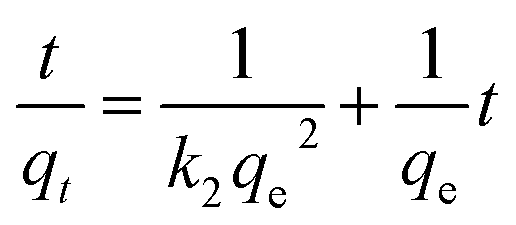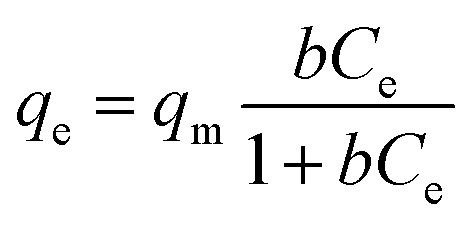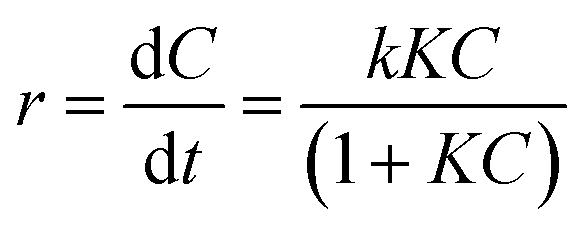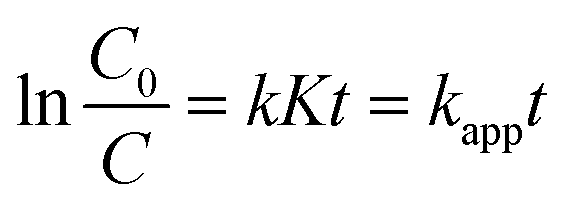DOI:
10.1039/C9RA01486A
(Paper)
RSC Adv., 2019,
9, 15561-15570
Alkylammonium thiostannate inorganic/organic hybrids as high-performance photocatalysts with a decoupled adsorption–photodegradation mechanism†
Received
27th February 2019
, Accepted 23rd April 2019
First published on 17th May 2019
Abstract
For traditional photocatalysts, the adsorption and successive surface reaction constitute a coupled and integrated process, owing to the limited number of catalytic active centres available. An attempt to boost the photocatalytic performance to optimize the adsorption and surface reaction process may be performed by exploring various photocatalyst infrastructures. Herein, we use a facile solvothermal method to synthesize a series of layered alkylammonium thiostannate hybrids, namely (baH)2Sn3S7, (haH)2Sn3S7 and (oaH)2Sn3S7 (ba = butylamine, ha = hexylamine, oa = octylamine). The hybrids showed broad UV-visible light absorption with appropriate band gaps. The inorganic/organic amphiphilic infrastructure of these hybrids enables them to exhibit prominent ion-exchange properties for Rhodamine B, with a large capacity over a wide pH range (1–11). And the adsorbed Rhodamine B is photodegraded within 30 minutes. A mechanistic study indicates that the adsorption and photodegradation steps are performed at the organic and inorganic layers within these hybrids, respectively, which are decoupled and independent. We conclude that the high-performance integrated adsorption–photodegradation ability is a consequence of the lipophilicity of intercalated alkylammonium and the photocatalysis performance of the 2D [Sn3S7]n2n− monolayers.
1. Introduction
Inorganic–organic hybrid chalcogenides have attracted considerable interest, where the organic components and inorganic motifs are bonded by ionic bonds, covalent bonds, hydrogen bonds or van der Waals forces to form chalcogenide-based open-framework structures, and exhibit different optical, electrical and magnetic properties from their bulk counterparts.1 Among these hybrids, II–VI transition metal chalcogenides are especially important. Their periodic structure enables them to display a strong quantum confinement effect, for example, α-ZnTe(en)1/2, β-ZnTe(en)1/2, and ZnTe(pda)1/2 (en = ethylenediamine, pda = propanediamine) exhibit a large blue shift (1.2–1.4 eV) of the absorption edge compared with bulk ZnTe.2 In addition, the amine-hybrid chalcogenides can act as intermediates to prepare ultrathin nanosheets, such as the reported large-area [ZnSe] monolayers with a four-atom-layer thickness that are stripped from the lamellar (Zn2Se2)(pa) intermediate (pa = n-propylamine). It has been proved that the prepared ZnSe monolayer exhibits a unique electronic structure, excellent stability, enhanced photocatalytic water splitting efficiency and photostability.3 Similarly, the ultrathin Zn1−xCdxS nanosheets with atomic-scale thickness, prepared by liquid exfoliation of lamellar Zn1−xCdxS(ha) (ha = hexylamine) complexes, were the first transferable two-dimensional (2D) white light-emitter obtained by engineering grain boundaries in atomically-thin semiconducting nanosheets.4
The pioneering work of R. L. Bedard demonstrated the synthesis of microporous germanium and the tin sulphide phase by a hydrothermal method in the presence of alkylammonium cations.5 Since then, the application of amines for the synthesis of chalcogenidometalates of group 13–15 elements by a solvothermal method has thrived, such as germanium, tin, arsenic, antimony, and indium chalcogenides. These compounds not only possess the ion exchange and catalysis properties, which are characteristic of microporous oxides, but also inherit the semiconductor properties of chalcogenides.6 The basic structural units of germanium and tin–sulphur compounds are MQ4 (M = Ge, Sn; Q = S, Se, and Te) tetrahedra, and multiple MQ4 tetrahedra are shared by corners or edges to form secondary structural units such as polytetrahedra [M2Q6]4−, [M3Q7]2−, [M4Q9]2−, and [M4Q10]4−. These secondary structural units form inorganic–organic hybrid chalcogenides in different structures of 0–3 dimensions by supramolecular assembly or polymerization.7 G. A. Ozin et al. focused on synthesizing a series of R2Sn3S7 (R-SnS-1, Me4N+, Et4N+, QUIH+, DABCOH+) and R2Sn4S9 (R = Pr3NH+, BuNH3+) type compounds, where the organic cations existed in the pores and/or the interlamellar spacing.8 Furthermore, layered A2Sn3S7 (A = cations) hybrids can be used as a novel kind of ion exchanger to remove heavy metal ions,9–11 which could effectively avoid the instability of the organic ion exchanger in aqueous solution and the pH sensitivity of the inorganic ion exchanger.12 Some hybrid ion exchangers have been reported, such as [(CH3CH2CH2)2NH2]5In5Sb6S19·1.45H2O,13 [(CH3)2NH2]2Ga2Sb2S7·H2O,14 [(CH3)2NH2]2[GeSb2S6],15 and [(CH3)2NH2]0.75[Ag1.25SnSe3].16 Recently, Huang et al. successfully synthesized a new thiostannate [Me2NH2]4/3[Me3NH]2/3Sn3S7·1.25H2O (denoted as FJSM-SnS) to adsorb Cs+ and Sr2+,9 UO2+,10 and rare-earth elements.11 FJSM-SnS possesses a microporous layered structure with 2D [Sn3S7]n2n− layers stacked in AA sequence along the c-axis, and alkylammonium as counter cations existing in the interlayers. It has been revealed that FJSM-SnS, as a superior ion-exchanger, exhibits a high exchange rate, high adsorption capacity and wide pH resistance during the ion-exchange process. Compared with inorganic ion-exchangers having K+ and NH4+ as counterions,17–19 the alkylammonium chalcogenides with tunable size and conformationally flexible alkylammonium are promising ion-exchangers.
Photocatalysis is a thorough and eco-friendly approach for the degradation of toxic, carcinogenic and non-biodegradable pollutants. A highly efficient photocatalyst is required to have a broad photoresponse, effective separation of the photogenerated electrons and holes, and prominent adsorption capacity for reactants. To date, some inorganic chalcogenides, such as CdS,20 SnS2,21 Bi2S3,22 MoS2,23 and ZnIn2S4,24 have been developed as visible-light photocatalysts owing to their tunable band gaps and promising photocatalytic potential. However, the limited adsorption capacity seriously prevents their high performance photocatalysis,25–27 which is a notorious and tough problem that hampers performance improvement. Especially in the treatment of water-soluble organic pollutants, inorganic photocatalysts cannot effectively adsorb hydrophobic contaminants, which degrades the photocatalytic efficiency to some extent. Therefore, pre-adsorption is a prerequisite for efficient photocatalysis.28 Based on the excellent adsorption performance of layered A2Sn3S7 on the capture of heavy metal ions, it was proposed that the oleophilic group of interlayered alkylammonium could have a positive effect on the adsorption of organic pollutants. Meanwhile, it has been reported that organic cationic dyes, such as Methylene Blue (MB) or Safranin T (ST), have been introduced into the crystalline structure of 2D thiostannate (trenH)2Sn3S7 (tren = tris(2-aminoethyl)amine) by ion exchange with an adsorption capacity of 45 mg g−1.29 This revealed that the oleophility of the interlayered alkylammonium would be favourable to enhance the adsorption of organic contaminants and, replacing shorter chain alkylammonium with longer chain alkylammonium in the hybrid should be reasonable to enhance this adsorption. Furthermore, if the hybrid possesses an ion-exchange and photocatalytic ability at the same time, the organic pollutants could be further thoroughly degraded in an environmental-friendly way. These hybrids that combine a high adsorption capacity with excellent photocatalytic ability would be a new concept photocatalyst with a distinctive photocatalytic mechanism. However, to date, there are only a few studies that have focused on investigating alkylammonium thiostannate hybrids as high performance photocatalysts.
Herein, we synthesized a series of layered thiostannates, namely (baH)2Sn3S7 (ba = butylamine), (haH)2Sn3S7 (ha = hexylamine) and (oaH)2Sn3S7 (oa = octylamine), which are denoted as A2Sn3S7 (A = baH, haH, oaH), by a facile, one-pot solvothermal method. Using the organic cation dye Rhodamine B (RhB) as a prototypical pollutant, the ion-exchange and photocatalytic properties of A2Sn3S7 (A = baH, haH, oaH) were explored. The results indicated that the three thiostannates could quickly adsorb RhB through the ion-exchange reaction with enormous exchange capacities of 1047 mg g−1 to 1305 mg g−1. In addition, A2Sn3S7 could maintain their robust layered framework and effective photoabsorption over a wide pH range (1–11) during the ion-exchange processes. More importantly, A2Sn3S7 showed an excellent photodegradation ability for the adsorbed RhB within 15–60 min with a remarkable recycle stability, which demonstrated that A2Sn3S7 is a novel high-performance photocatalyst with a decoupled adsorption and photocatalysis mechanism, based on its inorganic–organic layered structure.
2. Experimental
2.1. Material synthesis
All reagents and chemicals were purchased from commercial sources and used without further purification. For the synthesis of (baH)2Sn3S7: a mixture of SnCl4·5H2O (8 mmol, 2.806 g), S (32 mmol, 1.026 g) in butylamine (12 mL) and water (4 mL) was stirred under ambient conditions until homogeneously mixed. The resulting mixture was sealed in a 50 mL stainless steel reactor with a Teflon liner, heated at 180 °C for 12 h and then cooled to room temperature. The final products were obtained by centrifugation, then washed with ethanol and vacuum dried. The synthesis procedures for (haH)2Sn3S7 and (oaH)2Sn3S7 were similar as (baH)2Sn3S7, except for the solvents used were hexylamine and octylamine, and the reaction time was 3 days and 5 days, respectively.
2.2. Characterization
Powder X-ray diffraction (PXRD) patterns were obtained on a PANalytical X Pert PRO MPD X-ray Powder Diffractometer working at 40 kV and 40 mA using Cu Kα radiation (1.541844 Å) in the angular range of 2θ = 3.5–60° at room temperature. Elemental analyses of C, H and N were carried out with a German Elementary Vario EL instrument. Inductively coupled plasma (ICP) atomic emission spectral analyses were performed with a SPECTRO axial-view inductively coupled plasma spectrometer. The morphology of the products was characterized by a Hitachi S-8010 field emission scanning electron microscope. X-ray photoelectron spectroscopy (XPS) measurements were conducted on an ESCALAB 250Xi X-ray photoelectron spectrometer with a Mg target (1283 eV). The thermogravimetric (TG) analyses were performed on a Mietler Toledo TGA/DSC thermogravimetric analyser with a 5 °C min−1 ramp from 25 to 700 °C in N2 gas. UV-visible (UV-Vis) diffuse reflection spectra were obtained with a PerkinElmer Lambda 950 UV/VIS/NIR spectrometer by using BaSO4 as a standard (100% reflectance) at room temperature. The UV-Vis absorption spectra were collected in the range from 300 to 700 nm on a Shimadzu UV-2600 spectrophotometer with the slit width of 1.0 nm. The steady state fluorescence excitation and emission spectra were performed on an Edinburgh-FS5 fluorescence spectrophotometer with the excitation and emission slit widths of 3.0 nm.
2.3. Ion-exchange experiments
Isotherm adsorption. A set of RhB solutions with a concentration ranging from 20 to 450 mg L−1 were prepared beforehand. A2Sn3S7 samples (30 mg) were added into RhB solutions (100 mL) at 333 K in an incubator shaker in the dark. Until equilibrium, the concentration of RhB was determined by UV-Vis absorption spectroscopy. Appropriate dilution was conducted, if necessary.
Adsorption kinetics. A2Sn3S7 samples (30 mg) were added into RhB solution (100 mL; 20 mg L−1), then the mixture was shaken in an incubator shaker at 298 K in the dark. During the experiments, an aliquot of the suspension (2 mL) was taken from the solution every five minutes, and filtered by a Nylon 66 microporous membrane. Finally, the concentration of the dye solution was determined by UV-Vis absorption spectroscopy at the calibrated maximum wavelength of 554 nm for RhB.
pH-dependent adsorption tests. RhB aqueous solutions with a pH value ranging from 1 to 11 were prepared by adding NaOH or HCl solution. Then the typical ion-exchange experiments were carried out within 30 min. The A2Sn3S7 hybrid load was still 0.3 g L−1.
2.4. Photocatalytic activity tests
Photocatalysis. The visible-light driven photocatalytic degradation reactions of RhB were used as probes to evaluate the catalytic performances of A2Sn3S7. All reactions were carried out at 298 K in a 250 mL quartz reactor. Prior to the irradiation, RhB solutions (100 mL; 20 mg L−1) containing the A2Sn3S7 samples (30 mg) were magnetically stirred in the dark to reach adsorption–desorption equilibrium, then were irradiated by a 300 W Xenon lamp (CHF-XM300) located 10 cm away from the suspension. A cutoff filter was equipped to remove UV light below 420 nm.
Photocatalytic kinetics. During the photocatalytic experiments, an aliquot of suspension (2 mL) was added to a 5 mL centrifuge tube at regular intervals before adding a mixed extractant (2 mL; vethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :vdichloromethane = 3:1). After sufficient oscillation and centrifugation, the supernatant was discarded and the remainder was diluted to 2 mL using the mixed extractant. The RhB concentration was determined by a fluorescence spectrophotometer at the maximum photoluminescence wavelength of 570 nm for RhB.
:vdichloromethane = 3:1). After sufficient oscillation and centrifugation, the supernatant was discarded and the remainder was diluted to 2 mL using the mixed extractant. The RhB concentration was determined by a fluorescence spectrophotometer at the maximum photoluminescence wavelength of 570 nm for RhB.
Photocatalytic cycle. The photocatalytic reaction system was expand to 300 mL of 20 mg L−1 of RhB solution with 90 mg of photocatalyst (baH)2Sn3S7. After the first cycle of the photocatalytic reaction, the dispersion solution was firstly centrifuged with high-speed centrifugal (8000 rpm) to discard the supernatant. Due to the hydrophobicity of catalyst, there was small amounts lost. Next, butyl amine was added as eluent to wash photocatalytic product away. Because of its lipophilicity, the photocatalyst could be sufficiently dispersed in organic amine, and settled to the bottom of the centrifuge tube during the high-speed centrifugal sedimentation, which effectively reduced the loss of catalyst during the washing process. After centrifugation, the eluent was discarded and the catalyst was washed with anhydrous ethanol until the pH value was neutral. After drying, the second cycle experiment was conducted until the fourth cycle experiment.
Adsorption–photocatalytic integration. RhB solutions (100 mL; 20 mg L−1) containing A2Sn3S7 samples (30 mg) were irradiated under visible light once magnetically stirred. The filter liquor (2 mL) was sampled and the RhB concentration was detected by UV-Vis absorption spectroscopy, immediately.
Photocatalytic mechanism. Ethylene diamine tetraacetic acid (1.0 mM; EDTA) as a quencher of holes, iso-propanol (1.0 mM; IPA) as a quencher of ·OH and p-benzoquinone (1.0 mM; BQ) as a quencher of ·O2− were employed in the photocatalytic process to trap active species of hole, ·OH, and ·O2− radicals, respectively, for the photocatalytic mechanism investigation.
3. Results and discussion
3.1. Structural characterization
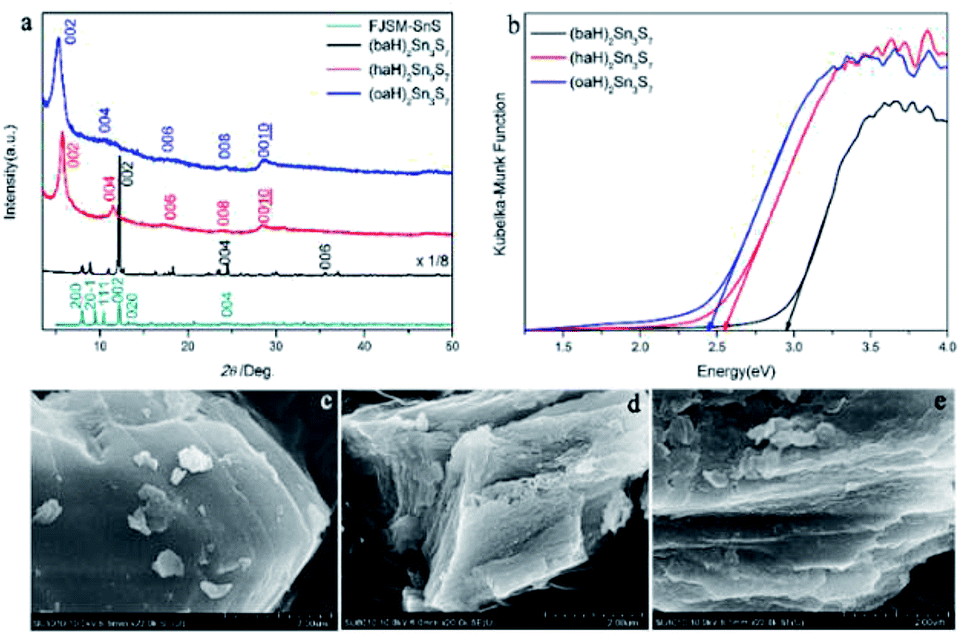
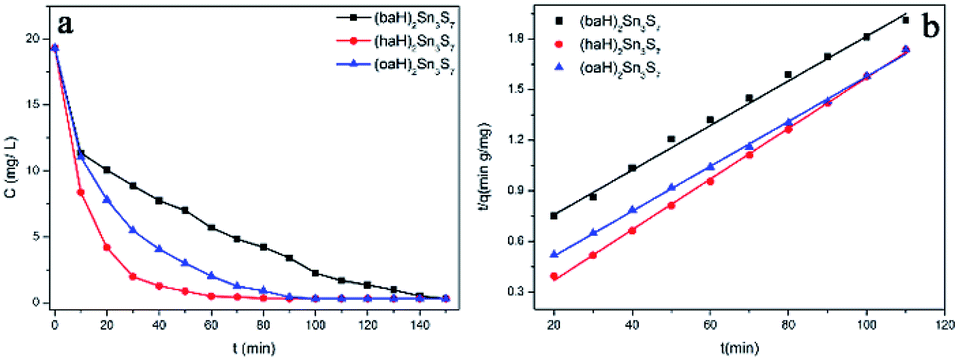
The XRD patterns of the as-prepared A2Sn3S7 are shown in Fig. 1a. The three thiostannates featured layered structures, which presented characteristic 00l diffraction peaks. Using FJSM-SnS as a reference,9 the strongest peaks of (baH)2Sn3S7 (2θ = 12.18°, 7.26 Å) could be assigned to the 002 diffraction, and the other diffraction peaks located at 24.47° (3.63 Å) and 37.04° (2.42 Å) corresponded to 004 and 006, respectively. Compared with (baH)2Sn3S7, the diffraction peak intensity of (haH)2Sn3S7 and (oaH)2Sn3S7 appeared weaker. A series of diffraction peaks located at 2θ = 5.73° (15.41 Å), 11.54° (7.66 Å), 17.24° (5.14 Å), 23.94° (3.85 Å), and 28.46° (3.13 Å) for (haH)2Sn3S7 and 5.29° (16.68 Å), 10.58° (8.36 Å), 18.71° (4.74 Å), 24.45° (3.63 Å), and 28.50° (3.13 Å) for (oaH)2Sn3S7, corresponded to the 002, 004, 006, 008, 0010 diffraction, respectively. These structures were similar to those of the tin sulphides FJMS-SnS,9 Cs-SnS-1,30 and DABCOH-SnS-1,31,32 which featured a microporous layered structure with 2D [Sn3S7]n2n− layers stacked in AA sequence along the c-axis, and alkylammonium as counter cations existing in the interlayers. The host–guest interactions consisted of anion–cation electrostatic forces and hydrogen bonding. The interlayer distances of (baH)2Sn3S7, (haH)2Sn3S7 and (oaH)2Sn3S7 increased as the organic amine chain grew, which was estimated to be 7.26 Å, 15.41 Å and 16.68 Å, respectively. Compared with the alkylammonium ion length, it was concluded that the interlayered alkylammonium ion adopted an all-trans interdigital configuration within the hybrid. These results indicated that the open inorganic framework of the [Sn3S7]n2n− layers could adjust their lattice parameters to adopt the various counter cations. The UV-Vis absorption spectra were used to determine the band gaps of A2Sn3S7, which were approximately 2.95 eV, 2.56 eV and 2.45 eV for (baH)2Sn3S7, (haH)2Sn3S7 and (oaH)2Sn3S7, respectively (Fig. 1b). This demonstrated that the hybrids could absorb the UV-Vis light effectively. Furthermore, the scanning electron microscopy (SEM) analyses provided visual evidence for the layered nature of the A2Sn3S7 hybrid (Fig. 1c–e), in accordance with the results of XRD mentioned above.
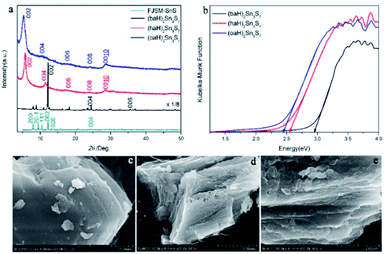 |
| | Fig. 1 The PXRD patterns of A2Sn3S7 (a); solid-state optical adsorption spectra (b), SEM patterns of (baH)2Sn3S7 (c), (haH)2Sn3S7 (d), and (oaH)2Sn3S7 (e). | |
Meanwhile, the elemental contents of C, H, N and Sn listed in Table S1 in ESI† were used to confirm the formula of these thiostannates. All element data were consistent with the value calculated from the formula A2Sn3S7, which demonstrated (baH)2Sn3S7, (haH)2Sn3S7 and (oaH)2Sn3S7 compounds were successfully synthesized, and the three compounds could be represented by A2Sn3S7 (A = baH, haH, oaH).
To verify the valence state of the Sn and S elements of the hybrids, high resolution X-ray photoelectron spectra (XPS) was performed to obtain the Sn 3d and S 2p spectra, all the three hybrids exhibited the similar spectra and those of the (baH)2Sn3S7 were shown in Fig. S1 ESI.† The evolution of Sn 3d with two strong peaks was observed at 495.3 and 486.8 eV, which were attributed to Sn 3d3/2 and Sn 3d5/2, respectively. The high-resolution S 2p core level analysis at binding energies of 162.5 and 161.5 eV corresponded to S 2p1/2 and S 2p3/2. The binding energies (Table S2 in ESI†) of the A2Sn3S7 revealed that all the Sn species were present in the 4+ oxidation state, while all the S species were present in the 2− state. Therefore, XPS analysis further supported the formula of A2Sn3S7.
To test the thermal stability of A2Sn3S7 (A = baH, haH, oaH), TGA analyses were carried out and depicted in Fig. S2 of ESI.† The thermogravimetric curve demonstrated that (baH)2Sn3S7 exhibited the best thermal stability, as it could totally maintain the integrity of structure and composition below 220 °C, which was related to its excellent crystallinity. (haH)2Sn3S7 and (oaH)2Sn3S7 were almost stable below 100 °C owing to their poor crystallinity, although their thermal stability could still satisfy the requirements of ion-exchangers and photocatalysts in conventional wastewater treatment.
3.2. Kinetic and isotherm studies of ion exchange
RhB was selected as a prototype for the kinetic and isotherm studies of ion exchange. The kinetics of RhB ion exchange at 298 K were studied. Fig. 2a shows that the RhB concentration decreased sharply within the first few minutes, then decreased slightly to near zero, which demonstrated that A2Sn3S7 (A = baH, haH, oaH) could effectively adsorb RhB. Among the three thiostannates, (haH)2Sn3S7 exhibited the fastest adsorption rate while (baH)2Sn3S7 possessed the slowest rate. Fig. 2b shows the kinetic fitting curves of A2Sn3S7 for RhB adsorption with initial concentrations of 20 mg L−1. The pseudo-second order kinetic model was used to fit the experiment data.
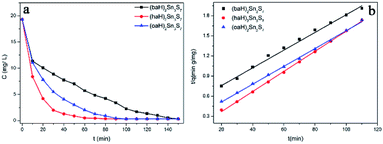 |
| | Fig. 2 Kinetics of RhB ion-exchange of A2Sn3S7, RhB concentration C (mg L−1) vs. the time (min) (a); pseudo-second-order model for A2Sn3S7 (b). | |
The equation is expressed as:
| |
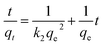 | (1) |
where
qt (mg g
−1) is the adsorption capacity at time
t (min), while
qe (mg g
−1) is the amount adsorbed at equilibrium.
k2 is the kinetic rate constant (g mg
−1 min
−1). According to the linear fitting results, all the correlation coefficients (
R2) exceeded 0.99, which implied that the RhB adsorption on A
2Sn
3S
7 could be perfectly fitted with the pseudo second-order kinetic model, and the rate-limiting step of the adsorption was chemical adsorptions. The mechanism of adsorption was more likely to be ion exchange. The calculated kinetics parameters are given in
Table 1. The kinetic rate constants
k2 were 0.34 × 10
−3, 3.19 × 10
−3, and 0.71 × 10
−3 g mg
−1 min
−1 for (baH)
2Sn
3S
7, (haH)
2Sn
3S
7, and (oaH)
2Sn
3S
7, respectively, namely
k2[(haH)
2Sn
3S
7] >
k2[(oaH)
2Sn
3S
7] >
k2[(baH)
2Sn
3S
7]. This phenomenon could be attributed to the combined effect of interlayer distance and dispersion of A
2Sn
3S
7 in an aqueous solution. Although the expansion of interlayer distance would facilitate the ion exchange between RhB cation ions and alkylammonium ions, the longer chain length of the organic amine would enhance the affinity with organic RhB cation and weaken the dispersibility of A
2Sn
3S
7 in aqueous solution at the same time, consequently, retarding the integrated ion-exchange rate.
Table 1 Adsorption kinetics parameters for A2Sn3S7
| |
Pseudo-second-order |
| k2 (10−3 g mg−1 min−1) |
qe (mg g−1) |
R2 |
| (baH)2Sn3S7 |
0.34 |
76.33 |
0.9939 |
| (haH)2Sn3S7 |
3.19 |
66.67 |
0.9992 |
| (oaH)2Sn3S7 |
0.71 |
75.24 |
0.9989 |
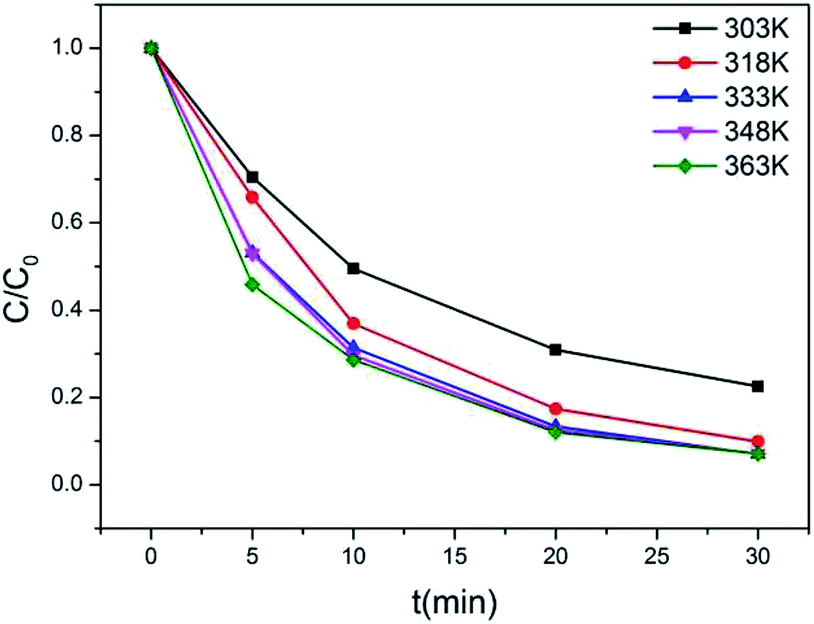
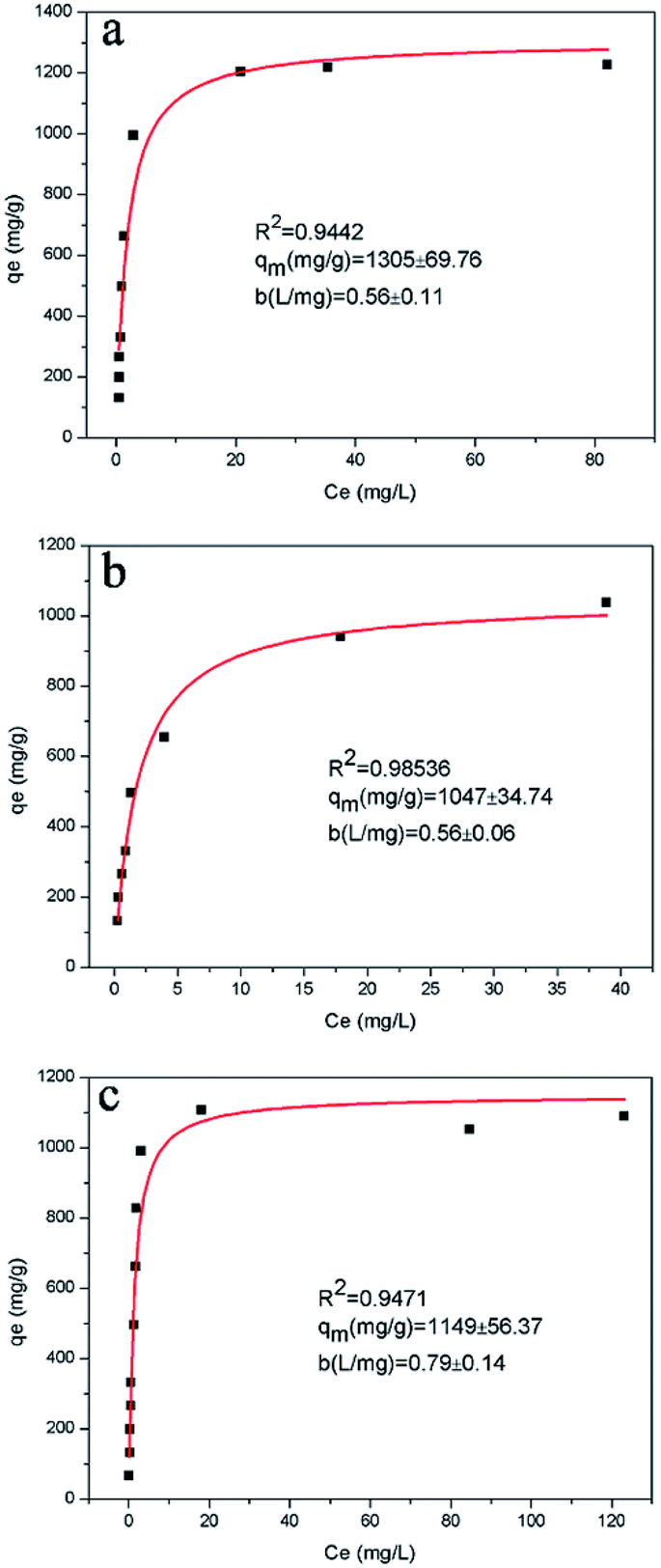
According to the results of the kinetic experiments, A2Sn3S7 (A = baH, haH, oaH) could adsorb RhB by an ion-exchange reaction. To evaluate the adsorption capacity of A2Sn3S7, isotherm experiments with an initial concentration ranging from 20 to 450 mg L−1 of RhB solution in neutral conditions was carried out. However, we found that the adsorption rate reduced with the increase of concentration of RhB. In order to seek for the suitable adsorption temperature, (haH)2Sn3S7 was used to study the relationship between adsorption rate and temperature (Fig. 3). The results showed that the adsorption rate accelerated significantly at 333 K, almost consistent with the effects at 348 K and 363 K. Therefore, the final isothermal adsorption temperature was determined to be 333 K. The Langmuir equilibrium equation is expressed as:
| |
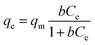 | (2) |
where
qe (mg g
−1) is the adsorption capacity at equilibrium,
Ce (mg L
−1) is the equilibrium concentration;
qm (mg g
−1) is the maximum adsorption capacity, and
b (L mg
−1) is the Langmuir affinity constant related to the binding strength. The equilibrium curve of RhB is shown in
Fig. 4, which describes the capacity of RhB-exchange (
qe)
vs. the concentration of RhB (
Ce) at equilibrium. Nonlinear fitting indicated that the Langmuir isotherm model was appropriate to describe the adsorption behaviour of A
2Sn
3S
7 towards RhB with
R2 = 0.9442, 0.9854 and 0.9471 corresponding to (baH)
2Sn
3S
7, (haH)
2Sn
3S
7 and (oaH)
2Sn
3S
7, respectively. The Langmuir isotherm model is based on the assumptions that the adsorption surface is homogeneous and each site can capture only one ion, which is a monolayer-type adsorption. The value of
qm was calculated to be 1305 ± 69.76 mg g
−1, 1047 ± 34.74 mg g
−1 and 1149 ± 56.37 mg g
−1 for (baH)
2Sn
3S
7, (haH)
2Sn
3S
7 and (oaH)
2Sn
3S
7, respectively. The order of
qm values was in agreement with the trend of
qe in the kinetic experiments (76.33 for (baH)
2Sn
3S
7, 66.67 for (haH)
2Sn
3S
7, and 75.24 mg g
−1 for (oaH)
2Sn
3S
7). Therefore, the adsorption capacity was also related to the active adsorption site of the ion-exchanger. In addition, to demonstrate the excellent adsorption performance of A
2Sn
3S
7, the maximal adsorption capacity towards RhB was compared with some common adsorbents (see Table S3 in ESI
†).
33–40 The comparative results revealed that the A
2Sn
3S
7 (A = baH, haH, oaH) exhibited a very high RhB ion-exchange capacity which was mainly attributed to the affinity of interlayered alkylammonium and the electrostatic interaction of 2D [Sn
3S
7]
n2n− layers with the organic RhB cation.
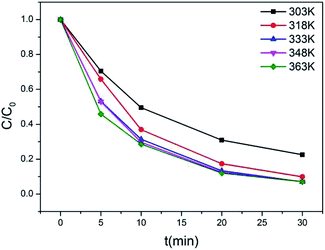 |
| | Fig. 3 Kinetics of RhB (20 mg L−1) ion-exchange of (haH)2Sn3S7 at different temperature. | |
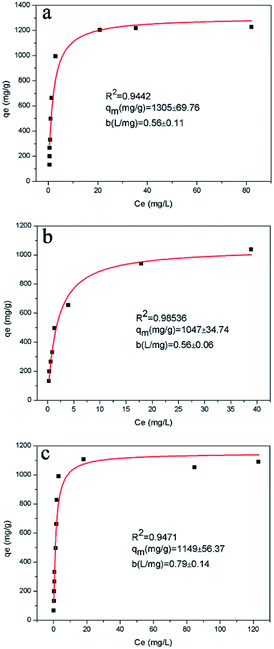 |
| | Fig. 4 RhB equilibrium curves for A2Sn3S7 at 333 K, Langmuir equilibrium isotherms qe vs. Ce for (baH)2Sn3S7 (a), (haH)2Sn3S7 (b), and (oaH)2Sn3S7 (c). | |
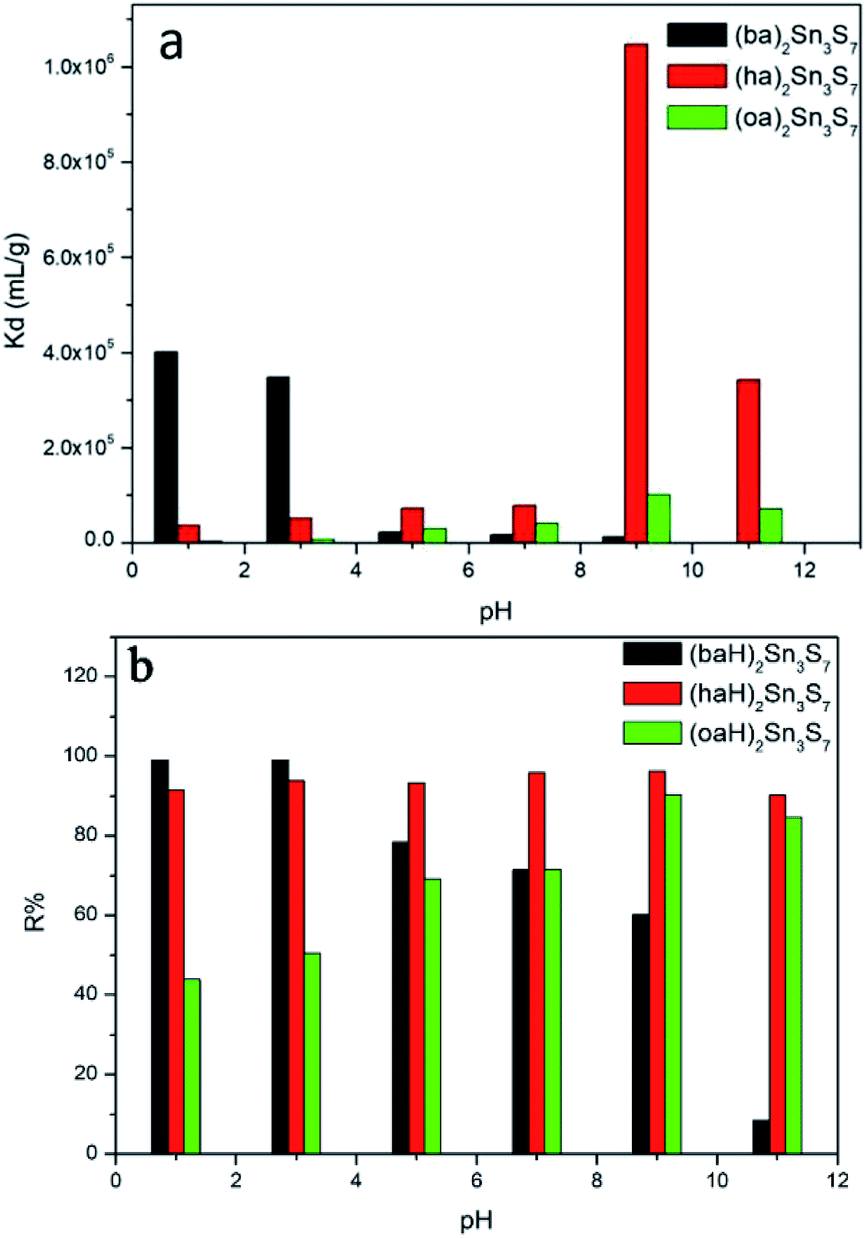
The pH resistance is another important evaluative parameter for a good adsorbent. In general, a material with Kd (distribution coefficient) greater than 104 mL g−1 is regarded as an excellent adsorbent. Fig. 5a shows the Kd of A2Sn3S7 in a RhB solution with different pH. Since the Kd value of (haH)2Sn3S7 in the solution of pH ≈ 9 and 11 were too large, we used a third of that value to plot. As shown in Fig. 5a, the Kd value of (baH)2Sn3S7 was determined as 4.0 × 105 mL g−1 when pH ≈ 1, then the value decreased with the increase of pH until pH ≈ 9 (Kd = 1.32 × 104 mL g−1). It was possible that in a strong alkaline solution, the electrostatic repulsive-force between –COO− of RhB and 2D [Sn3S7]n2n− layers prevented the RhB cation from entering into the narrow amine layers of (baH)2Sn3S7. For (haH)2Sn3S7 and (oaH)2Sn3S7, the trend was reversed. In the solution with pH ≈ 1, the Kd were determined as 3.57 × 104 mL g−1 for (haH)2Sn3S7 and 2.89 × 103 mL g−1 for (oaH)2Sn3S7, owing to the high concentration of H+ as the competitive ions. With the increase of pH, both adsorbents displayed outstanding adsorption capability. Especially when pH ≈ 9, the Kd reached a maximum of 3.57 × 106, and 1.01 × 105 mL g−1, owing to the decrease of the H+ concentration, then when the pH reached 11, the Kd slightly decreased to 1.02 × 105 and 7.19 × 104 mL g−1 for (haH)2Sn3S7 and (oaH)2Sn3S7, respectively, owing to the partial structure destruction. Therefore, the pH resistance data demonstrated that A2Sn3S7 (A = baH, haH, oaH) was a good adsorbent over a wide pH solution range (pH = 1–11). Meanwhile, the RhB removal efficiency (R%) vs. pH was shown in Fig. 5b, which further illustrated that (baH)2Sn3S7 was more suitable to remove RhB in acidic conditions, while (oaH)2Sn3S7 preferred to eradicate RhB in basic conditions and the hybrids were structural stable and maintained an excellent ion-exchange performance over a wide pH range (Fig. S3 in ESI†).
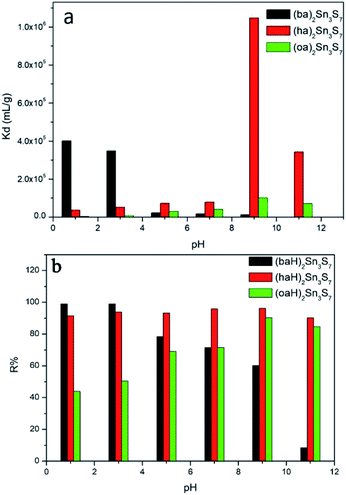 |
| | Fig. 5 Kd of A2Sn3S7 in RhB solution (a) and removal rates R% of RhB (b) at different pH. | |
3.3. Photocatalytic activities
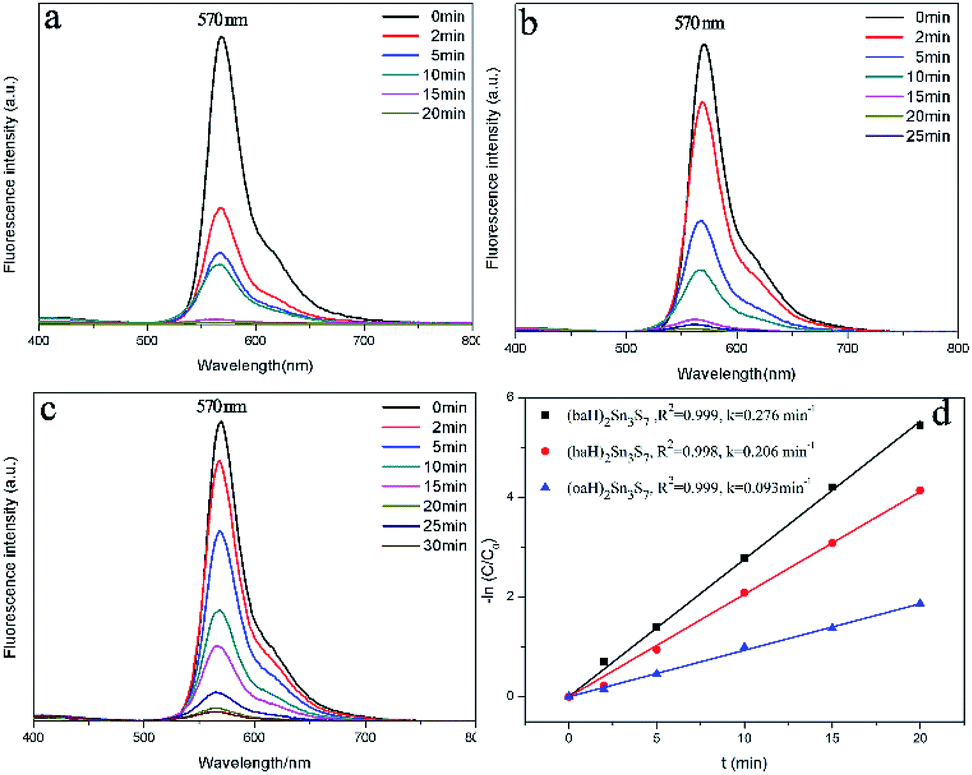
In addition to the high capacity and wide pH resistance of A2Sn3S7, the photocatalytic ability to degrade organic RhB cation is discussed. All the photocatalytic experiments were carried out at 298 K in a 250 mL quartz reactor. RhB solutions (100 mL; 20 mg L−1) containing the A2Sn3S7 sample (30 mg) were magnetically stirred in the dark to reach the adsorption–desorption equilibrium, then irradiated by a 300 W Xenon lamp (λ > 420 nm). During the kinetic experiments, the UV-Vis spectrophotometer failed to detect the concentration variation of RhB in the aqueous solution because RhB had been strongly adsorbed between the amine layers of A2Sn3S7.
To detect the RhB concentration at different reaction time, we adopted a new method, called dispersive liquid–liquid microextraction (DLLME).41,42 To be more specific, the mixture extraction agent consisting of absolute ethyl alcohol and dichloromethane were used to extract the adsorbed but undegraded RhB in the interlayers. The extraction process was repeated for twice to guarantee the complete collection of undegraded RhB molecules, detected by the fluorescence of the extraction solution (Fig. S4 in ESI†). The fluorescence spectra were obtained, with excitation wavelength at 294 nm and both excitation and emission slit widths set to 3 nm, to capture the emission band intensity of RhB during the photocatalytic process. The change of fluorescence intensity at 570 nm made it possible to determine a variation of the concentration of RhB. Fig. 6a–c shows the fluorescence spectra change of RhB in the extracting solution. From the fluorescence spectra, (baH)2Sn3S7 was able to thoroughly degrade RhB within 15 min, (haH)2Sn3S7 needed 20 min and (oaH)2Sn3S7 needed approximately 30 min to remove RhB by a photocatalytic reaction. The Langmuir–Hinshelwood (L–H) apparent first-order kinetics model was adopted to describe the photocatalytic ability of A2Sn3S7. The L–H equation is described as follows:
| |
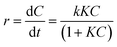 | (3) |
where
r is the degradation rate of the reactant (mg L
−1 min
−1),
C is the concentration of the reactant (mg L
−1),
t is the visible light irradiation time (min),
k is the reaction rate constant (mg L
−1 min
−1), and
K is the adsorption coefficient of the reactant (L mg
−1). When the initial concentration (
C0) is very low (
C0 = 20 mg L
−1 for RhB in the present experiments),
eqn (3) could be simplified to an apparent first-order model:
| |
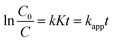 | (4) |
where
kapp is the apparent first-order rate constant (min
−1).
Fig. 6d shows that a linear relation between ln(
C0/
C) and time was observed for all the photocatalysts, which indicated that the photocatalytic degradation of RhB was well fitted to pseudo-first-order kinetics, and the apparent rate
kapp was 0.276 min
−1 for (baH)
2Sn
3S
7, 0.206 min
−1 for (haH)
2Sn
3S
7, and 0.093 min
−1 for (oaH)
2Sn
3S
7. The results revealed that RhB could be thoroughly degraded by an A
2Sn
3S
7 (A = baH, haH, oaH) photocatalyst in a short period of time. In addition to the high adsorption capacity, which provided the continuous supply of target pollutants, the semiconductor property of the 2D [Sn
3S
7]
n2n− layers could respond to visual light and produce a separated photogenerated electron and hole to degrade the organic contaminant. Essentially, the high-performance integrated adsorption–photodegradation ability was ascribed to the unique hybrid structure of A
2Sn
3S
7.
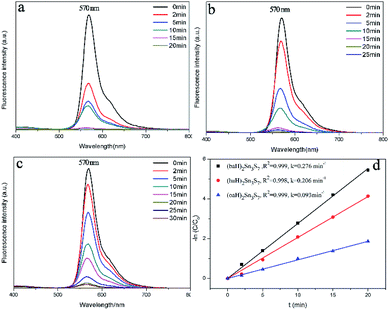 |
| | Fig. 6 (a) (baH)2Sn3S7, (b) (haH)2Sn3S7, (c) (oaH)2Sn3S7 fluorescence spectral changes of the RhB extracting solution; (d) pseudo-first-order model for A2Sn3S7 in the photocatalytic process. | |
To test the stability and reusability of A2Sn3S7, (baH)2Sn3S7 was reused four times for the photocatalytic reaction under the same conditions. The results are shown in Fig. 7. There was no significant decrease in the photocatalytic activity of (baH)2Sn3S7 after the four consecutive photocatalytic degradation cycles, which indicated that (baH)2Sn3S7 was stable and could be used repeatedly. Moreover, the XRD demonstrated that the structure of (baH)2Sn3S7 after four consecutive cycles was consistent with the original structure (Fig. S5 in ESI†). The colored photos of (baH)2Sn3S7 powder undergo ion exchange, photocatalysis and elution in a cycle experiment were shown in Fig. S6 in ESI.† Thus, (baH)2Sn3S7 showed remarkable stability and reproducibility for photocatalysis.
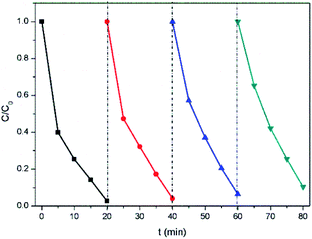 |
| | Fig. 7 Recycling test of (baH)2Sn3S7 in photocatalytic degradation of RhB. | |
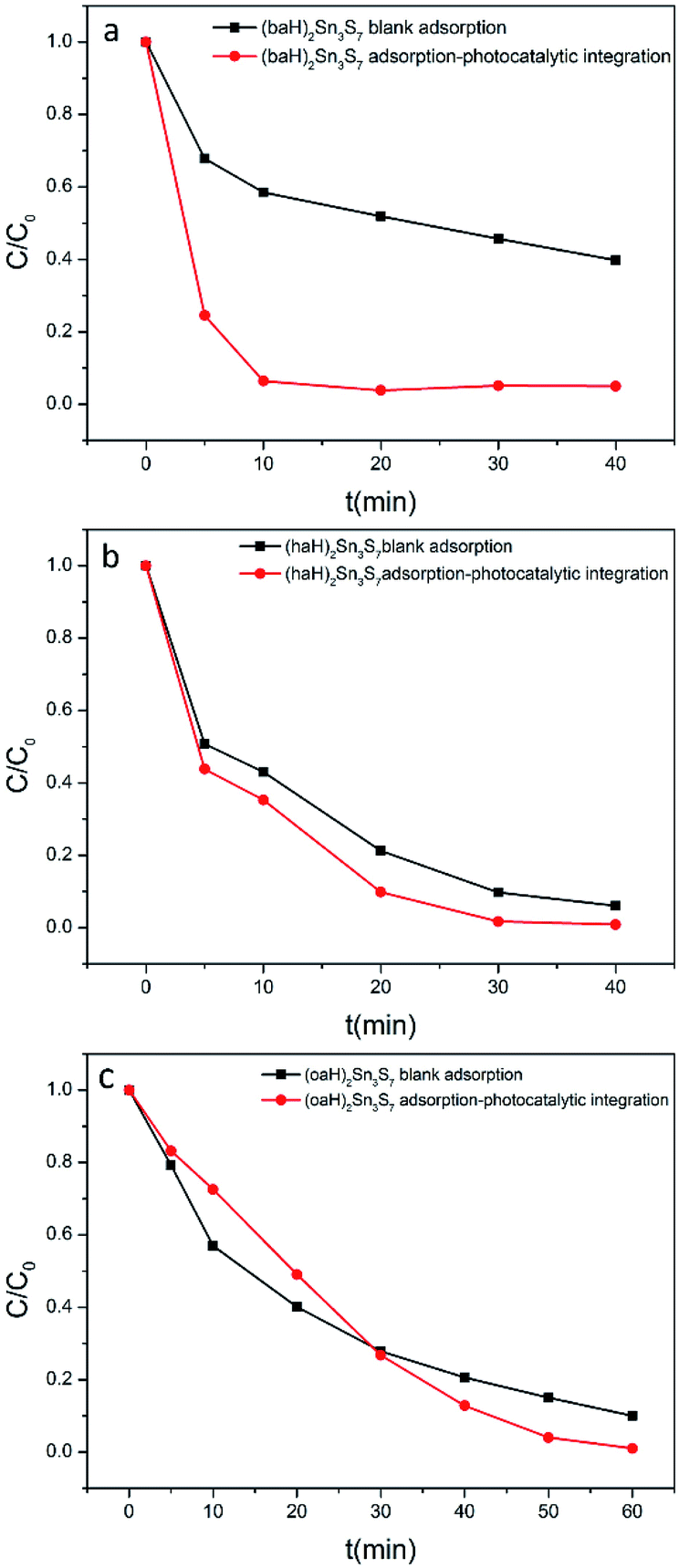
In a real sewage treatment process, the effects of adsorption and photocatalysis synergistically occur. To test the overall performance of A2Sn3S7, integrated adsorption–photocatalysis experiments were conducted. In fact, the RhB degradation efficiency in the integrated experiment is actually determined by the adsorption efficiency and photocatalytic efficiency of A2Sn3S7. If the adsorption capacity can meet the demand of photocatalytic degradation, the degradation efficiency will be greatly improved (Fig. 8). In a dark environment, the time for the total adsorption of RhB was 150 min, 60 min and 90 min when using (baH)2Sn3S7, (haH)2Sn3S7 and (oaH)2Sn3S7 as adsorbents, respectively (Fig. S7 in ESI†). However, in the integrated adsorption–photocatalysis experiments, the time for RhB to be thoroughly degraded was 15 min, 30 min and 60 min for (baH)2Sn3S7, (haH)2Sn3S7 and (oaH)2Sn3S7, respectively. While, if the adsorption amount is insufficient, the degradation efficiency will decrease in a short time. Exactly as (oaH)2Sn3S7, in the first 30 min, the degradation efficiency was slower owning to the poor adsorption. This definitely demonstrated that the degradation of RhB could be accelerated when adsorption and photocatalysis occurred synergistically, which indicated that A2Sn3S7 photocatalysts with remarkable adsorbability could realize a rapid photocatalytic degradation of RhB under visible light.
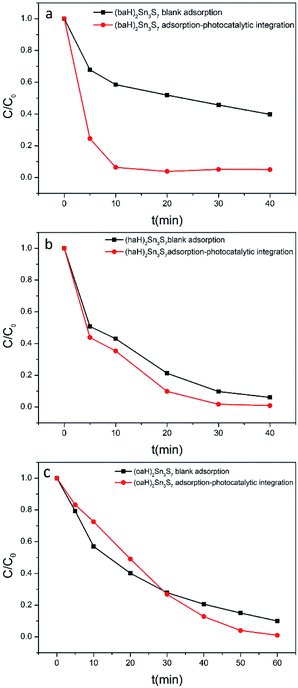 |
| | Fig. 8 Kinetics of RhB in dark environment and integrated adsorption–photocatalysis experiments. | |
3.4. Photocatalytic mechanism
According to the UV-visible spectra of these hybrids, it can be found that the absorption edge was in the order of (baH)2Sn3S7 (2.95 eV, 420 nm) > (haH)2Sn3S7 (2.56 eV, 484 nm) > (oaH)2Sn3S7 (2.45 eV, 506 nm). It is very possible that this order was related with the interaction between adjacent [Sn3S72−] layers with their different spacing, given the identical intralayer structure of the [Sn3S72−] layers. Upon the visible light illumination (>420 nm), the photodegradation rate sequence for Rhodamine B is same like the above order (Fig. 8), although the adsorption rate followed the different order, which indicates that the band edge excitons within the (baH)2Sn3S7 is more efficient for the photodegradation reaction, compared with that hot excitons in the (haH)2Sn3S7 and (oaH)2Sn3S7. Of course, more depth investigation should be carried out to reveal the details of the relation between the light absorption, exciton energy and the photodegradation performance of these hybrids. This feature was also an implication of the independence between the adsorption and photodegradation process for this unique inorganic/organic hybrid.
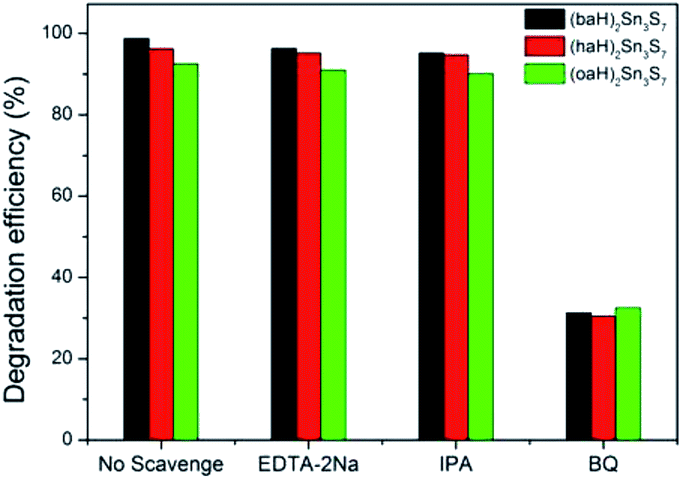
To investigate the mechanism of the photocatalytic process, the trapping experiments of the main active species h+, ·OH, and ·O2− radicals were designed by adding 1.0 mmol L−1 EDTA (a quencher of holes), 1.0 mmol L−1 IPA (a quencher of ·OH) and 1.0 mmol L−1 BQ (a quencher of ·O2−), respectively, into the system.
The degradation activity was slightly changed in the presence of EDTA or IPA, which indicated that h+ and ·OH had a negligible effect on the photodegradation of RhB. However, when BQ was added, the degradation efficiency sharply reduced, which meant the photocatalytic process was restrained43 (Fig. 9). Therefore, the ·O2− radicals were the decisive active species in the degradation of RhB, and the degradation mechanism of A2Sn3S7 could be concluded as follows:
| | |
0(A2Sn3S7) − hν → h+ (S−) + e− (Sn3+)
| (5) |
| | |
·O2− + RhB+ → degradation products
| (7) |
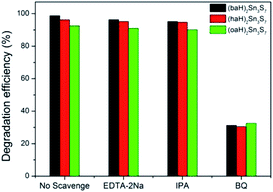 |
| | Fig. 9 Photocatalytic activities of the A2Sn3S7 for the degradation of RhB solution in the presence of different trapping agents. | |
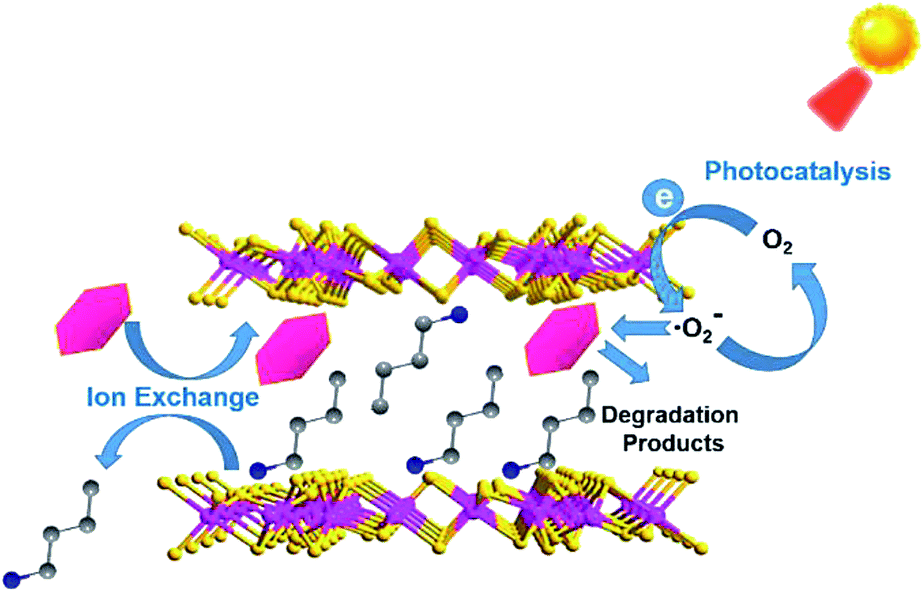
Thus, there was a possible photocatalytic mechanism proposed according to the aforementioned experimental results and this is summarized and shown in Scheme 1. The reason why A2Sn3S7 (A = baH, haH, oaH) could realize a rapid photodegradation of RhB was attributed to their unique layered structures. RhB could exchange with interlayered alkylammonium to realize its rapid accumulation within the interlayers of the hybrid. Furthermore, under illumination, photogenerated electrons from the (Sn3S7)n2n− anion layers could be quickly captured by dissolved oxygen to produce ·O2− radical, which could then attack adjacent adsorbed RhB molecules within the nanometer scale. Finally, the carcinogenic and nonbiodegradable organic dye RhB was thoroughly photodegraded. This hybrid outperformed traditional photocatalysts with decoupled adsorption and photocatalytic properties, and can be used as an adsorbent in the dark and as a photocatalyst in daylight, which is well compatible for the intermittent feature of solar light.
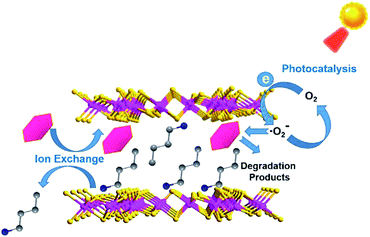 |
| | Scheme 1 Possible photocatalytic mechanism in A2Sn3S7. | |
4. Conclusions
In conclusion, a series of layered thiostannates A2Sn3S7 (A = baH, haH, oaH), with integrated high adsorption and fast photocatalysis, could be synthesized by a facile, one-pot solvothermal method. The structure of layered A2Sn3S7 (A = baH, haH, oaH) was characterized by the periodic arrangement of 2D [Sn3S7]n2n− layers and interlayered alkylammonium. Based on the unique hybrid structure, A2Sn3S7 (A = baH, haH, oaH) would be able to efficiently degrade organic cationic pollutants in sewage water, like RhB. The ion-exchange studies targeted at RhB revealed that A2Sn3S7 exhibited a remarkable ion-exchange capacity and wide pH resistance. The photocatalytic experiments demonstrated that A2Sn3S7 exhibited excellent photocatalytic activity and reusability. For conventional photocatalysts, the effect of photocatalysis is more decisive than adsorption, which means that these photocatalysts are only available in the daytime. However, the novel A2Sn3S7 (A = baH, haH, oaH) photocatalysts synthesized in this paper can decouple the adsorption from photocatalysis to realize their independent functions. During the day, A2Sn3S7 (A = baH, haH, oaH) acts as photocatalyst to rapidly and thoroughly degrade organic pollutants, and at night, it can serve as a superior ion-exchanger to adsorb contaminants to prepare for the next photocatalysis cycle. It is expected that these multifunctional A2Sn3S7 materials could be developed to deal with larger organic cationic pollutants, like dyes and pigments. More research is needed to degrade other nonbiodegradable organic contaminants. These hybrids outperform traditional photocatalysts with decoupled adsorption and photocatalytic properties, and can be used as adsorbents in the dark and as photocatalysts in daylight, which makes them compatible with the intermittent nature of solar light.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Notes and references
- K. Y. Wang, M. L. Feng, L. J. Zhou, J. R. Li, X. H. Qi and X. Y. Huang, Chem. Commun., 2014, 50, 14960–14963 RSC.
- X. Y. Huang and J. Li, J. Am. Chem. Soc., 2000, 122, 8789–8790 CrossRef CAS.
- Y. F. Sun, Z. H. Sun, S. Gao, H. Cheng, Q. H. Liu, J. Y. Piao, T. Yao, C. Z. Wu, S. L. Hu, S. Q. Wei and Y. Xie, Nat. Commun., 2012, 3, 1057 CrossRef PubMed.
- J. F. Xie, S. Li, R. X. Wang, H. Zhang and Y. Xie, Chem. Sci., 2014, 5, 1328–1335 RSC.
- R. L. Bedard, S. T. Wilson, L. D. Vail, J. M. Bennett and E. M. Flanigen, in Zeolites: Facts, Figures, Future, Proceedings of the 8th International Zeolite Conference, ed. P. A. Jacobs and R. A. van Santen, Elsevier, Amsterdam, 1989, pp. 375–387 Search PubMed.
- W. S. Sheldrick and M. Wachhold, Coord. Chem. Rev., 1998, 176, 211–322 CrossRef CAS.
- T. Jiang, A. Lough, G. A. Ozin, R. L. Bedard and R. Broach, J. Mater. Chem., 1998, 8, 721–732 RSC.
- T. Jiang, A. Lough, G. A. Ozin and R. L. Bedard, J. Mater. Chem., 1998, 8, 733–741 RSC.
- X. H. Qi, K. Z. Du, M. L. Feng, J. R. Li, C. F. Du, B. Zhang and X. Y. Huang, J. Mater. Chem. A, 2015, 3, 5665–5673 RSC.
- M. L. Feng, D. Sarma, X. H. Qi, K. Z. Du, X. Y. Huang and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 12578–12585 CrossRef CAS PubMed.
- X. H. Qi, K. Z. Du, M. L. Feng, Y. J. Gao, X. Y. Huang and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 4314–4317 CrossRef CAS PubMed.
- M. L. Feng, K. Y. Wang and X. Y. Huang, Chem. Rec., 2016, 16, 582–600 CrossRef CAS PubMed.
- N. Ding and M. G. Kanatzidis, Chem. Mater., 2007, 19, 3867–3869 CrossRef CAS.
- N. Ding and M. G. Kanatzidis, Nat. Chem., 2010, 2, 187–191 CrossRef CAS PubMed.
- M. L. Feng, D. N. Kong, Z. L. Xie and X. Y. Huang, Angew. Chem., Int. Ed. Engl., 2008, 47, 8623–8626 CrossRef CAS PubMed.
- J. R. Li and X. Y. Huang, Dalton Trans., 2011, 40, 4387–4390 RSC.
- J. L. Mertz, Z. H. Fard, C. D. Malliakas, M. J. Manos and M. G. Kanatzidis, Chem. Mater., 2013, 25, 2116–2127 CrossRef CAS.
- M. J. Manos, C. D. Malliakas and M. G. Kanatzidis, Chem.–Eur. J., 2007, 13, 51–58 CrossRef CAS PubMed.
- P. Sengupta, N. L. Dudwadkar, B. Vishwanadh, V. Pulhani, R. Rao, S. C. Tripathi and G. K. Dey, J. Hazard. Mater., 2014, 266, 94–101 CrossRef CAS PubMed.
- T. Y. Zhai, X. S. Fang, L. Li, Y. Bando and D. Golberg, Nanoscale, 2010, 2, 168–187 RSC.
- G. W. Li, R. Su, J. C. Rao, J. Q. Wu, P. Rudolf, G. R. Blake, R. A. deGroot, F. Besenbacher and T. T. M. Palstra, J. Mater. Chem. A, 2016, 4, 209–216 RSC.
- L. L. Long, A. Y. Zhang, Y. X. Huang, X. Zhang and H. Q. Yu, J. Mater. Chem. A, 2015, 3, 4301–4306 RSC.
- W. J. Zhou, Z. Y. Yin, Y. P. Du, X. Huang, Z. Y. Zeng, Z. X. Fan, H. Liu, J. Y. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed.
- L. Su, X. J. Ye, S. G. Meng, X. L. Fu and S. F. Chen, Appl. Surf. Sci., 2016, 384, 161–174 CrossRef CAS.
- Y. M. Xu and C. H. Langford, J. Phys. Chem., 1995, 99, 11501–11507 CrossRef CAS.
- T. Torimoto, S. Ito and S. Yoneyama, Environ. Sci. Technol., 1996, 30, 1275–1281 CrossRef CAS.
- M. Takeuchi, T. Kimura, M. Hidaka, D. Rakhmawaty and M. Anpo, J. Catal., 2007, 246, 235–240 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS.
- M. S. Hvid, P. Lamagni and N. Lock, Sci. Rep., 2017, 7, 45822 CrossRef CAS PubMed.
- G. A. Marking and M. G. Kanatzidis, J. Mater. Chem., 1995, 7, 1915–1921 CrossRef CAS.
- T. Jiang, A. Lough, G. A. Ozin and R. L. Bedard, J. Mater. Chem., 1998, 8, 733–741 RSC.
- T. Jiang, A. Lough, G. A. Ozin, R. L. Bedard and R. Broach, J. Mater. Chem., 1998, 8, 721–732 RSC.
- C. Yang, S. C. Wu, J. H. Cheng and Y. C. Chen, J. Alloys Compd., 2016, 687, 804–812 CrossRef CAS.
- K. Han, W. Wei, Z. F. Jiang, J. W. Lu, J. J. Zhu and J. M. Xie, Colloids Surf., A, 2016, 509, 539–549 CrossRef.
- T. Bhowmik, M. K. Kundu and S. Barman, RSC Adv., 2015, 5, 38760–38773 RSC.
- J. N. Tiwari, K. Mahesh, N. H. Le, K. C. Kemp, R. Timilsina, R. N. Tiwari and K. S. Kim, Carbon, 2013, 56, 173–182 CrossRef CAS.
- X. J. Bian, X. F. Lu, Y. P. Xue, C. C. Zhang, L. R. Kong and C. Wang, J. Colloid Interface Sci., 2013, 406, 37–43 CrossRef CAS PubMed.
- S. Y. Wang, B. Yang and Y. P. Liu, J. Colloid Interface Sci., 2017, 507, 225–233 CrossRef CAS PubMed.
- X. H. Wang, J. J. Ding, S. W. Yao, X. X. Wu, Q. Q. Feng, Z. H. Wang and B. Y. Geng, J. Mater. Chem. A, 2014, 2, 15958–15963 RSC.
- X. Q. Gao, F. Xiao, C. Yang, J. D. Wang and X. T. Su, J. Mater. Chem. A, 2013, 1, 5831–5834 RSC.
- M. Rezaee, Y. Assadi, M. R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- N. Xiao, J. Deng, K. H. Huang, S. Q. Ju, C. H. Hu and J. Liang, Spectrochim. Acta, Part A, 2014, 128, 312–318 CrossRef CAS PubMed.
- J. M. Wu and T. W. Zhang, J. Photochem. Photobiol., A, 2004, 162, 171–177 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The full elements analyses, XPS, TGA of A2Sn3S7, comparison of the adsorption capacities on various adsorbents for RhB. PXRD of (baH)2Sn3S7 catalyst in cycle process. See DOI: 10.1039/c9ra01486a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Jun Lu
*a and
Jun Lu