3 minutes (Femto 300, Diener Electronic, Germany) to prepare the surface for chemical functionalization.Three antibodies provided by Prestodiag (Villejuif, France) were used for surface spot functionalization: two polyclonal antibodies (LiM14 and LiM16) specific to Listeria monocytogenes and one polyclonal antibody (Lis12) specific to Listeria innocua. One monoclonal antibody (KLH), directed against Keyhole Limpet Hemocyanin (obtained from L. Bellanger CEA Marcoule, France), i.e. not directed against bacterial targets, was used as a negative control in all experiments.
The biochip surface functionalization was prepared with an electro-polymerization process described in detail elsewhere.19,42 Electropolymerization of the solution (1 μM of antibodies coupled with NHS-pyrrole, 10 μM of free NHS-pyrroles in spotting buffer) was carried out using a Microgrid spotter (BioRobotics). Biochips were then washed with phosphate buffer solution (PBS) and final surface passivation was performed using 1% (w/v) bovine serum albumin (Sigma Aldrich, Saint Quentin Fallavier, France) for 15 min at room temperature, ensuring very low non-specific binding.43 The biochips were washed again with PBS and kept in PBS at 4 °C for up to several weeks.44,45
Before being introduced into the RO-SPRI system, described in detail the next section, the non-coated faces of the prism were thoroughly washed and cleaned to avoid any optical defects.
2.3 Apparatus set-up
The set-up was designed to demonstrate and validate the use of resolution-optimized prism-based SPRI (RO-SPRI) for bacterial detection. The system was coupled to a differential interference contrast (DIC) microscope for simultaneous observation during the experiments.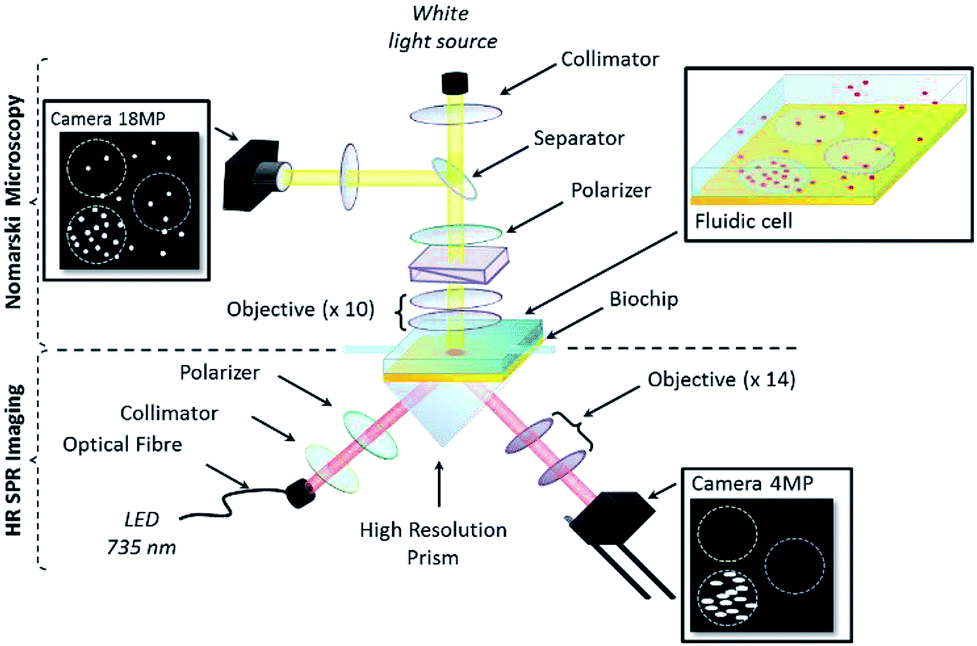 | ||
| Fig. 1 Dual DIC microscopy and RO-SPR imaging apparatus. | ||
Note that in prism-based SPRI, the image plane usually is not perpendicular to the optical axis of the system, i.e. there is a skew between the camera image plane and the sensor surface, resulting in only a narrow band being in focus at any time in the image, with resolution dropping off towards the edges due to progressive loss of focus.40 The amount of skew is dependent on the geometry of the prism and dictates the width of the “in-focus” band at the centre of the image. In RO-SPRI, a 15 images sequence is captured corresponding to a series of focal plane step-changes designed to scan the “in-focus” band across the image. The images in the sequence are then processed to combine them into a single “RO-SPRI image” that is in focus across the entire image thereby having uniform spatial resolution. In the experiments described below, one RO-SPRI image was generated every 3 minutes.
Note also that due to the propagative nature of the surface plasmons, the spatial resolution in the direction parallel to plasmon propagation is ultimately limited by the propagation length of the surface plasmons.46 In our case, spatial resolution in the direction parallel to plasmon propagation is ∼6 μm. Even if better resolution can be reached using other settings, these choices appeared to be a satisfactory compromise between spatial and temporal resolution for the targeted application.
2.4 Fluidic chamber
The bacterial suspensions were injected on the biochip surface through a custom-made temperature regulated fluidic chamber, enabling bacterial growth during experiments. As shown in Fig. 2, a 200 μL fluidic chamber was fabricated from a 1.5 mm thick PDMS film and then glued to a 2 mm thick glass plate after plasma activation (40 W, 0.6 mbar, 75% O2, 25% Ar for 30 s, Femto 300, Diener Electronic, Germany). Fluids were injected through 600 μm diameter PE tubes.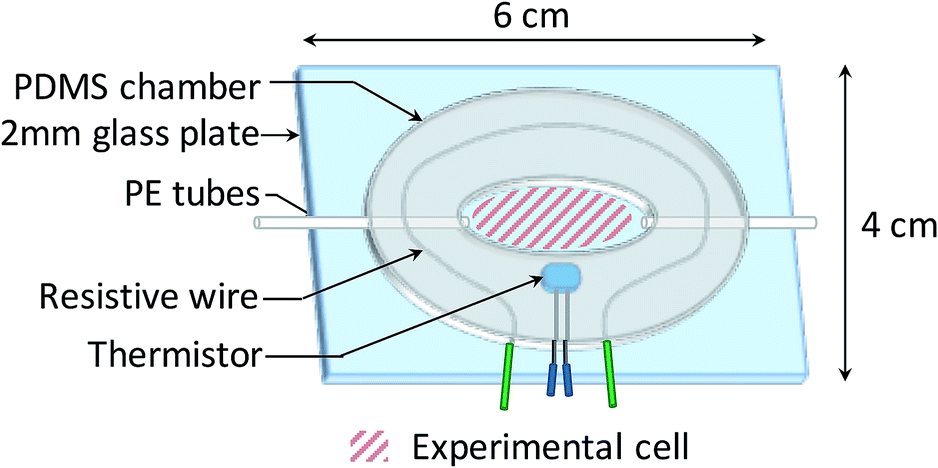 | ||
| Fig. 2 Schematic diagram of the fluidic chamber. | ||
The temperature regulation was carried out by a heating circuit made of resistive wires contouring the chamber, supplied with a variable voltage. The voltage was regulated using a PID controller tuned by the measurement of the temperature through a thermistor. The variability of the temperature was ±0.15 °C, which is adequate for microbiology applications.47 Note that SPR signal variations induced by a 0.15 °C temperature variation (about 0.02% reflectivity) are much smaller than the SPR reflectivity changes that indicate the presence of bacteria on the sensor surface (>1% reflectivity).48,49 The operating temperature for the experiments was set at 37 °C.
2.5 Data processing
3 Results and discussion
In the experiments, several concentrations of L. monocytogenes and L. innocua were injected into the fluidic chamber and RO-SPRI and DIC images were acquired over time using the dual imaging apparatus.3.1 Image comparison between RO-SPRI images and DIC microscopy images
A comparison of images and event detection, carried out on both types of images, is presented Fig. 3. The images correspond to an experiment with an original concentration of L. monocytogenes at 2 × 102 CFU mL−1, starting at 400 min after injection into the fluidic chamber. | ||
| Fig. 3 Results from an experiment with an injection of L. monocytogenes at 2 × 102 CFU mL−1, images were taken 400 min after injection. (a) Differential RO-SPRI image (b) differential microscopy (DIC) image, (c) and (d) results of the event detection using ImageJ on (a) and (b), respectively – events detected both in the RO-SPRI and DIC images are colored in blue. | ||
Under these conditions, high-intensity events indicating the presence of individual bacteria in the RO-SPRI micrographs (Fig. 3a) were 2.7 ± 0.5 μm × 7.9 ± 0.6 μm in size on average along the directions perpendicular and parallel to plasmon propagation, respectively. L. monocytogenes is a bacterium of average size 2 μm × 0.5 μm × 0.5 μm, the discrepancy along one of the two planar dimensions being due to loss of spatial resolution associated with plasmon propagation. In the DIC microscopy images, the bacteria have an estimated size of 2.0 ± 0.2 μm × 0.7 ± 0.2 μm, the variability being due both to the variability in the actual size of the bacteria and to the low magnification of the objective which was fixed at 10× to obtain a field of view compatible with the RO-SPRI system.
The event counting data processing (Fig. 3c and d) showed that more particles were detected in the DIC images. This is due to the difference in the depth of field between the two methods: the evanescent field associated with the surface plasmons has a penetration depth in the dielectric of less than 300 nm at a wavelength of 740 nm, while the DIC microscope has a depth of field of around 8 μm for the 10× objective. As the images were taken while the bacterial solution was still in the chamber, bacteria in the volume could be observed in the DIC images outside the penetration depth of the SPRI system, explaining the difference in the number of bacteria observed with each imaging system. For larger densities of bacteria, the difference could also be related to the lower spatial resolution of the RO-SPRI system causing close events to merge and not to be discernible. This is of minor concern for early diagnostics, i.e. for low concentration detection, provided there is no factor promoting aggregation.
3.2 Quality control of the bacterial growth conditions in the fluidic chamber
The biocompatibility of the fluidic chamber was validated by assessing the doubling time of L. monocytogenes during experiments from the DIC microscopy images. As the depth of field of this method is of several microns, the number of bacteria detected over time is a direct assessment of growth in the solution. In the absence of flow, the evolution of the number of bacteria, N(t), in DIC microscope images on a surface S, depends solely on diffusion and gravity and can be calculated with: N(t) = C0exp(t/τ) × S × v × t, where v is the sedimentation speed and τ the growth rate of the bacteria.
The event detection data calculated from the DIC differential images of bacterial density over time were fitted to a two-parameter model (t) = Aexp(t/τ)t, where A =C0Sv and the doubling time τ2 = τln2 (Fig. 4a). As a control or bacterial viability and growth in each experiment, model parameters were estimated for an initial injection rate of 2 × 102 CFU mL−1 (±10%), yielding a doubling time of 40.5 ± 2.7 min. This values are in line with what is reported in the literature for growth in optimal conditions.56 No difference in growth efficiency was seen between the antibody spots. This analysis shows that our fluidic chamber enables close to optimal growth conditions for the bacteria of interest. The residuals of the modelling process, defined as the difference between the model and the experimental data, are available in the ESI.† Considering this doubling time, it is assessed the concentration in solution, 400 min after injection, has doubled ten fold since injection.
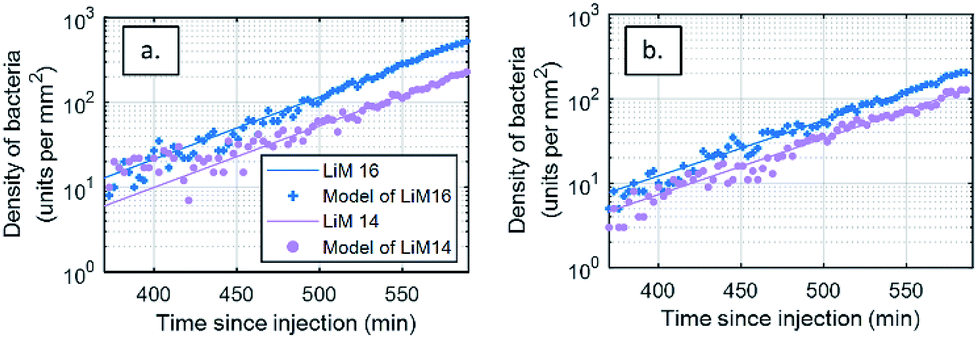 | ||
| Fig. 4 Modelling of Listeria monocytogenes bacterial growth from event-detection data in both types of images from areas functionalized with antibodies targeting L. monocytogenes (LiM16 and LiM14). Number of bacteria respectively detected on images (a) DIC and (b) RO-SPRI images on both specific antibody areas and their model fits – injection at 102 CFU mL−1. | ||
The same analysis was also performed on SPRI images (Fig. 4b), leading to a doubling time of 46.7 ± 1.8 min. This difference with the DIC-based analysis can be attributed to the limitation of the SPRI system to differentiate between individual events that are closer than the spatial resolution of the system, leading to an apparent lower increase in the number of bacteria.
3.3 Early detection of bacteria
The objective of the experiments was to detect bacterial presence as soon as possible, quantified by the “onset of positive detection” defined as the time at which the signal-to-noise ratio (SNR) exceeds 3. The noise floor for SNR calculation was estimated from the KLH negative control.Both SPRI sensorgrams-based analysis and RO-SPRI-based event counting analysis were carried out for the detection of two species of Listeria: L. monocytogenes and L. innocua. The two species were first detected separately at different concentrations. A third type of experiment consisted in the simultaneous detection of both species L. monocytogenes and L. innocua injected at a concentration of C0 = 2 × 104 CFU mL−1 (±10%).
A comparison between the SPRI sensorgram-based analysis and the RO-SPRI-based event counting is presented in Fig. 5 for the detection of L. monocytogenes and L. innocua injected at C0 = 3 × 104 CFU mL−1 (±10%) (Fig. 5). For SPRI sensorgrams, results are generally presented in terms of reflectivity variations: with the imaging components in our apparatus, an intensity variation of 100 grey levels (I − I0 = 100) corresponded to a reflectivity variation of ∼1% (Δ%R ∼ 1%).
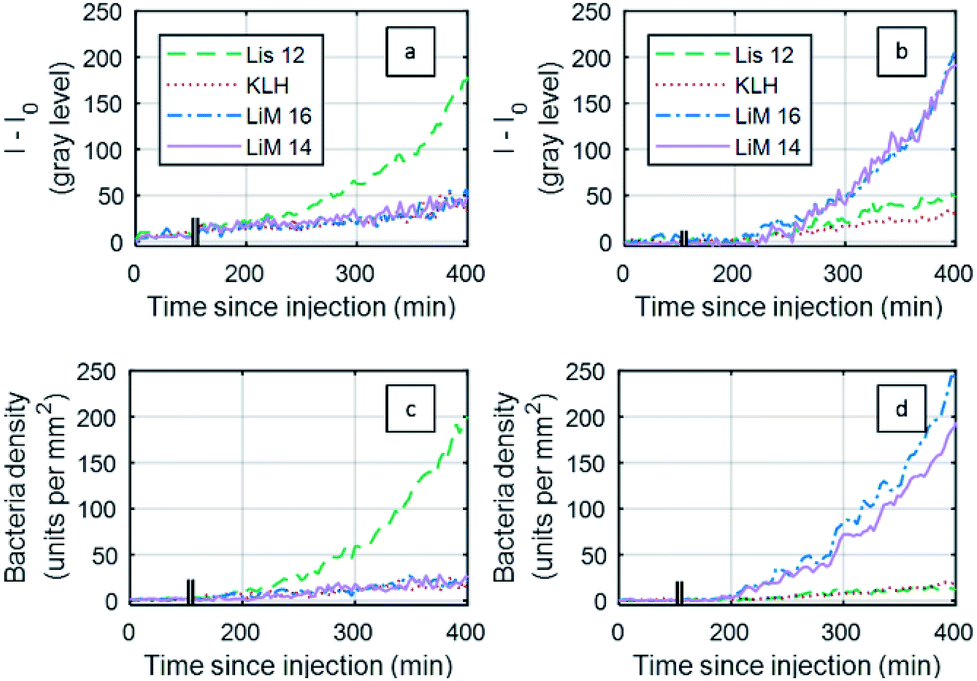 | ||
| Fig. 5 Experimental results for the detection of Listeria innocua (a and c) and Listeria monocytogenes (b and d) RO-SPRI: (a and b) SPRI sensorgrams: (c and d) fluid injected at C0 = 3 × 104 CFU mL−1 ±10%. | ||
In the SPRI sensorgram-based analysis (Fig. 5a), the negative control areas (KLH) showed a drift of 20% between 200 min and 400 min following injection, while in the RO-SPRI-based event counting analysis, the same negative control area (KLH) showed a non-specific signal of less than 13% over the same interval. This difference in the negative control is due to the different processing strategies: in SPRI sensorgrams, intensity changes inside a region of interest also depend on refractive index changes in the fluid medium close to the surface (change of composition due to the consumption of nutrients by the bacteria, light source intensity and/or variations, etc.). For RO-SPRI-based event detection, even if the background signal varies, the individual bacterial event counting is unaffected.
In the case of RO-SPRI-based event counting, the “time to onset of positive detection” (SNR > 3) corresponded to a surface density of between 10 and 20 bacteria per mm2 (between 3 and 6 units per antibody spot), for an average value of 15 ± 4 bacteria per mm2.
Results of “time to onset of positive detection” estimations for both types of analyses are presented in Table 1. For SPRI sensorgrams, calculated onsets of positive detection, at ∼9 h for an initial concentration of 2.0 × 102 CFU mL−1 and ∼5 h for an initial injection at 3 × 104 CFU mL−1, are consistent with the literature.32,57–59 From RO-SPRI data, the surface density at these times of onset of positive detection on SPRI sensorgrams are respectively estimated at 62 ± 10 bacteria per mm2, and 75 ± 20 bacteria per mm2.
| Initial concentration (CFU mL−1) | Strain | Number of experiments | Time to onset of positive detection (in min) | |||||
|---|---|---|---|---|---|---|---|---|
| SPRI sensorgrams | Event counting using RO-SPRI | |||||||
| Lis12 | LiM16 | LiM14 | Lis12 | LiM16 | LiM14 | |||
| 2 × 102 | L. monocytogenes | n = 2 | — | 518 ± 2 | 528 ± 6 | — | 432 ± 15 | 440 ± 17 |
| 3 × 104 | L. monocytogenes | n = 2 | — | 306 ± 6 | 306 ± 6 | — | 236 ± 6 | 236 ± 6 |
| 3 × 104 | L. innocua | n = 2 | 312 ± 8 | — | — | 246 ± 9 | — | — |
| 3 × 104 | L. monocytogenes & L. innocua | n = 1 | 318 | 316 | 312 | 247 | 247 | 243 |
For RO-SPRI-based event counting, the time to onset of positive detection were reduced by at least 66 min and up to 86 min compared to the sensorgram method. This gain is coherent with the difference between the surface densities at the onset of positive detection of both methods.
4 Conclusion
In this work, we demonstrated a label-free, real-time biosensor using a prism-based, resolution-optimized SPRI system, optimized for the detection of individual bacteria over a relatively wide field of view. This innovative apparatus can detect individual bacteria in a field of view of 1.5 mm2, a surface area wide enough to image several antibody-functionalized spots, enabling parallel measurement of different bacteria–antibody interactions. By coupling the system to a DIC microscope, we validated the viability of the fluidic chamber bacterial growth conditions.The system proved to be effective for the detection of two different species of Listeria, both separately and simultaneously. The data analysis showed a gain of at least one hour compared to classical SPRI analysis based on sensorgrams, with an even higher gain of 1 h 30 min for low concentration injection of 2.0 × 102 (±10%) CFU mL−1. The improvement in the times to onset of positive detection between the two SPRI methods is clear. Indeed, the presence at the surface of even a very small number of bacteria creates a measurable RO-SPRI signal that is not differentiable from noise in SPRI sensorgrams. The ability of our system to detect the presence of bacteria at surface densities as low as 15 ± 4 bacteria per mm2 is very promising for early detection of bacteria.
To develop further our technique, experiments on complex samples closer to real life applications, such as food matrices, and for the detection of low concentration pathogens, should be perform. Such experiments were already successfully carried out on standard SPRI devices19 and look therefore like a realistic perspective.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Idex Université Grenoble Alpes, the French Agence National de la Recherche et de la Technologie (ANRT grant no 624/2013), and Labex ARCANE and CBH-EUR-GS (ANR-17-EURE-0003). LN2 is an international laboratory (Unité Mixte Internationale UMI 3463) jointly managed by the French CNRS and the Université de Sherbrooke as well as Université de Lyon (ECL, INSA de Lyon, CPE) and the Université Grenoble Alpes (UGA).Notes and references
- E. De Boer and R. R. Beumer, Int. J. Food Microbiol., 1999, 50, 119–130 CrossRef CAS PubMed.
- M. D. Kirk, I. Mckay, G. V. Hall, C. B. Dalton, R. Stafford, L. Unicomb and J. Gregory, Clin. Infect. Dis., 2008, 47, 392–400 CrossRef PubMed.
- E. Scallan, R. M. Hoekstra, F. J. Angulo, R. V. Tauxe, M. Widdowson, S. L. Roy, J. L. Jones and P. M. Griffin, Emerging Infect. Dis., 2011, 17, 1–21 CrossRef PubMed.
- Risk assessment of Listeria monocytogenes in ready-to-eat foods, WHO/FAO, 2004 Search PubMed.
- S. Jadhav, M. Bhave and E. A. Palombo, J. Microbiol. Methods, 2012, 88, 327–341 CrossRef CAS PubMed.
- B. Mandracchia, J. Palpacuer, F. Nazzaro, V. Bianco, R. Rega, P. Ferraro and S. Grilli, IEEE J. Sel. Top. Quantum Electron., 2019, 25, 1–6 Search PubMed.
- D. Rodriguez-Lazaro, P. Gonzalez-García, A. Gattuso, M. V. Gianfranceschi and M. Hernandez, Int. J. Food Microbiol., 2014, 184, 98–105 CrossRef CAS PubMed.
- R. Radhakrishnan and P. Poltronieri, Biosensors, 2017, 7, 1–12 CrossRef PubMed.
- V. Velusamy, K. Arshak, O. Korostynska, K. Oliwa and C. Adley, Biotechnol. Adv., 2010, 28, 232–254 CrossRef CAS PubMed.
- E. Howe and G. Harding, Biosens. Bioelectron., 2000, 15, 641–649 CrossRef CAS PubMed.
- J. Suehiro, A. Ohtsubo, T. Hatano and M. Hara, Sens. Actuators, B, 2006, 119, 319–326 CrossRef CAS.
- L. Yang, Y. Li and G. F. Erf, Anal. Chem., 2004, 76, 1107–1113 CrossRef CAS PubMed.
- S. M. Chiriacò, I. Parlangeli, F. Sirsi, P. Poltronieri and E. Primiceri, Electronics, 2018, 7, 347 CrossRef.
- I.-S. Park, W.-Y. Kim and N. Kim, Biosens. Bioelectron., 2000, 15, 167–172 CrossRef CAS PubMed.
- Z. Shen, M. Huang, C. Xiao, Y. Zhang, X. Zeng and P. G. Wang, Anal. Chem., 2007, 79, 2312–2319 CrossRef CAS PubMed.
- A. D. Taylor, J. Ladd, Q. Yu, S. Chen, J. Homola and S. Jiang, Biosens. Bioelectron., 2006, 22, 752–758 CrossRef CAS PubMed.
- Ö. Torun, İ. Hakkı Boyacı, E. Temür and U. Tamer, Biosens. Bioelectron., 2012, 37, 53–60 CrossRef PubMed.
- O. Lazcka, F. J. D. Campo and F. X. Muñoz, Biosens. Bioelectron., 2007, 22, 1205–1217 CrossRef CAS PubMed.
- S. Bouguelia, Y. Roupioz, S. Slimani, L. Mondani, M. G. Casabona, C. Durmort, T. Vernet, R. Calemczuk and T. Livache, Lab Chip, 2013, 13, 4024 RSC.
- D. A. Boehm, P. A. Gottlieb and S. Z. Hua, Sens. Actuators, B, 2007, 126, 508–514 CrossRef CAS.
- M. Varshney, Y. Li, B. Srinivasan and S. Tung, Sens. Actuators, B, 2007, 128, 99–107 CrossRef CAS.
- D. Puchberger-Enengl, S. Podszun, H. Heinz, C. Hermann, P. Vulto and G. A. Urban, Biomicrofluidics, 2011, 5, 044111 CrossRef PubMed.
- F. Cimaglia, E. De Lorenzis, V. Mezzolla, F. Rossi and P. Poltronieri, IEEE Sens. J., 2016, 16, 7045–7052 CAS.
- V. Templier, A. Roux, Y. Roupioz and T. Livache, TrAC, Trends Anal. Chem., 2016, 79, 71–79 CrossRef CAS.
- Y. Jung, J. Young Jeong and B. Hyun Chung, Analyst, 2008, 133, 697–701 RSC.
- A. K. Trilling, J. Beekwilder and H. Zuilhof, Analyst, 2013, 138, 1619–1627 RSC.
- B. Byrne, E. Stack, N. Gilmartin and R. O'Kennedy, Sensors, 2009, 9, 4407–4445 CrossRef CAS PubMed.
- J. B. Fasoli and R. M. Corn, Langmuir, 2015, 31, 9527–9536 CrossRef CAS PubMed.
- C.-J. Huang, J. Dostalek, A. Sessitsch and W. Knoll, Anal. Chem., 2011, 83, 674–677 CrossRef CAS PubMed.
- Y. Wang, W. Knoll and J. Dostalek, Anal. Chem., 2012, 84, 8345–8350 CrossRef CAS PubMed.
- H. J. Lee, A. W. Wark and R. M. Corn, Langmuir, 2006, 22, 5241–5250 CrossRef CAS PubMed.
- A. Morlay, A. Duquenoy, F. Piat, R. Calemczuk, T. Mercey, T. Livache and Y. Roupioz, Measurement, 2017, 98, 305–310 CrossRef.
- J. A. Hudson, R. J. Lake, M. G. Savill, P. Scholes and R. E. McCormick, J. Appl. Microbiol., 2001, 90, 614–621 CrossRef CAS PubMed.
- G. Marusov, A. Sweatt, K. Pietrosimone, D. Benson, S. J. Geary, L. K. Silbart, S. Challa, J. Lagoy, D. A. Lawrence and M. A. Lynes, Environ. Sci. Technol., 2012, 46, 348–359 CrossRef CAS PubMed.
- V. Chabot, C. M. Cuerrier, E. Escher, V. Aimez, M. Grandbois and P. G. Charette, Biosens. Bioelectron., 2009, 24, 1667–1673 CrossRef CAS PubMed.
- B. Mandracchia, V. Pagliarulo, M. Paturzo and P. Ferraro, Anal. Chem., 2015, 87, 4124–4128 CrossRef CAS PubMed.
- P. N. Abadian and E. D. Goluch, Anal. Methods, 2015, 7, 115–122 RSC.
- M. Piliarik, L. Párová and J. Homola, Biosens. Bioelectron., 2009, 24, 1399–1404 CrossRef CAS PubMed.
- F. Bardin, A. Bellemain, G. Roger and M. Canva, Biosens. Bioelectron., 2009, 24, 2100–2105 CrossRef CAS PubMed.
- L. Laplatine, L. Leroy, R. Calemczuk, D. Baganizi, P. N. Marche, Y. Roupioz and T. Livache, Opt. Express, 2014, 22, 22771–22785 CrossRef PubMed.
- W. W. Baldwin and P. W. Bankston, Appl. Environ. Microbiol., 1988, 54, 105–109 CAS.
- L. Grosjean, B. Cherif, E. Mercey, A. Roget, Y. Levy, P. N. Marche, M.-B. Villiers and T. Livache, Anal. Biochem., 2005, 347, 193–200 CrossRef CAS PubMed.
- B. Cherif, A. Roget, C. L. Villiers, R. Calemczuk, V. Leroy, P. N. Marche, T. Livache and M.-B. Villiers, Clin. Chem., 2006, 52, 255–262 CrossRef CAS PubMed.
- J. B. Fiche, J. Fuchs, A. Buhot, R. Calemczuk and T. Livache, Anal. Chem., 2008, 80, 1049–1057 CrossRef CAS PubMed.
- E. Mercey, R. Sadir, E. Maillart, A. Roget, F. Baleux, H. Lortat-Jacob and T. Livache, Anal. Chem., 2008, 3476–3482 CrossRef CAS PubMed.
- J. Homola, Springer Ser. Chem. Sens. Biosens., 2006, 4, 3–44 CAS.
- M. H. Zwietering, J. T. de Koos, B. E. Hasenack, J. C. de Witt and K. van't Riet, Appl. Environ. Microbiol., 1991, 57, 1094–1101 CAS.
- C. S. Moreira, A. M. N. Lima, H. Neff and C. Thirstrup, Sens. Actuators, B, 2008, 134, 854–862 CrossRef CAS.
- K. Özdemir and G. Turhan-Sayan, J. Lightwave Technol., 2003, 21, 805–814 CrossRef.
- J.-Y. Tinevez, N. Perry, J. Schindelin, G. M. Hoopes, G. D. Reynolds, E. Laplantine, S. Y. Bednarek, S. L. Shorte and K. W. Eliceiri, Methods, 2017, 115, 80–90 CrossRef CAS PubMed.
- W. Hu, G. He, T. Chen, C. Xian Guo, Z. Lu, J. Nimal Selvaraj, Y. Liu and C. Ming Li, Chem. Commun., 2014, 50, 2133–2135 RSC.
- L. Zhu, Z. Zhao, P. Cheng, Z. He, Z. Cheng, J. Peng, H. Wang, C. Wang, Y. Yang and Z. Hu, Adv. Mater., 2017, 29, 1700057 CrossRef PubMed.
- Z. Wang, T. Wilkop, D. Xu, Y. Dong, G. Ma and Q. Cheng, Anal. Bioanal. Chem., 2007, 389, 819–825 CrossRef CAS PubMed.
- E. Ouellet, C. Lausted, T. Lin, C. W. T. Yang, L. Hood and E. T. Lagally, Lab Chip, 2010, 10, 581–588 RSC.
- I. Stojanović, T. J. G. van der Velden, H. W. Mulder, R. B. M. Schasfoort and L. W. M. M. Terstappen, Anal. Biochem., 2015, 485, 112–118 CrossRef PubMed.
- J.-C. Augustin and V. Carlier, Int. J. Food Microbiol., 2000, 56, 29–51 CrossRef CAS PubMed.
- V. Nanduri, A. K. Bhunia, S.-I. Tu, G. C. Paoli and J. D. Brewster, Biosens. Bioelectron., 2007, 23, 248–252 CrossRef CAS PubMed.
- P. Leonard, S. Hearty, J. Quinn and R. O'kennedy, Biosens. Bioelectron., 2004, 19, 1331–1335 CrossRef CAS PubMed.
- V. Koubová, E. Brynda, L. Karasová, J. Škvor, J. Homola, J. Dostálek, P. Tobiška and J. Rošický, Sens. Actuators, B, 2001, 74, 100–105 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01466g |
| This journal is © The Royal Society of Chemistry 2019 |