
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of graphene-nanoplatelets on gelation and structural integrity of a polyvinyltrimethoxysilane-based aerogel†
Solmaz Karamikamkar ac,
Abdelnasser Abidlia,
Ehsan Behzadfarb,
Sasan Rezaeia,
Hani E. Naguibc and
Chul B. Park*a
ac,
Abdelnasser Abidlia,
Ehsan Behzadfarb,
Sasan Rezaeia,
Hani E. Naguibc and
Chul B. Park*a
aMicrocellular Plastics Manufacturing Laboratory, Department of Mechanical and Industrial Engineering, University of Toronto, Toronto, Ontario M5S 3G8, Canada. E-mail: park@mie.utoronto.ca
bDepartment of Chemical Engineering, Lakehead University, Thunder Bay, Ontario, Canada P7B 5E1
cSmart Polymers & Composites Lab, Department of Mechanical and Industrial Engineering, University of Toronto, Toronto, Ontario M5S 3G8, Canada
First published on 12th April 2019
Abstract
Aerogels suffer greatly from poor mechanical properties resulting from their particulate structure. They also experience noticeable pore shrinkage during drying due to their low structural integrity. These shortfalls limit their broad application. To enhance the mechanical properties and improve the structural integrity of silica-based aerogels, graphene nanoplatelets (GnPs), as a nanofiller, were embedded into the solution of polymerized vinyltrimethoxysilane (VTMS) to prepare P-VTMS-based silica/GnP (PE-b-Si/GnP) hybrid aerogel monoliths based on sol–gel synthesis and supercritical drying. The inclusion of GnPs in our polymer-based silica aerogel processes reinforced the nanostructure and suppressed PE-b-Si nanopore shrinkage during supercritical drying, thus acting as an effective anti-shrinkage nanofiller. Accordingly, the GnPs significantly contributed to the PE-b-Si solution's uniform gelation and to the change of the hydrophilic nature to a hydrophobic one even with 1 wt% addition. In this study, the influence of the GnP content on the sol–gel process, structure, and physical properties of PE-based silica aerogels is studied.
1. Introduction
Silica-based aerogels are a new mesoporous class of solid materials made of low-density solids (∼0.003–0.5 g cm−3), with a high degree of porosity (50–99.8%).1 They are derived from gel materials in which the liquid constituent is substituted with gas to leave the interconnected solid structures intact, without noticeable structural collapse. Thus, their high porosity and low density make them among the lightest of solid materials.2 Silica-based aerogels are very fragile, with both poor mechanical properties,7,8 and a hydrophilic nature,5 all of which dramatically limit their applications.A new class of aerogels called polymer-based silica aerogels has recently been created using polymerized precursors6 with both nonparticulate (co-continuous)7,8 and particulate8–10 structure with different morphologies though the same range of properties. Zu et al.8–10 created series of transparent superflexible aerogels from a vinylmethyldimethoxysilane (VMDMS) or vinylmethyldiethoxysilane (VMDES) by radical polymerization followed by hydrolytic polycondensation resulting in a doubly crosslinked morphology. They showed low-density aerogels (∼0.2 g cm−3) with flexible hydrocarbon backbone chains, and elastic polymethylsiloxanes crosslinks leading to excellent superflexibility in both bending and compression while maintaining the superinsulating properties (∼λ = 15.2 mW m−1 K−1). More specifically, in nonparticulate polymer-based silica aerogels, the conventional silica-based aerogel particulate structure has been eliminated by introducing spinodal decomposition to offset the long aging process and strengthen the particle-to-particle neck, meaning the connections between the particles are no longer a concern as the structure is not particulate. This nonparticulate class of aerogels, and particularly the PVTMS-based silica aerogel (PVPDMS),10 has shown great mechanical strength (∼2 MPa) and excellent thermal insulation characteristics (∼15–27 mW m−1 K−1) when compared with their conventional silica-based aerogel counterparts.11,12 Interestingly, the particulate aerogels that are similar to the PE-based materials (PVPSQ)12,13 can exhibit almost the same mechanical properties; although four days of aging is required to strengthen the gel resulting in a structure with a limited void fraction.16 The limitation of a low void fraction in this new class of aerogels (polymer-based particulate structure) can be improved using spinodal decomposition phase separation during polycondensation by increasing the amount of catalyst to accelerate hydrolytic polycondensation leading to a polymer-based co-continuous nanostructure.7,9
Several studies showed the reinforcement of the conventional salt-based silica aerogels with biopolymers namely, polysaccharides (such as cellulose,14 pectin,15 and chitosan16), or protein (such as silk fibroin17), as well as carbon materials (such as graphene,18,19 graphene nanoplatelets (GnPs), and graphene oxide (GO)20–23). Besides, some studies showed that the addition of GnPs or GO can introduce multifunctionalities to the new class of aerogels made of pre-polymerized silica-based precursors (∼30 wt% GnPs).10 Recently, GnPs have attracted a great deal of attention as a reinforcing nanofiller in polymer composites,24,25 which is due to their unique two-dimensional honeycomb layer structure,26,27 their excellent mechanical properties28 and their isotropic reinforcement capability in more than one direction. During the past several years, a number of polymers, including poly(vinyl alcohol),29,30 poly(vinylidene fluoride),31,32 polyurethane,33 cellulose,34 chitosan,35 and so forth, have been used to prepare GnP–polymer composites, in which the mechanical properties have been significantly enhanced. Several studies have been published on GnP and silica composites with various GnP contents.36 Watcharotone et al.37 created a mixture of hydrolyzed tetramethyl orthosilicate (TMOS) in a GnP suspension and then further dried it by removing the solvent at an ambient condition, resulting a GnP/silica material (11 wt% GnP content). Although, the presence of GnPs in the silica composite improved its structural integrity, as well as its mechanical and physical properties, the GnP amount used was always large (>10 wt%). Zu et al.10 also added 30 wt% GnP to polyvinylpolydimethylsiloxane/polyvinylpolymethylsiloxane (PVPDMS/PVPMS) wet gel and further dried the gel with freeze drying to create a GnP-functionalized particulate PVPDMS/PVPMS aerogel with a particle size in the range of 20–100 nm.10 Reportedly, the presence of GnPs has significantly improved the mechanical properties of the PVPDMS/PVPMS aerogel.10 However, there is no report on the anti-shrinkage effect of GnP in the aforementioned studies. Dervin et al.38 demonstrated a change in the physical characteristics of the conventional particulate silica aerogels, using GO leading to an increased surface area from 390 m2 g−1 (pure SiO2 aerogel) to 798 m2 g−1 (2 wt% GO–SiO2 aerogel) with a 19% decrease in volume shrinkage.
Despite the excellent improvements of the new class of polymer-based silica aerogels, shrinkage still occurs during the gelation and drying process, especially during ambient-pressure drying, which is unavoidable when making aerogels due to the large capillary pressure on the pore walls. To lower the capillary-pressure gradients on the pore walls during the drying process, the supercritical-fluid drying technique has been used extensively in aerogel production.3,4,39–42 In this process, the liquid in the pores is removed above the critical pressure and temperature, and this produced minimal liquid–vapor interfaces. Such a result could be achieved, to some extent, by using the supercritical- and/or freeze-drying technique. However, there was still noticeable shrinkage in the aerogel structure after the drying process,9,10 which might have been due to the low integrity and/or flexibility of the underlying gel structure, causing permanent or semi-permanent shrinkage during the gelation and/or drying processes.
Here, by investigating the effect of the spinodal decomposition process in creating a nonparticulate morphology in the GnPs' orientation and dispersion, we studied how the gelation reaction can participate in the inclusion of GnPs in the aerogel backbone during the sol–gel process to strengthen the body of the gel. The use of spinodal decomposition to create a nonparticulate gel network helps to offset the required long aging step in the binodal process, which is inevitable to strengthen the particle-to-particle neck.
2. Materials and methods
2.1. Materials
Di-tert-butyl peroxide (DTBP, 98%), ammonium hydroxide solution (ACS reagent, 28–30% NH3 basis), and VTMS (98%) were purchased from Sigma-Aldrich. Graphene nanoplatelets (GnPs, heXo-G V4) were provided by NanoXplore™. Denatured ethanol and distilled water were purchased from Fisher Scientific.2.2. Polymeric precursor synthesis
The molar ratio of the initiator to the VTMS (60 gr, 98%, Sigma-Aldrich) R was 0.1. Hence, the DTBP (6 g, 0.04 mol), acting as the thermal initiator, was dissolved in the VTMS (60 g, 0.4 mol) at the room temperature. A four-neck flask equipped with a condenser and a stirrer was then heated to 125 °C in a silicon oil bath. Next, the VTMS and the DTBP mixture were poured into the flask, and the reaction proceeded in a nitrogen atmosphere at a rotational speed of 200 rpm for 3 hours. To remove all of the unreacted monomer and initiator, the flask was placed in a vacuum oven at 125 °C for 1 hour at a reduced pressure. Finally, the polymer was poured into a closed container, and was cooled to the room temperature for further characterization. Silane functionalized polyethylene (polyvinyltrimethoxysilane, P-VTMS, PE-based silica) was made in a batch polymerization glass reactor. A P-VTMS with a molecular weight of ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was achieved with a DPI of 74.
000 was achieved with a DPI of 74.
2.3. Sol–gel synthesis for pure and GnP-reinforced PE-based silica wet gels (PE-b-Si/GnP)
The procedure of sol–gel synthesis to produce pure PE-based silica gel has been fully described in our previous studies.7,43 In summary, to develop a wet gel for a pure PE-b-Si gel, 4 g of a polymeric precursor, with 5.4 g of ammonia (catalyst), was added to 20 ml of ethanol, and then warmed at 40 °C. The mixture gelled after 90 minutes and provided sample 1.Samples 2, 3, and 4 were made followingly by suspending 0.5, 1, and 2 wt% GnP in the PE-b-Si solution in ethanol (PE-b-Si with GnP: PE-b-Si/GnP0.5, PE-b-Si/GnP1.0, and PE-b-Si/GnP2.0, respectively) prior to the gelation process at the theoretical density of 0.2 g ml−1. All of the mixtures were then gelled/crosslinked in a one-step sol–gel process by adding ammonium hydroxide in a 1:1 molar ratio to the silicon (Si) portion. They were then heated up in a vacuum oven at 40 °C for three hours until the gels were fully formed. The required gelation time is a function of the desired aerogel-density. The lower the density the longer the gelation time.
For the density of 0.2 g ml−1, the gelation time was as low as three hours for the fully formed gels to be obtained. The gels were then de-molded and bathed in a pure dehydrated ethanol for another four hours before the solvent exchange and supercritical drying processes occurred. In this procedure, the long aging step (7–20 days)8,13,44 have been eliminated as the obtained structure has no particles in its structure. The fully continuous network (nonparticulate) does not require for long aging procedure to strengthen the neck area between the particles as it has no particle in its structure.
Dispersion of GnPs in the polymer matrix (nonparticulate polymeric network in our case) has always been extremely challenging in different types of processing such as melt processing. Here, however, we use an in situ polymerization and solvent processing method to disperse and keep dispersed the GnPs in the matrix.45 During sol–gel transition, polycondensation polymerization of methoxy silane groups of P-VTMS in the presence of GnPs in the mixing chamber, and the high sheer forces induced by the polycondensation polymerization were helpful to break the agglomerates. During the sol–gel polymerization there is a very low chance of re-agglomeration in the composites compared to the melt processing46 method. In this study, the composite hybrid material presented a remarkable improvement in mechanical properties. Especially in compression modulus the nonparticulate material reinforced with GnPs have a significant improvement of a factor of 3. The wet-gel made in this process was further dried via supercritical drying method as fully described in next section (Fig. 1).
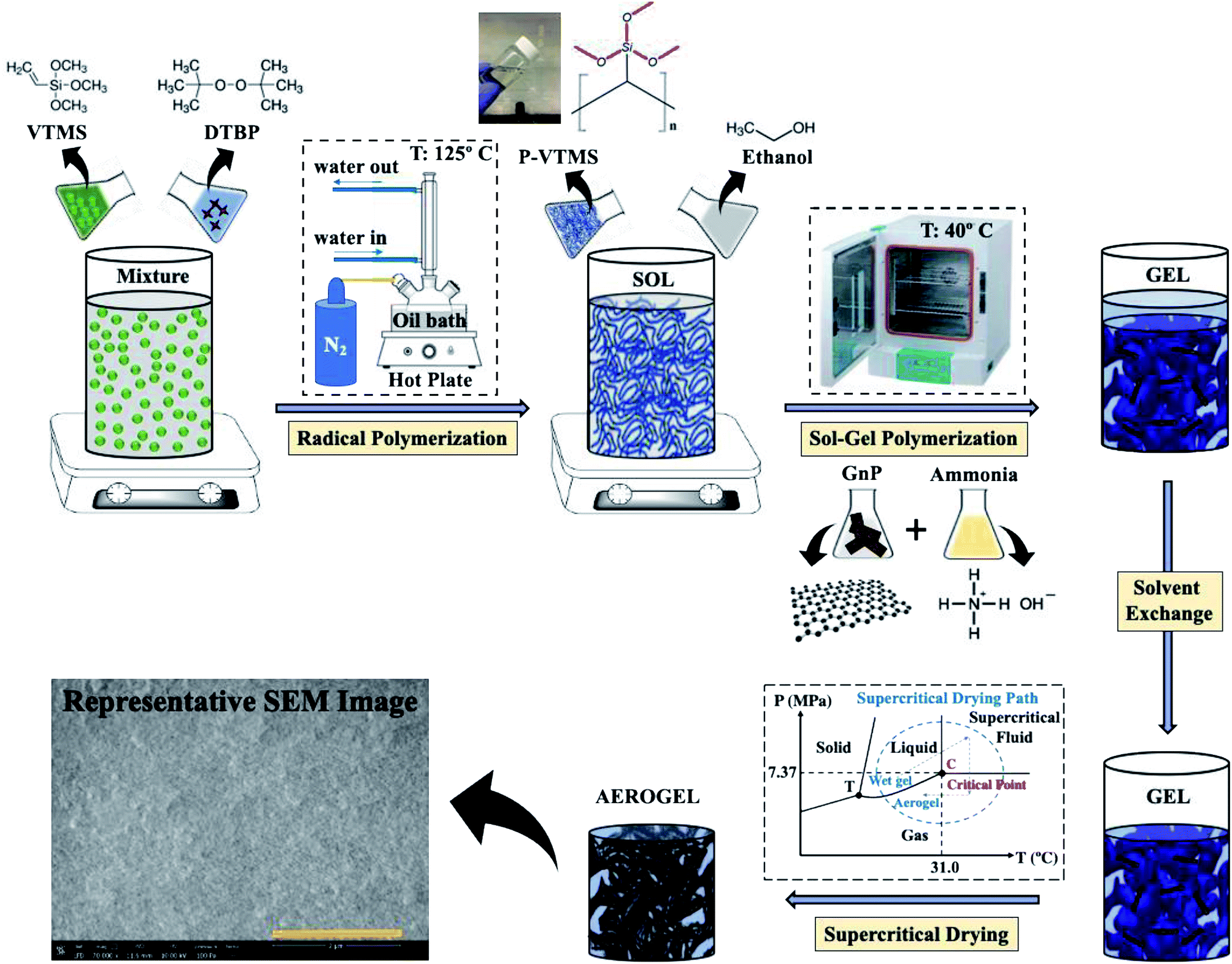 | ||
| Fig. 1 The schematic represents the aerogel processing steps, which consist of the polymerization of the VTMS at 125 °C and, later, the sol–gel polymerization of the P-VTMS both with/without GnP at 40 °C. Supercritical drying47 in a high-pressure chamber was the last step in this process. The scale bar in the SEM image represents 2 μm. | ||
2.4. Aerogel preparation/supercritical drying
To counteract the inevitably induced capillary forces during the solvent extraction to the pore walls, the samples first experienced the solvent exchange, and then the solvent extraction through the supercritical drying process. This approach helped, to some extent, to maintain the structural integrity of the solid parts after the drying process.The solvent exchange was accomplished by replacing the ethanol (solvent) with liquid CO2 in a high-pressure chamber for the gel samples. They were placed in a sealed stainless-steel chamber filled with pure dehydrated ethanol and were pressurized at 1500 psi (10.3 MPa), using a high-pressure syringe pump. At this pressure, the liquid CO2, which was connected to the syringe pump, was diffused into the gels and started to replace the initial ethanol solvent. Then, the chambers were regularly depressurized (80 ml every 2 h) to extract the ethanol and to further introduce the liquid CO2. Thus, the ethanol in the pores was gradually replaced with the liquid CO2 until all of it had been replaced by the liquid CO2 in the gels (7 days). The solvent exchange was done in a high-pressure chamber to avoid any capillary forces on the pore walls during the solvent extraction to maintain the structural integrity. When no more ethanol was exiting the chamber outlet, the supercritical drying process was started and removed the liquid CO2 from the gel pores by increasing the temperature from an ambient temperature to 40 °C. In this way, all possible capillary forces were minimized to prevent any cracks from forming in the structure during the process and to result in only negligible shrinkage. Fig. 1 summarizes the complete procedure undertaken to produce the final aerogel.
2.5. Characterization methods
The material's morphology and its physical, chemical, mechanical, and thermal properties were analyzed. The bulk densities (ρb) of the resultant aerogels were calculated in terms of their mass to volume ratios.The percentage of linear shrinkage (Ls%) was determined from the change in the height and diameter of both the alcogel and the aerogel below equation, where L is the length of the aerogel (either vertical or horizontal) and L′ is the length of the alcogel (either vertical or horizontal).

The samples' pore size and surface areas were characterized by nitrogen adsorption–desorption isotherm analysis performed at 77 K, using Autosorb iQ (Quantachrome Instruments). The samples were outgassed for 15 h at room temperature at a pressure of 10−3 Pa prior to the nitrogen adsorption analysis. Applying this analytical method, the adsorption and desorption isotherms of the N2 and the pore size distribution, as well as the samples' surface areas, were determined using the Barrett–Joyner–Halenda (BJH) theory.48
The aerogel's microstructure was studied using environmental scanning electron microscopy (ESEM) (Quanta FEG-250). The samples were coated with platinum before they were placed in the ESEM at a low vacuum setting, with water vapor acting as the working environment to decrease the charging effect on them. The sol–gel transition point was continuously monitored by an in situ methodology through small amplitude oscillatory test. The instrument used here was the discovery-HR3 rheometer (TA Instruments, USA), the titanium cone plate geometry with a radius of 50 mm and a cone angle of 0.0403 radian was used. The chemical structure of the synthesized material was studied using Fourier transient infrared (FTIR) spectroscopy. The FTIR (Perkin Elmer Spectrum) was conducted on the pure PE-based silica aerogel and on the GnP-reinforced PE-based silica aerogel samples. This was done with different GnP contents to measure the spectral transmittance and to evaluate the chemical bonds. The spectral transmittance was collected by averaging 8 scans in a spectral range of 4000–550 cm−1 (wavelength from 2.5 to 18 μm), with a spectral resolution of 4 cm−1. Before running the FTIR test, the airborne H2O and CO2 background noise were registered. The surface composition and the functional groups of the GnP and PE-b-Si/GnP were evaluated using X-ray photoelectron spectroscopy (XPS) measurements, which were taken in an ESCALAB 250 (Thermo Fisher Scientific, USA) X-ray photoelectron spectrometer microprobe. The spectra were recorded using a monochromatic Al-Kα X-ray source at 100 W. Curve fitting of the XPS peaks was conducted using a Gauss–Lorentzian peak shape, after performing a Shirley background correction.
The thermal properties and hydrophobicity characteristics of the GnP-reinforced aerogels were also characterized. The aerogel's thermal conductivity was studied using a thermal conductivity analyzer (HotDisk) (Transient Plane Source, TPS2500S). A C5501 Kapton insulated sensor, with a diameter of 6.4 mm along the transient plane, was used as the source method. The sensor was placed between two solid aerogel samples with the same dimensions to measure their total conductivity. The sensor measured both the heat generated and the temperature's increase within the samples by measuring the voltage's stepwise change over time. The aerogels' compression modulus was determined by a uniaxial compression test, using an Instron Microtester (5848) with a 500 N load cell. A 25 mm × 6 mm cylindrical sample with a compression rate of 1.2 mm min−1 was used, according to the ISO 604-2002. The thermogravimetric analysis (TGA) was performed on a Shimadzu TG50 analyzer to quantitatively characterize the samples' thermal stability with GnP present.
The hydrophobic behaviour of the samples' surface (wettability) was studied using water contact angel (WCA) measurements for both pure and GnP-reinforced samples. Prior to the measurements, deionized water was degassed to avoid nanobubble formation at the solid surface.49 A droplet of distilled water was deposited on a smooth surface of both pure and GnP-reinforced samples and the water wetting behavior was observed. The observed results are shown in the ESI.†
3. Results and discussion
The main contribution of this study is to show the effects of GnP inclusion in the underlying aerogel backbone on (i) the gelation duration and the corresponding sol–gel rheology; (ii) the ultimate chemistry of the resulting material; and (iii) the structural integrity and the mechanical properties of the aerogel during gelation and drying. In this regard, the results were categorized into three main paths: (i) sol–gel rheology characterization; (ii) chemical characterization; which included FTIR, XPS, and TGA; and (iii) physical characterization; which included pore size, pore distribution, surface area, gelation duration, linear shrinkage percentages, skeletal porosity, and mechanical strength.3.1. Sol–gel rheology
The gel is a viscoelastic material composed of interpenetrating liquid and solid phases. We observe that the addition of a small amount of GnP drastically improves the homogeneity and uniformity of gel films formed in the mold. The sol–gel point in hydrogels is when there is a crossover between G′ and G′′. The gel point is the time at which a three-dimensional network of GnP is created within the matrix.50Rheological measurements reveal that GnP addition promotes the sol–gel transition at a fixed temperature (here at 40 °C) thereby increasing the viscosity at the sol–gel transition as shown in Fig. 2. The GnP reduces the gel point time up to 25%. We believe that these observations can be explained by the classical theory of gelation51 which predicts that the sol–gel transition is promoted by increasing the amount of cross linking. The resulting increase in viscosity reduces the amount of shear experienced by the gelling system. The formation of such a network is a typical characteristic of polymeric composites of well-dispersed GnP reinforcements.
 | ||
| Fig. 2 The rheological behaviour of PE-b-Si solution with and without GnP in terms of G′ and G′′ changes over time at 40 °C and strain rate of 0.1% for all samples. | ||
3.2. Chemical characterization
cm−1 was attributed to the presence of the silanol groups (Si–OH) on the surface of the PE-b-Si materials. The absorption band at 1440, 2850 and around 2950 cm−1, indicated the bending of the methylene groups, the CH3 groups, and the C–H stretching vibration band, respectively. This originated in the PE backbone, which was covalently bonded to the silica matrix through the Si–C bonds.53–55 The band at around 1610 cm−1 could be attributed to the deformation vibrations of the adsorbed water molecules after their exposure to air.55,56
 | ||
| Fig. 3 FTIR spectra of the prepared pure PE-b-Si aerogel sample (PE-b-Si/0% GnP). | ||
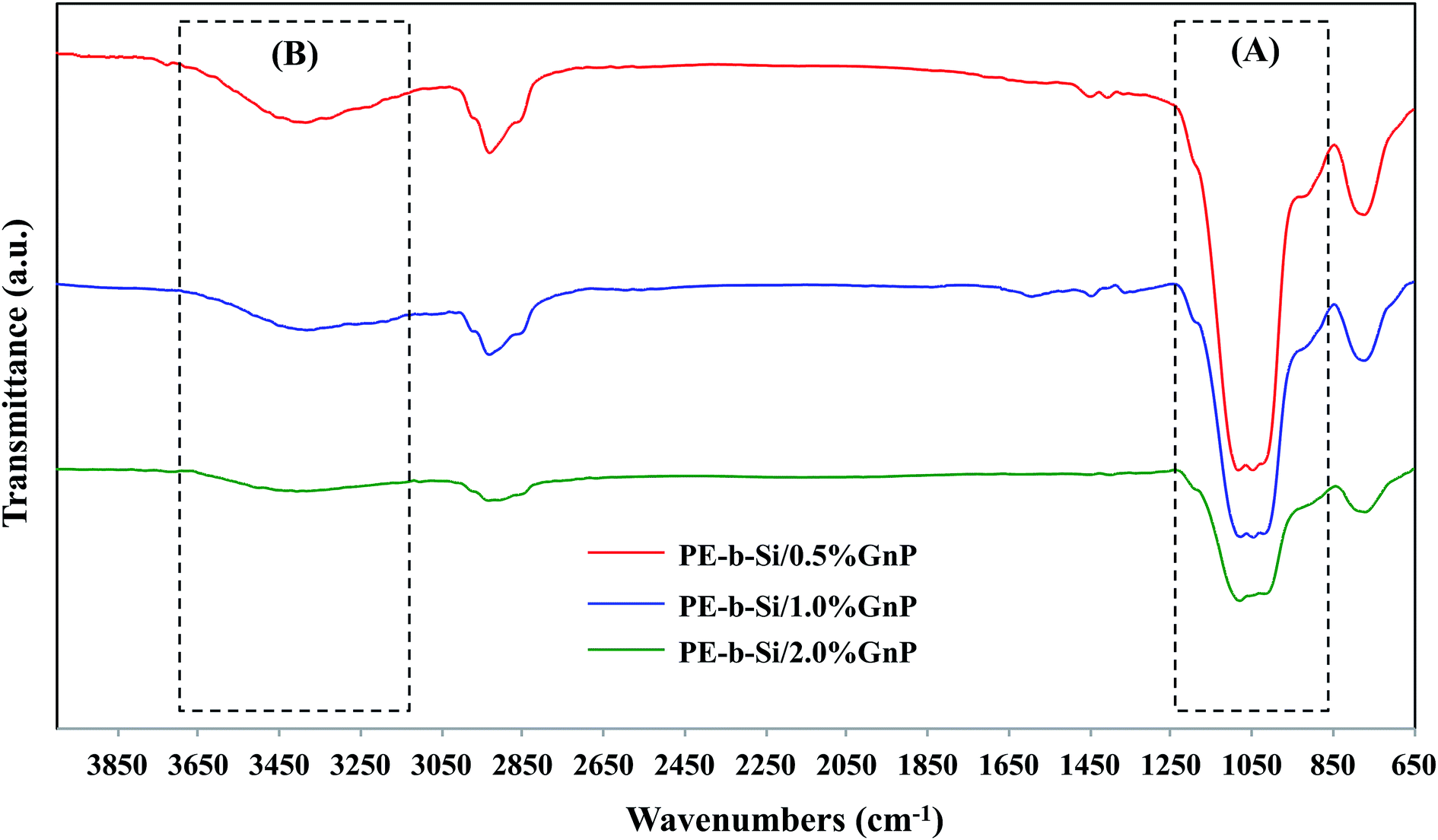 | ||
| Fig. 4 FTIR spectra of the prepared PE-b-Si/GnP aerogel samples (PE-b-Si/0.5% GnP, PE-b-Si/1.0% GnP, PE-b-Si/2.0% GnP). | ||
On the other hand, the GnP-embedded PE-b-Si aerogel samples exhibited similar FTIR spectra (Fig. 4), which indicated the successful maintenance of the silica's structural integrity. Due to the low GnP content (0.5–2.0 wt%), no increase in the C–H stretch vibrations was observed, which usually arises from the methylene group of the GnP aromatic ring (e.g. C–H band of sp2-hybridized carbon). However, there was a constant decline in the intensity of the characteristic peaks for the oxygen-based components with an increased GnP content; namely, the Si–OH and Si–O–Si bands (Fig. 4A), as well as in the hydroxyl groups (OH) at around 3400 cm−1 (Fig. 4B). This behavior indicated a strong interaction of the GnP with the oxygenated segments in the PE-b-Si matrix, including the bridging (Si–O–Si) and the terminal (Si–O–H) components. We surmised that the GnP bonded to these oxygen centers via the terminal hydroxyl groups (that is, the silanol groups) and through a scission in the oxygen-based covalent bonds in the silica matrix. Such an interaction contributed to maintaining the aerogels' structural integrity. Thus, we were given clear evidence of a successful intercalation and of an abundant dispersion of the GnP species within both the aerogel walls and the bulk and porous network. It did so by enhancing the intra- and inter-crosslinking effect between the PE-b-Si polymeric chains.
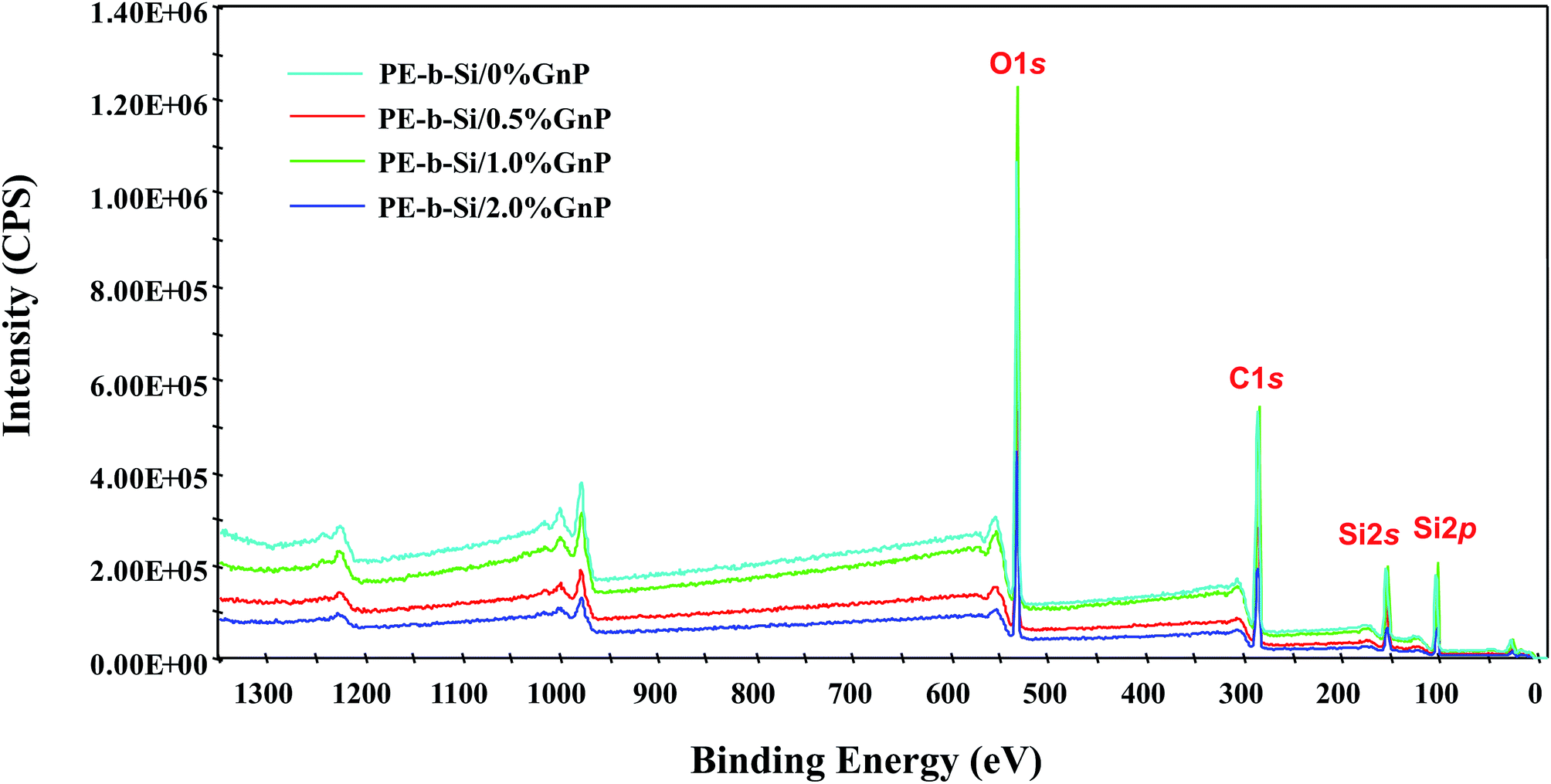 | ||
| Fig. 5 Survey of XPS spectra in the prepared PE-b-Si and the PE-b-Si/GnP aerogel samples. | ||
These results are summarized in Table 1, which shows the relative carbon and oxygen content (C:O ratios) obtained via the XPS measurements. First, the quantitative analysis of the XPS spectra shows a slight decline in C:O ratio, which corresponded to an extremely small amount of added oxygen coming from the GnP. However, with a further GnP addition, there was a slight increase in the C:O ratio, which corresponds to the higher carbon content that was due to the GnP aromatic carbon rings. These results further demonstrated that there was an extremely low number of oxygen-containing functional groups in the pristine GnP.
:O) in the PE-b-Si and PE-b-Si/GnP aerogel samples
| Atomic% (samples) | ||||
|---|---|---|---|---|
| Element | 0% G | 0.5% G | 1% G | 2% G |
| Si 2p (PE-b-Si/GnP) | 16.24 | 16.00 | 15.26 | 15.23 |
| C 1s (PE-b-Si/GnP) | 50.86 | 50.58 | 51.92 | 52.09 |
| O 1s (PE-b-Si/GnP) | 32.91 | 33.42 | 32.83 | 32.69 |
| C:O ratio |
1.54 | 1.51 | 1.58 | 1.59 |
These findings are in great agreement with the XPS data obtained for the pristine GnP samples, which indicated that the pure GnP had an extremely low oxygen content (<3%, Fig. S1†). In addition, a very low content of GnP was incorporated into the PE-b-Si aerogel (0.5–2.0%). This meant that, interestingly, the contribution of the GnP-sourced oxygen components to the final structural integrity and the bonding of the GnP to the polymer backbone were highly crucial, even at extremely low concentrations. The C 1s′ core-level XPS of the prepared PE-b-Si aerogel, clearly showed that two main components corresponded with the carbon atoms in the different functional groups (Fig. 6). The first peak was attributed to the Si–C functionality bonds centred at 284.7 eV.57 These could have included a small amount of sp2-hybridized weak carbon bonds in the condensed and non-polymerized edges of the silica backbone (that is, the vinyl groups) and the non-hydrolyzed VTMS substrates. The second peak was attributed to the sp3-hybridized carbon bonds C (C–C, C–H) at 286.6 eV on the PE backbone, which showed that the polymerized VTMS chain was covalently bonded to the Si centers.58 The second peak could also include some C–O contamination formed at the surface of the samples.59 After the GnP was added, a constant increase in the first peak's intensity with an increased GnP content was observed, with a slight shift to a higher binding energy (284.8 eV). This was attributed to the Si–C bonds that overlapped the C components of the GnP's ring C; namely, the sp2-hybridized C–C in the aromatic ring carbon.60 There was an increase in the number and in the contribution of the GnP-derived sp3 C bonds.61,62 As a reference sample, the data was consistent with the C 1s′ spectrum of the pristine GnP (see ESI† in Fig. S1†). It had a major peak that was ascribed to the GnP sp2-hybridized carbon atoms. Similarly, shifting was also observed in the second C peak (286.8 eV) for the GnP-modified samples. This was due to the change in the electronic environment induced by the GnP defected C–C and the oxidized carbon species.63 We believe that the GnP induced a strong electronic effect via, but not limited to, the strong electrostatic interactions between the oxygen lone pairs on the PE-b-Si's backbone, with the electrons in aromatic rings on the GnP sheets' surfaces. This was consistent with the successful integration of the GnP sheets into the PE-b-Si matrix, and led to a stable and well-dispersed GnP–polymer composite.64
 | ||
| Fig. 6 C 1s core-level spectra decomposed by the Lorentzian–Gaussian fitting for the prepared GnP-embedded samples: (A) PE-b-Si/0% GnP; (B) PE-b-Si/0.5% GnP; (C) PE-b-Si/1% GnP; and (D) PE-b-Si/2% GnP. | ||
The silicon Si 2p core level spectrum of the GnP-free samples, shown in Fig. 7, can be resolved into two components, located at 102.5 and 103.1 eV, and can be ascribed to the Si–O(H) and the Si–Ox, respectively.65,66 The first peak was attributed to the silicon bond with hydroxyl-based oxygen atoms (that is, the silanol groups: Si–O–H).67–69 Meanwhile, the second, weaker peak was attributed to the Si–O for the silicon bonded to multiple oxygen atoms in the bridging Si–O–Si species.68,70,71 Fig. 7 shows that a decrease occurred in both peaks when the GnP content was increased in the samples. This phenomenon indicated that the GnP bonding occurred in both the terminal silanol and in the bridging oxygen species, thus lowering the amount of Si–O–H and Si–O–Si components, respectively. Despite the low amount of oxygen that the pristine GnP added, we believe that such oxygenated components tend to form very strong hydrogen bonds with the PE-b-Si matrix. This would have occurred throughout possible Si–O–H⋯O (GnP) and Si–O⋯H–O (GnP) species,72 in which, a slight shift towards a higher binding energy was also observed. This was due to the electronic environmental change that was caused by the intercalation and dispersion of the GnP components inside the PE-b-Si matrix. However, this did not exclude the presence of a small amount of Si–Si interactions within the aerogel bulk material. The Si–Si bonding signals could be fitted at ∼103.1 eV, thus corresponding to the sp3 bonding signals.73
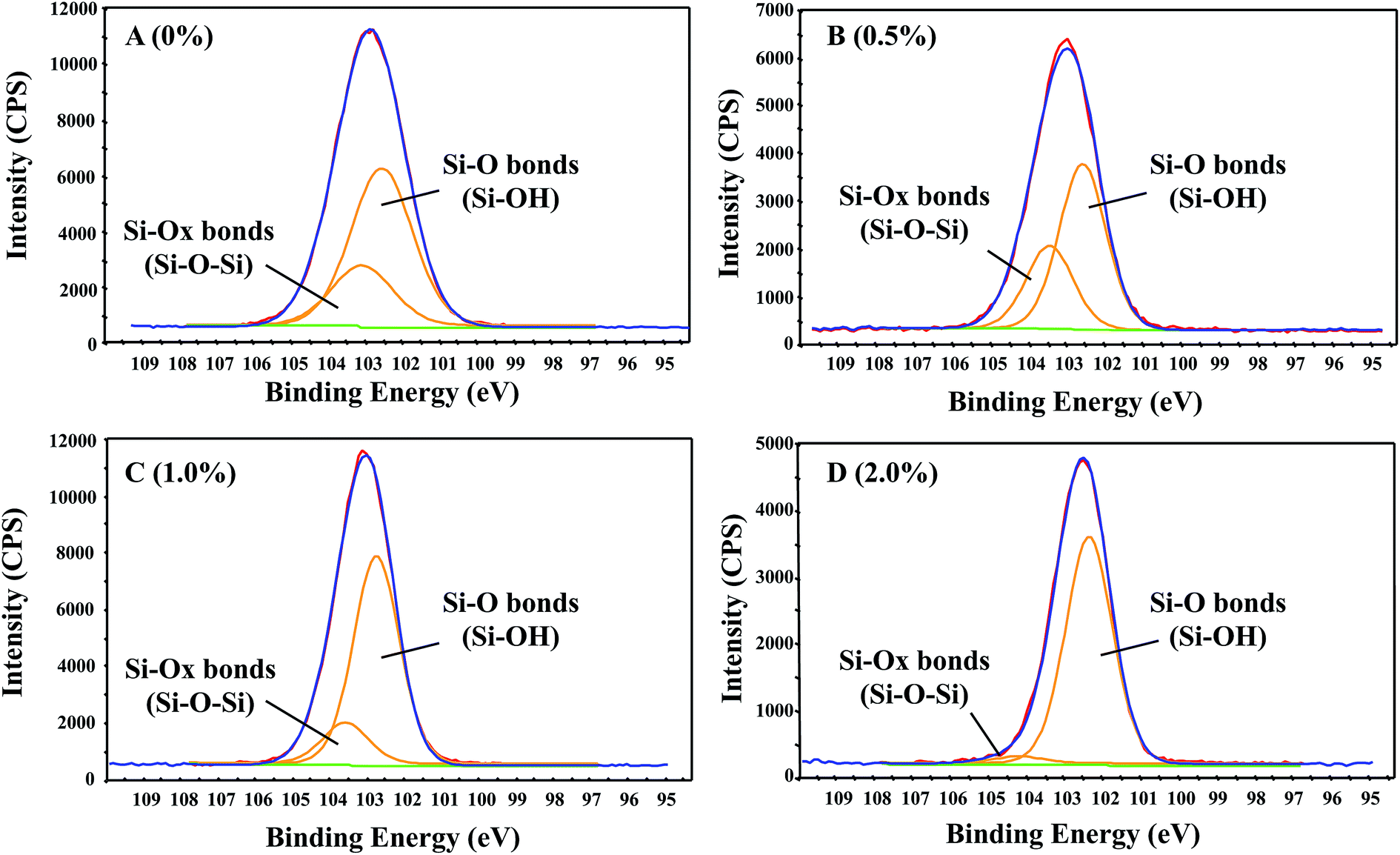 | ||
| Fig. 7 The Si 2p core-level spectra decomposed by the Lorentzian–Gaussian fitting for the prepared PE-b-Si/GnP aerogel samples: (A) PE-b-Si/0% GnP; (B) PE-b-Si/0.5% GnP; (C) PE-b-Si/1% GnP; and (D) PE-b-Si/2% GnP. | ||
The oxygen O 1s core level spectrum of the GnP-free samples had two main peaks at 531.7 and 532.6 eV. In agreement with the Si 2p core level spectrum, the first peak was attributed to the O–Si in the bridging of the Si–O–Si segments,68,74,75 while the second peak was attributed to the terminal hydroxyl groups in the aerogel bulk (Si–O–H).66,75,76
The O 1s core level spectra of the GnP-modified PE-b-Si provided more insights into the mechanism of the GnP interaction with the PE-b-Si matrix (Fig. 8). Interestingly, we initially observed that adding GnP (0.5 wt%) led to a decrease in the intensity of both peaks. This confirmed the above-noted conclusion regarding the GnP interaction with both the silanol and the bridging oxygen components. However, by increasing the GnP content (1 wt%), there was an increase in the second peak with a simultaneous increase in the first peak. This was attributed to the preferable interaction of the GnP with the bridging oxygen segments (Si–O–Si). A further GnP (2 wt%) addition revealed a significant decrease in the first peak, which indicated a higher GnP bonding degree with the terminal hydroxyl groups (silanol functions). In conclusion, GnP can be bonded with different oxygenated species on the PE-b-Si network that have different bonding kinetics. On the other hand, both peaks shifted to a higher binding energy after the GnP addition, which could also be attributed to the hydrogen bonding of the PE-b-Si matrix with traces of the oxygenated species on the GnP's surfaces. These results further confirm those obtained from the FTIR data, and they show a strong GnP interaction with the oxygenated segments in the PE-b-Si matrix, including the bridging (Si–O–Si) and the terminal (Si–O–H) components.
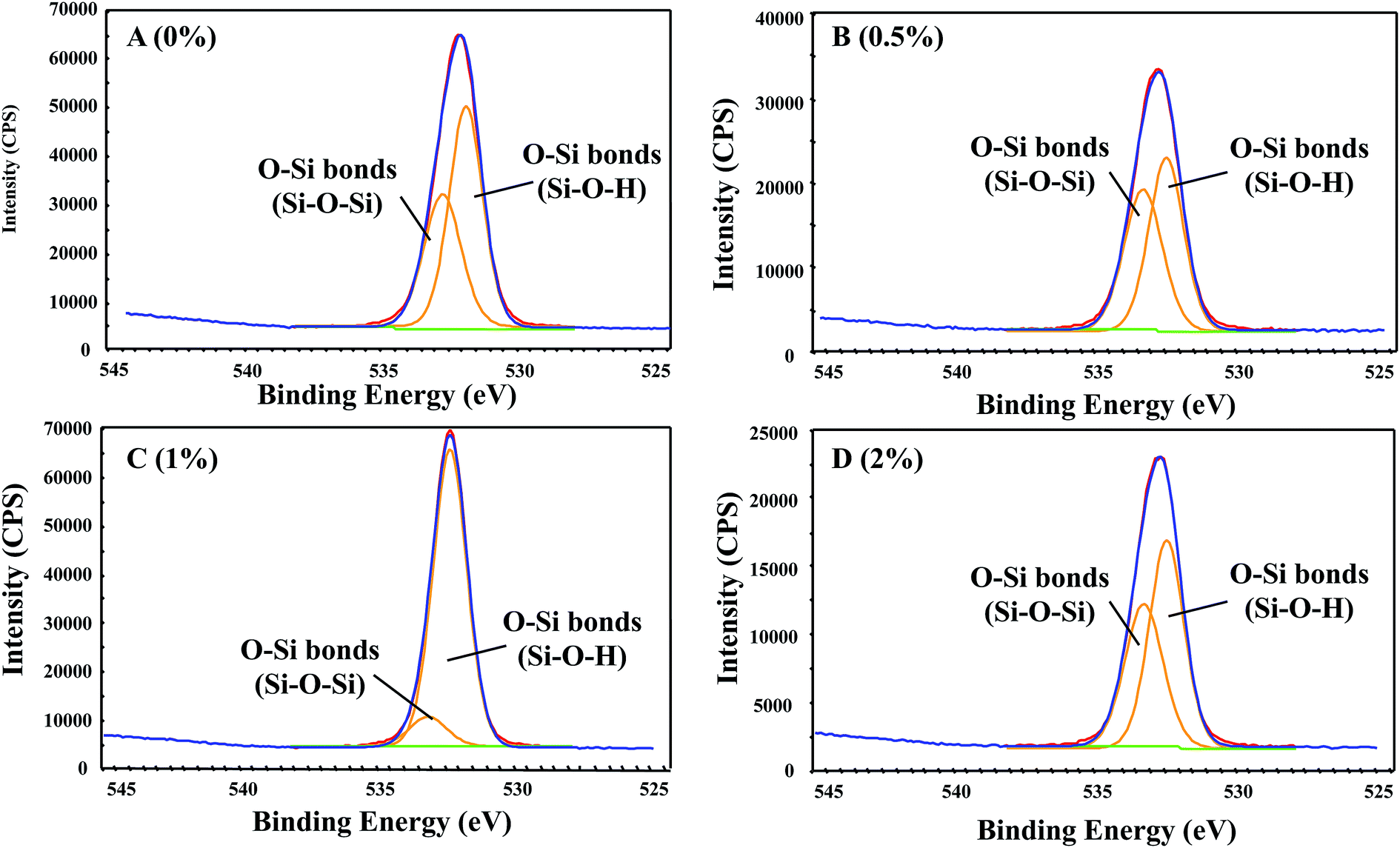 | ||
| Fig. 8 The O 1s core-level spectra decomposed by the Lorentzian–Gaussian fitting for the prepared GnP-embedded samples: (A) PE-b-Si/0% GnP; (B) PE-b-Si/0.5% GnP; (C) PE-b-Si/1% GnP; and (D) PE-b-Si/2% GnP. | ||
According to previous studies, the interaction of the GnP with oxygen could lead to the formation of a highly stable species (e.g. epoxy),77,78 or to physiosorbed O2 molecules.79 However, there is no evidence herein of any formation of such a covalently-bonded species. On the other hand, a charged oxygen species and oxygen radicals (atomic oxygen) with small polarizabilities interacted efficiently on the GnP surface.80 Additionally, if we consider the presence of the VTMS monomers or the non-hydrolyzed polymer edges that contain vinyl groups (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), these segments can contribute to the GnP binding trough an π–π stacking interaction and the CH–π interactions that occur between the electron-rich and electron-poor regions with the aromatic carbon ring on the GnP's surface.72 It is noteworthy that the GnP network can present electrostatic repulsions, which prevent its aggregation and improve its dispersion into the PE-b-Si matrix.72
C), these segments can contribute to the GnP binding trough an π–π stacking interaction and the CH–π interactions that occur between the electron-rich and electron-poor regions with the aromatic carbon ring on the GnP's surface.72 It is noteworthy that the GnP network can present electrostatic repulsions, which prevent its aggregation and improve its dispersion into the PE-b-Si matrix.72
In summary, oxygen interactions with GnP in the prepared aerogel occurred mainly through strong electrostatic coordination, hydrogen bonding, and the London-dispersion forces or van der Waals interactions. These continued to be the key factor contributing to GnP's intramolecular self-assembly and successful noncovalent incorporation into the polymer matrix. Consequently, the interfacial contact between the PE-b-Si and GnP72 was improved.
3.3. Physical, thermal, and mechanical properties
Four samples of cross-linked hybrid aerogels with and without GnP prior to the sol–gel reaction were analyzed. This experiment was designed to study how the GnP integration improved the mechanical properties through a structural reinforcement and if it reduced the linear shrinkage. As reported in Fig. 9, the effect of the GnP content on the PE-b-Si aerogels' linear shrinkage, both with and without the GnP after the drying process, was significantly noticeable. We observed that the use of GnPs resulted in a reduction in the volumetric shrinkage. The reduction in the shrinkage percentage in the thickness direction (perpendicular to the mold base) was more noticeable compared to that in the radial direction (parallel to the mold base), indicating that the samples with GnP experienced anisotropic shrinkage, while pure samples without GnP experienced shrinkage from all directions. Thus, GnPs mostly strengthened the gel backbone in the thickness direction. Furthermore, the additional shear forces created by the direction of gas escape inside the mold, during the sol–gel process, likely induced a specific GnP orientation in the thickness direction (perpendicular to the mold-base). Because of the preferred orientation of the GnPs in the solution, the gels were strengthened in the direction of the GnP orientation resulting in anisotropic shrinkage. | ||
| Fig. 9 The shrinkage percentages of the pure PE-based silica aerogel and the GnP-reinforced aerogels obtained with the VTMS precursor in both diameter and height directions. A schematic of GnP-embedded aerogel is inserted in the graph. | ||
This showed that the GnPs were able to physically interact with the aerogel backbone and had become part of the solid underlying structure, which successfully eliminated shrinkage during the solvent exchange and drying step. The observed extensive shrinkage in the pure PE-b-Si aerogel was strongly attributed to the reactivity of the unreacted hydroxyl groups (–OH) together in the skeletons during the drying step, which induced vicinity between hydroxyl groups and later induced chemical reactions between them resulting in irreversible shrinkage. However, the final shrinkage in the samples with the GnP-embedded PE-b-Si aerogels was not as noticeable. This is due to the induced interactions between the unreacted hydroxyl groups with the GnPs. The observed irreversible shrinkage in the pure PE-b-Si aerogel was caused by the condensation of the neighboring –OH groups on the skeletons during the temporal contraction during the drying process. This had been highly apparent due to the small pore size. This observation is in agreement with the observation in Zu et al.9 work as well.
Some studies have shown a decreased linear shrinkage of up to 21%; however, this was done by means of a long aging time at a very high temperature (100 °C), with a dramatic increase in the density, which resulted in an xerogel structure rather than an aerogel.9 It was also observed that the linear shrinkage via the samples' diameter did not considerably change after the GnP was added, which may have suggested the GnP's orientation. However, the linear shrinkage through in the height direction (perpendicular to the mold base) was dramatically suppressed from 15% to only 5%, when only 2 wt% GnP was added. This suggested that the GnP were most probably vertically oriented, perpendicular to the mold base around the skeleton. This could have prevented the shrinkage forces during the drying process through a height direction.
Due to the special reticulated structure on both molecular and nanoscales, PE-b-Si aerogels have excellent mechanical properties7 when compared with their material family.9 As shown in Fig. 10, the compression modulus of the neat PE-b-Si aerogel was about 7.1 MPa at a density of 0.235 g cm−3, which is comparable to the values reported by Zu et al.81 for neat PE-b-Si aerogel (same material family). The main reason for the good mechanical properties of the neat PE-b-Si aerogels is the existence of a flexible aliphatic hydrocarbon (PE portion) network in the skeleton cross-linked with a stiff and strong siloxane link (Si portion), which leads to the good load resistance during deformation. This was comparable to that of traditional silica82 and polymethylsilsesquioxane (PMSQ) aerogels.9
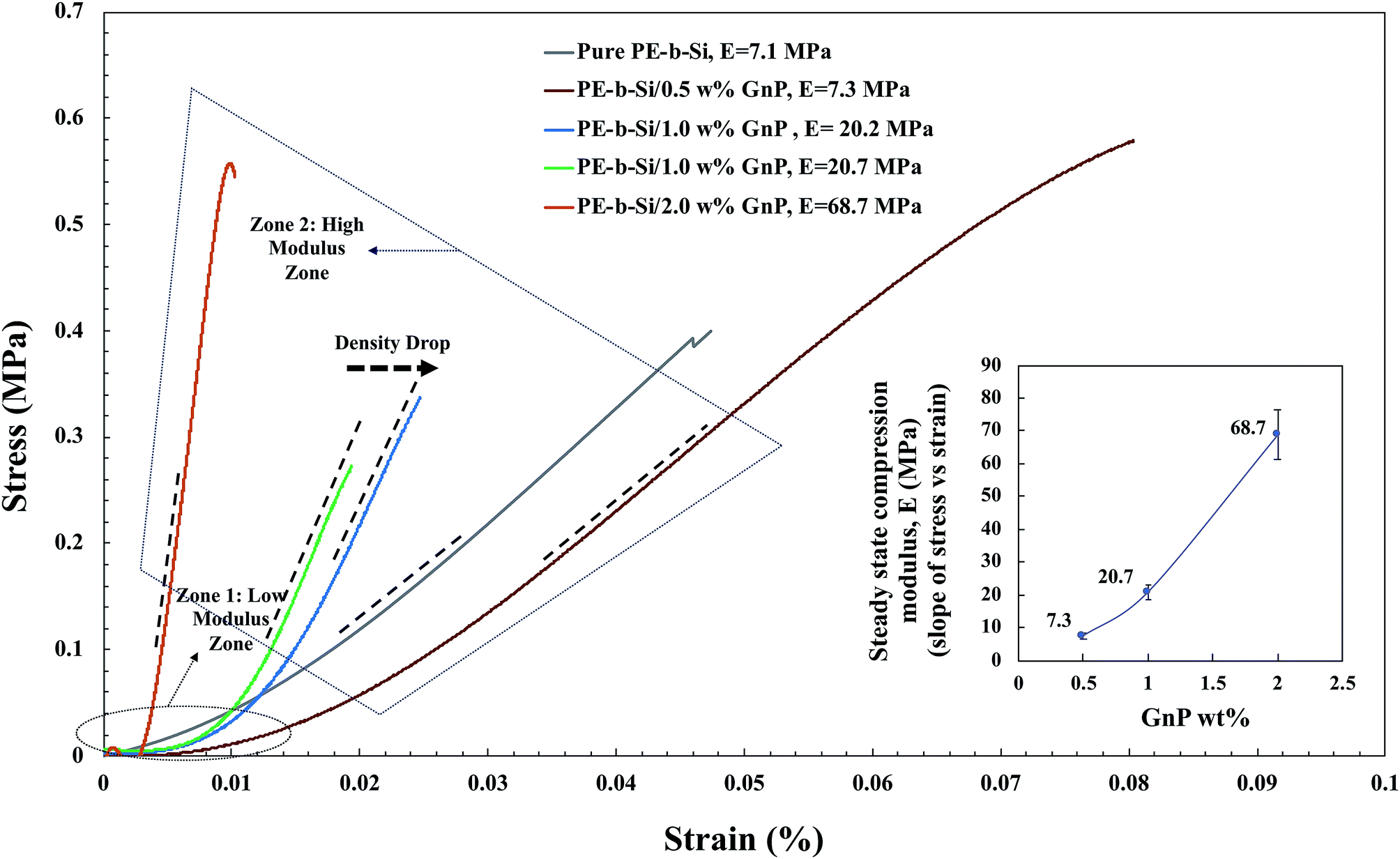 | ||
| Fig. 10 Stress versus strain curves for all samples. The inserted graph represents the slope of the stress–strain curve in the high modulus zone for different percentages of GnP (0.5–2 wt%). The values of the compression modulus (E) for GnP added samples were obtained as the slope of the stress–strain cure in the high modulus zone after GnP percolation threshold. | ||
However, once the GnP was embedded in the solid skeleton, there was a significant increase in the compression modulus. It increased from 7.1 MPa with no GnP, to 68.7 MPa in the steady state zone with only 2 wt% GnP added, even when the PE-b-Si/2.0 wt% GnP aerogel had a lower density of 0.186 g cm−3, than the PE-b-Si/1.0 w% GnP aerogel at the density of 0.195 g cm−3. It seems that the presence of GnPs in the matrix results in the stress–strain curve having two distinct zones. The first was a gradually increasing low-modulus zone and the second was a steady-state high-modulus zone, as shown in Fig. 10. This observation suggests that the deformation in the second zone was completely governed by the percolation threshold of the GnP network. However, the neat samples did not exhibit any transition from a first to a second zone.
Although the samples with a larger void fraction were expected to be deformed more than the samples with a lower void fraction, the samples with a higher GnP content (with a larger void fraction) exhibited a much smaller critical strain at which the transition occurred. For example, the sample with 2.0 wt% GnP reached the percolation threshold at a smaller strain compared to the samples with 1.0 wt% and 0.5% GnPs, despite the larger void fraction. It can also be observed that the samples with the same 1.0 wt% GnP content at different densities had almost the same steady-state compression moduli in the second zone, although the samples with a lower density had a larger critical strain. Obviously, a lower density at the same GnP content would mean a less compact aerogel structure, rendering the percolation threshold to be at a larger critical strain. However, the significantly reduced critical strain with increased GnP content from 1.0 wt% to 2.0 wt%, even at a larger void fraction, suggests that the critical strain was more sensitive to the GnP content than the density of the sample.
The excellent mechanical properties of the PE-b-Si/GnP aerogels can be ascribed to two possible factors. First, there was only a negligible number of unreacted –OH groups in the GnP-embedded aerogel skeletons due to their interactions with GnP (Fig. 4B), which reduced the irreversible shrinkage. Second, it is possible that the embedded GnP content was well exfoliated in the solid backbone during the fabrication step (sol–gel transition). The exfoliation of GnP might have been induced by the energy released during the hydrolysis/condensation and the increase in the pH level. The sol–gel reaction of silica-based materials with ammonium hydroxide was exothermic, accompanied by an increase in the solution pH.83 The exfoliation rate of GnPs must have been highly dependent on the pH change, that is, a higher pH (pH of 8–9) would induce a faster exfoliation rate.84 The well-exfoliated dispersed GnPs would act as a scaffold holding the PE-b-Si matrix. During the compression test, the GnP particles were forced to connect with each other and to establish a strong network at the critical strain. If the GnP particles are well exfoliated, the critical strain can be very small, regardless of the density. This confirms that a strong aerogel material at a very low density could be achieved at only 2 wt% GnP addition.
In our study, we synthesized the pure PE-b-Si aerogels11 and the GnP-embedded PE-b-Si aerogels by varying the GnP content from 0.5 to 2.0 wt%. The samples' mesoporous morphology, which was controlled by our network's self-assembly, were characterized into an in situ-ordered GnP-embedded morphology. The pore sizes, pore shapes, and pore classifications were analyzed based on the nitrogen adsorption/desorption isotherms characterizations. They were further analyzed based on the comparison of the obtained graph shapes with the reference graphs on the IUPAC technical report on the gases' physisorption.85 Both samples with and without GnP showed the type V isotherm shape (Fig. S2†). This type V isotherm was attributed to a relatively weak adsorbent–adsorbate interaction, which meant that the molecular clustering was followed by pore filling at higher p/p0 (the p represents equilibrium relative pressure; the p0 is the saturation pressure against the p). As Fig. 11 shows, the hysteresis loop for the pure PE-b-Si aerogel represented a type H2 shape. This type occurs when the sample (adsorbent) has mesopores with a small width and a cylindrical pore shape in a narrow range of pore necks. In this kind if pore structures the network effects are important. However, the hysteresis loop for the GnP-embedded PE-b-Si represented a type H1 physisorption isotherm shape, which is attributed to materials exhibiting a narrow range of uniform mesopores.85 All of the GnP-added aerogel isotherms were classified as type H1, with hysteresis in the desorption loops. The hysteresis loops occur when the pore cavities are larger in diameter than their openings; that is, the pores have an ink-bottle shape.85 The samples with a different GnP content showed the same hysteresis loops shape, which confirmed that the pore shape was independent of the GnP percentage at 0.5–2%, and that their pore width was in the mesoporous region.86 This analysis also suggested that the pores had been created through pore-blocking/percolation. This explained the GnP orientation/percolation around the pores during the gelation phase that resulted from the forces created by the escape of gas during the sol–gel reaction.
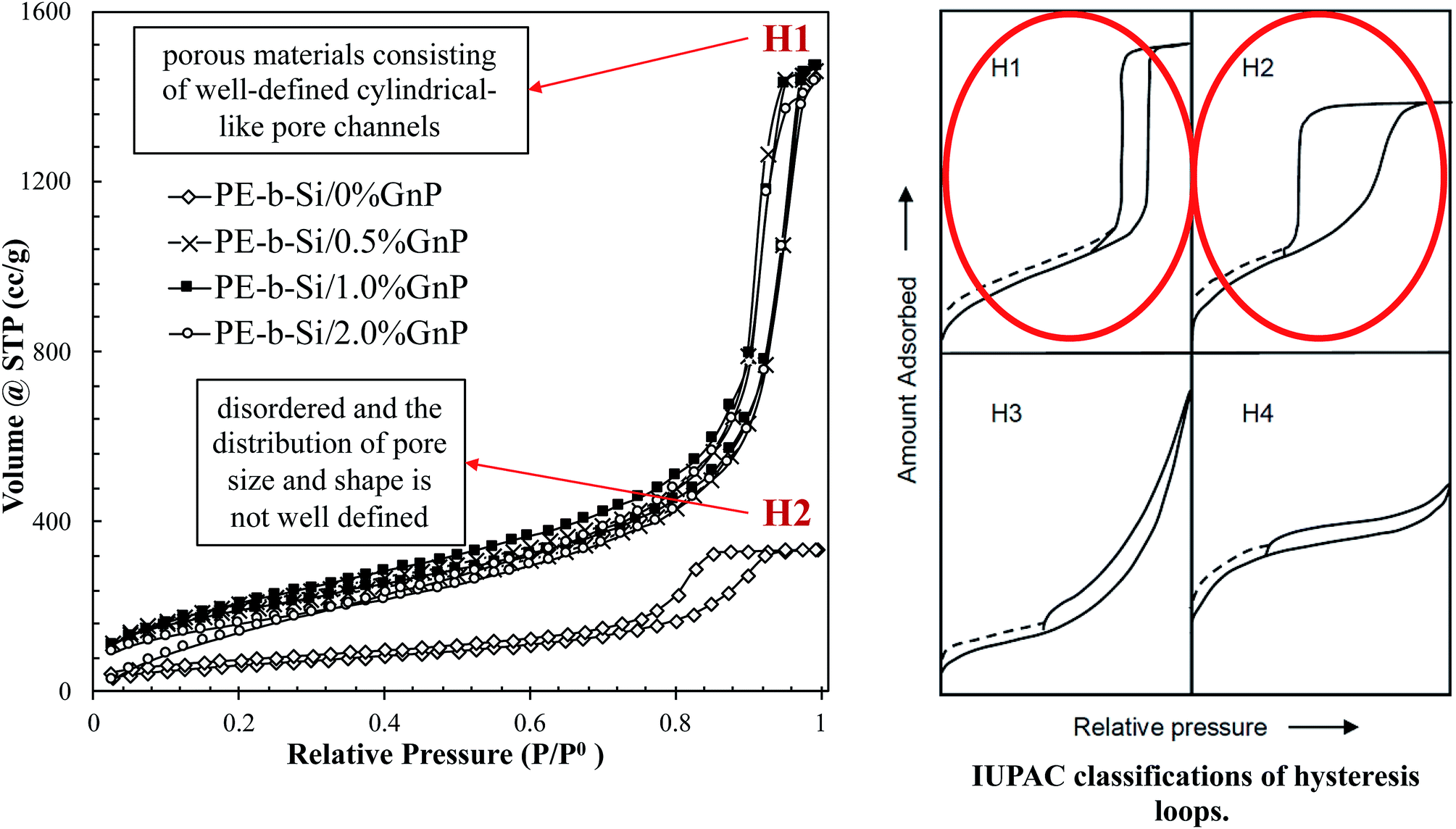 | ||
| Fig. 11 Nitrogen adsorption/desorption analysis for the PE-b-Si samples with various GnP content of 0–2 wt%, isotherms of N2 at 77 K compared with the IUPAC classification of hysteresis loops.85 | ||
The samples' porous morphologies, in terms of pore width and surface area, were measured using nitrogen adsorption/desorption analysis. Fig. 12 shows the Barrett–Joyner–Halenda (BJH) method used to measure the pore width and the pore size distributions of all the samples. The average pore size changed dramatically from 10 nm for pure PE-b-Si aerogel to 33 nm for PE-b-Si/2.0% GnP. This showed that the pore size had been defined by the GnP's presence and orientation. This increase in the pore size after an addition of 2.0 wt% GnP showed how the pore structure had resisted the shrinkage forces and maintained its structural integrity during the drying process.
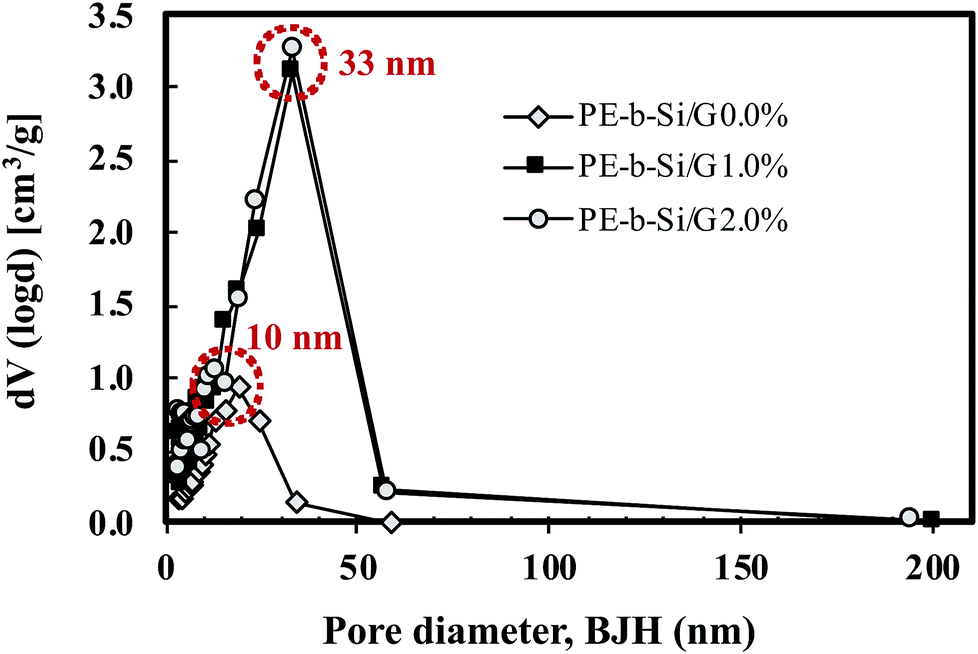 | ||
| Fig. 12 BJH pore size distributions for the PE-b-Si samples with various GnP content of 0–2 wt%. | ||
Also, as is summarized in Table 2, the samples' total surface areas were calculated using the Brunauer–Emmett–Teller (BET) analysis of nitrogen adsorption. When the GnP percentages increased, the BET surface areas also increased. This was because the pore shapes and sizes controlled the surface areas, which were controlled by the GnP content and its arrangements. Therefore, at a high GnP percentage, the number of pores was less, while their sizes were larger compared with the pure PE-b-Si aerogel. Furthermore, when the dilution ratio increased, it diminished the total surface area of the pure PE-b-Si aerogel. This was because the number of pores had increased in the same volume.
3.4. Thermal insulation and stability
The onset temperature of the non-reinforced aerogels was not dramatically changed by incorporating GnP into the underlying structure of the PE-b-Si aerogel. This outcome suggested that the thermal stability of the silica composites was not greatly affected by the addition, or by subsequent GnP content increases. The TGA plot of the GnP-embedded PE-b-Si aerogels confirmed that their thermal stability had scaled both with and without the GnP addition (Fig. 13). The TGA curves showed that, regardless of the GnP content, the weight loss was similar in all of the samples. The GnP-embedded samples experienced a 5% weight loss at 100 °C, which was due to the water evaporation; a 10% weight loss occurred between 300 °C and 500 °C, which could have been caused by the desorption of the adsorbed water; and a 20% weight loss occurred between 500 °C and 600 °C, which indicated a decomposition in the vinyl groups of the P-VTMS. These aerogels had a great thermal stability range, with only a small weight loss before the temperature reached 500 °C. Since all the samples had been tested in the same condition, the results could be compared in terms of moisture absorbency. It was clear that the samples without GnP were much more moisture sensitive and had absorbed about 25% of the environmental moisture compared with the GnP-embedded samples. The TGA results also indicated that the aerogels consisted of ∼70% silica and ∼30% polymerized VTMS plus the GnP (0–2 wt%). | ||
| Fig. 13 TGA curves obtained with the PE-b-Si/GnP aerogels for the pure PE-b-Si aerogel and the GnP-loaded aerogels: PE-b-Si/0.5% GnP, PE-b-Si/1% GnP, and PE-b-Si/2% GnP. | ||
To study the effect of the pore size and surface area on the thermal conductivity, we measured the pore size and surface area using the Density-Functional-Theory (DFT) model as this method is more accurate than BET measurements in type V hysteresis isotherm (Fig. S2†).85 The BET multi-pore surface area measurement relies upon a pressure range of 0.1 < p/p0 < 0.3. The surface area calculations using the BET model is reported here as a tool to compare the results with other studies. However, for precise discussion, it is necessary to look at the DFT measurements. And, since the samples show a type V isotherm (Fig. S2†), the growth of the monolayer of N2 molecule might not be completed at p/p0 lower than 0.3. Since the precise BET measurement can be done only when 0.1 < p/p0 < 0.3, the BET data would not be reliable. Therefore, the DFT values are also reported in Table 3.
| Aerogel samples | Total thermal conductivity (mW mK−1) | Water contact angle (degree) | adDFT, Dv (d) (nm) | bSDFT (m2 g−1) |
|---|---|---|---|---|
| a DFT pore diameter.b DFT specific surface area. | ||||
| PE-b-Si/0% GnP | 21 ± 0.5 | 57 ± 3 | 25 ± 0.7 | 540 |
| PE-b-Si/0.5% GnP | 29 ± 0.7 | 69 ± 2 | 26 ± 0.4 | 550 |
| PE-b-Si/1.0% GnP | 31 ± 0.6 | 120 ± 2 | 40 ± 0.4 | 659 |
| PE-b-Si/2.0% GnP | 37 ± 0.8 | 123 ± 3 | 40 ± 0.3 | 664 |
As for the thermal conductivity discussion; the thermal conductivity test of the GnP-embedded PE-b-Si and pure PE-b-Si aerogels confirmed that their thermal conductivity had scaled when the GnP is added which might have been due to the increase in solid conductivity (Table 3). The pore sizes of all the samples were below the mean free path of air resulting in similar gas conductivity in all of the samples. The thermal conductivity results showed that, regardless of the GnP content, the thermal insulation was reduced in all of the samples with GnP. At the theoretical density of 0.2 g cm−3 both samples with 0.5 wt% and 1 wt% GnP have been tested for thermal conductivity characteristics. It was observed that thermal conductivity of samples with 1% GnP was slightly higher than the sample containing 0.5% GnP at 0.2 g cm−3 density. This behaviour was also repeated comparing samples with 1% GnP and 2% GnP at the same range of density. Since the pore sizes are below the mean free path of air (68 nm), it is believed that the slight increase in the thermal conductivity of the samples with GnP is due to an increase in the solid thermal conductivity.
4. Conclusion
We prepared a PE-b-Si aerogel both with and without GnP as a pore definer and shrinkage inhibitor by using a straightforward sol–gel polymerization method. Mesoporosity studies showed that GnP could be integrated in a PE-b-Si matrix. This was due to the physical compatibility between them, which effectively and dramatically strengthened the aerogel skeleton. The GnP content noticeably reduced the induced linear shrinkage during drying step (from 15% to 5%) and increased the pore surface area (from 570 m2 g−1 to 694 m2 g−1). The outcome of this study is the creation of a new material with special characteristics that give it several functionalities. As its pore sizes (32 nm) are still below the mean free path of air (68 nm), it is valuable for use in thermal insulation applications eliminating the gas thermal conductivity through the pores.This study produced a unique nonparticulate morphology with very small pore sizes and a large surface area using an in situ structural engineering methodology for the selective inclusion of reticulate PVTMS-based silica aerogel (produced with spinodal decomposition43) with only 2 wt% GnP. Thus, we are able to create effective stress-transfer pathways within the three-dimensional (3-D) GnP-reinforced PVTMS-based silica aerogels (PE-b-Si/GnP). The 3-D bonded PE-b-Si/GnP was synergistically strengthened, and it had complete structural deformations and a great compressive mechanical strength (68.7 MPa at a compressive strain of 90%) in contrast to our pure PE-based silica aerogel (∼7.2 MPa at one compression cycle in the same-density range43). It also had the greatest minimal structural shrinkage reported to date for this aerogel family.
Given its excellent combinations of mechanical and thermal insulation properties, it is expected that the PE-b-Si/GnP will be used in a broad range of applications. Its ordered architecture opens the door to its use in the fabrication of new 3-D multifunctional and mechanically durable nanoporous cellular aerogels for emerging applications such as thermal superinsulation, efficient oil and water separation, strain sensing, supercapacitors, and durable nano-devices.
Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
S. K. appreciates the Natural Sciences and Engineering Research Council of Canada (NSERC), Ontario Graduate Scholarship, William Dunbar Memorial Scholarship, and Barbara and Frank Milligan Graduate Fellowship. The research work is financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and Consortium of Cellular and Microcellular Plastics (CCMCP).References
- H. Schäfer, B. Milow and L. Ratke, Synthesis of inorganic aerogels via rapid gelation using chloride precursors, RSC Adv., 2013, 3(35), 15263 RSC.
- A. Lamy-Mendes, R. F. Silva and L. Durães, Advances in carbon nanostructure–silica aerogel composites: a review, J. Mater. Chem. A, 2018, 6(4), 1340 RSC.
- M. Alshrah, H. E. Naguib and C. B. Park, Reinforced resorcinol formaldehyde aerogel with Co-assembled polyacrylonitrile nanofibers and graphene oxide nanosheets, Mater. Des., 2018, 151, 154 CrossRef CAS.
- M. Alshrah, M. P. Tran, P. Gong, H. E. Naguib and C. B. Park, Development of high-porosity resorcinol formaldehyde aerogels with enhanced mechanical properties through improved particle necking under CO2 supercritical conditions, J. Colloid Interface Sci., 2017, 485, 65 CrossRef CAS PubMed.
- M. Moner-Girona, E. Martínez, A. Roig, J. Esteve and E. Molins, Mechanical properties of silica aerogels measured by microindentation: influence of sol–gel processing parameters and carbon addition, J. Non-Cryst. Solids, 2001, 285(1), 244 CrossRef CAS.
- S. Karamikamkar, S. Rezaie, H. E. Naguib and C. B. Park, Bio-inspired polyethylene-based composite reinforced by thermoplastic polyurethane (TPU) fiber for aerogel production, AIP Conf. Proc., 2019, 2065(1), 030060 CrossRef.
- S. Rezaei and C. B. Park, A Class of Hybrid Aerogels with an Ultralight Nonparticulate Reticulated Structure and a Method of Producing the Same, US Patent, Non-provisional patent application No. 15/946,960, April 6, 2018, Chinese patent application no. 15946960.
- G. Zu, K. Kanamori, T. Shimizu, Y. Zhu, A. Maeno and H. Kaji, et al., Versatile Double-Cross-Linking Approach to Transparent, Machinable, Supercompressible, Highly Bendable Aerogel Thermal Superinsulators, Chem. Mater., 2018, 30(8), 2759 CrossRef CAS.
- G. Zu, T. Shimizu, K. Kanamori, Y. Zhu, A. Maeno and H. Kaji, et al., Transparent, Superflexible Doubly Cross-Linked Polyvinylpolymethylsiloxane Aerogel Superinsulators via Ambient Pressure Drying, ACS Nano, 2018, 12(1), 521 CrossRef CAS PubMed.
- G. Zu, K. Kanamori, A. Maeno, H. Kaji and K. Nakanishi, Superflexible Multifunctional Polyvinylpolydimethylsiloxane-Based Aerogels as Efficient Absorbents, Thermal Superinsulators, and Strain Sensors, Angew. Chem., Int. Ed., 2018, 57(31), 9722 CrossRef CAS PubMed.
- J. C. Williams, M. A. B. Meador, L. McCorkle, C. Mueller and N. Wilmoth, Synthesis and Properties of Step-Growth Polyamide Aerogels Cross-linked with Triacid Chlorides, Chem. Mater., 2014, 26(14), 4163 CrossRef CAS.
- J. Fricke, Aerogels - highly tenuous solids with fascinating properties, J. Non-Cryst. Solids, 1988, 100(1), 169 CrossRef CAS.
- K. Kanamori, M. Aizawa, K. Nakanishi and T. Hanada, New Transparent Methylsilsesquioxane Aerogels and Xerogels with Improved Mechanical Properties, Adv. Mater., 2007, 19(12), 1589 CrossRef CAS.
- J. Fu, C. He, J. Huang, Z. Chen and S. Wang, Cellulose nanofibril reinforced silica aerogels: optimization of the preparation process evaluated by a response surface methodology, RSC Adv., 2016, 6(102), 100326 RSC.
- S. Zhao, W. J. Malfait, A. Demilecamps, Y. Zhang, S. Brunner and L. Huber, et al., Strong, Thermally Superinsulating Biopolymer–Silica Aerogel Hybrids by Cogelation of Silicic Acid with Pectin, Angew. Chem., Int. Ed., 2015, 54(48), 14282 CrossRef CAS PubMed.
- M. R. Ayers and A. J. Hunt, Synthesis and properties of chitosan–silica hybrid aerogels, J. Non-Cryst. Solids, 2001, 285(1), 123–127 CrossRef CAS.
- H. Maleki, S. Montes, N. Hayati-Roodbari, F. Putz and N. Huesing, Compressible, Thermally Insulating, and Fire Retardant Aerogels through Self-Assembling Silk Fibroin Biopolymers Inside a Silica Structure—An Approach towards 3D Printing of Aerogels, ACS Appl. Mater. Interfaces, 2018, 10(26), 22718 CrossRef CAS PubMed.
- D. Loche, L. Malfatti, D. Carboni, V. Alzari, A. Mariani and M. F. Casula, Incorporation of graphene into silica-based aerogels and application for water remediation, RSC Adv., 2016, 6(71), 66516 RSC.
- S. Tajik, B. Nasernejad and A. Rashidi, Preparation of silica-graphene nanohybrid as a stabilizer of emulsions, J. Mol. Liq., 2016, 222, 788 CrossRef CAS.
- W. L. Zhang and H. J. Choi, Silica-Graphene Oxide Hybrid Composite Particles and Their Electroresponsive Characteristics, Langmuir, 2012, 28(17), 7055 CrossRef CAS PubMed.
- Y. Lei, Z. Hu, B. Cao, X. Chen and H. Song, Enhancements of thermal insulation and mechanical property of silica aerogel monoliths by mixing graphene oxide, Mater. Chem. Phys., 2017, 187, 183 CrossRef CAS.
- H.-Y. Mi, X. Jing, H.-X. Huang, X.-F. Peng and L.-S. Turng, Superhydrophobic Graphene/Cellulose/Silica Aerogel with Hierarchical Structure as Superabsorbers for High Efficiency Selective Oil Absorption and Recovery, Ind. Eng. Chem. Res., 2018, 57(5), 1745 CrossRef CAS.
- L. Hong-li, H. Xiang, L. Hong-yan, L. Jing and L. Ya-jing, Novel GO/silica composite aerogels with enhanced mechanical and thermal insulation properties prepared at ambient pressure, Ferroelectrics, 2018, 528(1), 15 CrossRef CAS.
- S. M. Hamidinejad, R. K. M. Chu, B. Zhao, C. B. Park and T. Filleter, Enhanced Thermal Conductivity of Graphene Nanoplatelet–Polymer Nanocomposites Fabricated via Supercritical Fluid-Assisted in Situ Exfoliation, ACS Appl. Mater. Interfaces, 2018, 10(1), 1225 CrossRef CAS PubMed.
- M. Hamidinejad, B. Zhao, R. K. M. Chu, N. Moghimian, H. E. Naguib and T. Filleter, et al., Ultralight Microcellular Polymer–Graphene Nanoplatelet Foams with Enhanced Dielectric Performance, ACS Appl. Mater. Interfaces, 2018, 10(23), 19987 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett and G. Evmenenko, et al., Preparation and characterization of graphene oxide paper, Nature, 2007, 448(7152), 457 CrossRef CAS PubMed.
- O. C. Compton and S. T. Nguyen, Graphene Oxide, Highly Reduced Graphene Oxide, and Graphene: Versatile Building Blocks for Carbon-Based Materials, Small, 2010, 6(6), 711 CrossRef CAS PubMed.
- P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim and J. Xu, et al., Ultrastrong and Stiff Layered Polymer Nanocomposites, Science, 2007, 318(5847), 80 CrossRef CAS PubMed.
- Y. Xu, W. Hong, H. Bai, C. Li and G. Shi, Strong and ductile poly(vinyl alcohol)/graphene oxide composite films with a layered structure, Carbon, 2009, 47(15), 3538 CrossRef CAS.
- L. Zhang, Z. Wang, C. Xu, Y. Li, J. Gao and W. Wang, et al., High strength graphene oxide/polyvinyl alcohol composite hydrogels, J. Mater. Chem., 2011, 21(28), 10399 RSC.
- B. Zhao, S. Wang, C. Zhao, R. Li, S. M. Hamidinejad and Y. Kazemi, et al., Synergism between carbon materials and Ni chains in flexible poly(vinylidene fluoride) composite films with high heat dissipation to improve electromagnetic shielding properties, Carbon, 2018, 127, 469 CrossRef CAS.
- B. Zhao, C. Zhao, R. Li, S. M. Hamidinejad and C. B. Park, Flexible, Ultrathin, and High-Efficiency Electromagnetic Shielding Properties of Poly(Vinylidene Fluoride)/Carbon Composite Films, ACS Appl. Mater. Interfaces, 2017, 9(24), 20873 CrossRef CAS PubMed.
- X. Wang, Y. Hu, L. Song, H. Yang, W. Xing and H. Lu, In situ polymerization of graphene nanosheets and polyurethane with enhanced mechanical and thermal properties, J. Mater. Chem., 2011, 21(12), 4222 RSC.
- N. D. Luong, N. Pahimanolis, U. Hippi, J. T. Korhonen, J. Ruokolainen and L.-S. Johansson, et al., Graphene/cellulose nanocomposite paper with high electrical and mechanical performances, J. Mater. Chem., 2011, 21(36), 13991 RSC.
- H. Fan, L. Wang, K. Zhao, N. Li, Z. Shi and Z. Ge, et al., Fabrication, mechanical properties, and biocompatibility of graphene-reinforced chitosan composites, Biomacromolecules, 2010, 11(9), 2345 CrossRef CAS PubMed.
- M. Hamidinejad, B. Zhao, A. Zandieh, N. Moghimian, T. Filleter and C. B. Park, Enhanced Electrical and Electromagnetic Interference Shielding Properties of Polymer–Graphene Nanoplatelet Composites Fabricated via Supercritical-Fluid Treatment and Physical Foaming, ACS Appl. Mater. Interfaces, 2018, 10(36), 30752 CrossRef CAS PubMed.
- S. Watcharotone, D. A. Dikin, S. Stankovich, R. Piner, I. Jung and G. H. B. Dommett, et al., Graphene−Silica Composite Thin Films as Transparent Conductors, Nano Lett., 2007, 7(7), 1888 CrossRef CAS PubMed.
- S. Dervin, Y. Lang, T. Perova, S. H. Hinder and S. C. Pillai, Graphene oxide reinforced high surface area silica aerogels, J. Non-Cryst. Solids, 2017, 465, 31 CrossRef CAS.
- J. P. Randall, M. A. B. Meador and S. C. Jana, Tailoring mechanical properties of aerogels for aerospace applications, ACS Appl. Mater. Interfaces, 2011, 3(3), 613 CrossRef CAS PubMed.
- M. Alshrah, L. H. Mark, C. Zhao, H. E. Naguib and C. B. Park, Nanostructure to thermal property relationship of resorcinol formaldehyde aerogels using the fractal technique, Nanoscale, 2018, 10(22), 10564 RSC.
- X. Wang and S. C. Jana, Synergistic Hybrid Organic–Inorganic Aerogels, ACS Appl. Mater. Interfaces, 2013, 5(13), 6423 CrossRef CAS PubMed.
- X. Wang and S. C. Jana, Tailoring of morphology and surface properties of syndiotactic polystyrene aerogels, Langmuir, 2013, 29(18), 5589 CrossRef CAS PubMed.
- S. Rezaei, A. Jalali, A. M. Zolali, M. Alshrah, S. Karamikamkar and C. B. Park, Robust, Ultra-Insulative and Transparent Polyethylene-based Hybrid Silica Aerogel with a Novel Non-particulate Structure, J. Colloid Interface Sci., 2019 DOI:10.1016/j.jcis.2019.04.028.
- T. Shimizu, K. Kanamori, A. Maeno, H. Kaji, C. M. Doherty and P. Falcaro, et al., Transparent, Highly Insulating Polyethyl- and Polyvinylsilsesquioxane Aerogels: Mechanical Improvements by Vulcanization for Ambient Pressure Drying, Chem. Mater., 2016, 28(19), 6860 CrossRef CAS.
- K. Müller, E. Bugnicourt, M. Latorre, M. Jorda, Y. Echegoyen Sanz and J. M. Lagaron, et al., Review on the Processing and Properties of Polymer Nanocomposites and Nanocoatings and Their Applications in the Packaging, Automotive and Solar Energy Fields, Nanomaterials, 2017, 7(4), 74 CrossRef PubMed.
- A. Afshar, M. Salami Hosseini and E. Behzadfar, Numerical Study of the Agglomerates Dispersion Behavior in Shear and Elongational Flow Fields in Viscous Media Using Population Balance Modeling (PBM), Sci. Iran., 2014, 21(6), 2107 Search PubMed.
- A. C. Pierre and G. M. Pajonk, Chemistry of Aerogels and Their Applications, Chem. Rev., 2002, 102(11), 4243 CrossRef CAS PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms, J. Am. Chem. Soc., 1951, 73(1), 373 CrossRef CAS.
- P. Cherukupally, E. J. Acosta, J. P. Hinestroza, A. M. Bilton and C. B. Park, Acid–Base Polymeric Foams for the Adsorption of Micro-oil Droplets from Industrial Effluents, Environ. Sci. Technol., 2017, 51(15), 8552 CrossRef CAS PubMed.
- C. Dawedeit, S. Ho Kim, T. Braun, M. A. Worsley, S. A. Letts and K. Jen Wu, et al., Tuning the rheological properties of sols for low-density aerogel coating applications, Soft Matter, 2012, 8(13), 3518 RSC.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, 1953, p. 696 Search PubMed.
- J. G. Reynolds, P. R. Coronado and L. W. Hrubesh, Hydrophobic aerogels for oil-spill clean up – synthesis and characterization, J. Non-Cryst. Solids, 2001, 292(1), 127 CrossRef CAS.
- E. F. Vansant, P. Van Der Voort and K. C. Vrancken, Characterization and Chemical Modification of the Silica Surface, Elsevier Science, 1995, 1st edn ISBN: 9780080528953 Search PubMed.
- J.-Z. Zheng, X.-P. Zhou, X.-L. Xie and Y.-W. Mai, Silica hybrid particles with nanometre polymer shells and their influence on the toughening of polypropylene, Nanoscale, 2010, 2(10), 2269 RSC.
- S. Zong, W. Wei, Z. Jiang, Z. Yan, J. Zhu and J. Xie, Characterization and comparison of uniform hydrophilic/hydrophobic transparent silica aerogel beads: skeleton strength and surface modification, RSC Adv., 2015, 5(68), 55579 RSC.
- L.-J. Wang, S.-Y. Zhao and M. Yang, Structural characteristics and thermal conductivity of ambient pressure dried silica aerogels with one-step solvent exchange/surface modification, Mater. Chem. Phys., 2009, 113(1), 485 CrossRef CAS.
- O. Pop-Georgievski, D. Kubies, J. Zemek, N. Neykova, R. Demianchuk and E. M. Chánová, et al., Self-assembled anchor layers/polysaccharide coatings on titanium surfaces: a study of functionalization and stability., Self-assembled anchor layers/polysaccharide coatings on titanium surfaces: a study of functionalization and stability, Beilstein J. Nanotechnol., 2015, 6, 617 CrossRef CAS PubMed.
- R. Bywalez, H. Karacuban, H. Nienhaus, C. Schulz and H. Wiggers, Stabilization of mid-sized silicon nanoparticles by functionalization with acrylic acid, Nanoscale Res. Lett., 2012, 7(1), 76 CrossRef PubMed.
- A. R. Marcondes, M. Ueda, K. G. Kostov, A. F. Beloto, N. F. Leite and G. F. Gomes, et al., Improvements of ultra-high molecular weight polyethylene mechanical properties by nitrogen plasma immersion ion implantation, Braz. J. Phys., 2004, 34(4B), 1667 CrossRef CAS.
- Y.-S. Ye, Y.-N. Chen, J.-S. Wang, J. Rick, Y.-J. Huang and F.-C. Chang, et al., Versatile Grafting Approaches to Functionalizing Individually Dispersed Graphene Nanosheets Using RAFT Polymerization and Click Chemistry, Chem. Mater., 2012, 24(15), 2987 CrossRef CAS.
- A. Sinitskii, A. Dimiev, D. A. Corley, A. A. Fursina, D. V. Kosynkin and J. M. Tour, Kinetics of Diazonium Functionalization of Chemically Converted Graphene Nanoribbons, ACS Nano, 2010, 4(4), 1949 CrossRef CAS PubMed.
- G. Gao, D. Liu, S. Tang, C. Huang, M. He and Y. Guo, et al., Heat-Initiated Chemical Functionalization of Graphene, Sci. Rep., 2016, 6, 20034 CrossRef CAS PubMed.
- H. Huang, Y. Xia, X. Tao, J. Du, J. Fang and Y. Gan, et al., Highly efficient electrolytic exfoliation of graphite into graphene sheets based on Li ions intercalation–expansion–microexplosion mechanism, J. Mater. Chem., 2012, 22(21), 10452 RSC.
- Graphene Science Handbook: Electrical and Optical Properties, CRC Press, 2016, 1st edn, ISBN: 9781466591325 Search PubMed.
- Y. Wang, B. Wang, Y. Lei, N. Wu, C. Han and Y. Gou, et al., Scalable in situ growth of SnO2 nanoparticle chains on SiC ultrathin fibers via a facile sol–gel-flame method, Appl. Surf. Sci., 2015, 335, 208 CrossRef CAS.
- B. Wang, Y. Wang, Y. Lei, S. Xie, N. Wu and Y. Gou, et al., Vertical SnO2 nanosheet@SiC nanofibers with hierarchical architecture for high-performance gas sensors, J. Mater. Chem. C, 2015, 4(2), 295 RSC.
- G. Kowalski, M. Tokarczyk, P. Dąbrowski, P. Ciepielewski, M. Możdżonek and W. Strupiński, et al., New X-ray insight into oxygen intercalation in epitaxial graphene grown on 4H-SiC(0001), J. Appl. Phys., 2015, 117(10), 105301 CrossRef.
- J. Binner and Y. Zhang, Characterization of silicon carbide and silicon powders by XPS and zeta potential measurement, J. Mater. Sci. Lett., 2001, 20(2), 123 CrossRef CAS.
- Z. Wen, G. Lu, S. Cui, H. Kim, S. Ci and J. Jiang, et al., Rational design of carbon network cross-linked Si–SiC hollow nanosphere as anode of lithium-ion batteries, Nanoscale, 2013, 6(1), 342 RSC.
- B. Yu, Z. Jiang, X.-Z. Tang, C. Y. Yue and J. Yang, Enhanced interphase between epoxy matrix and carbon fiber with carbon nanotube-modified silane coating, Compos. Sci. Technol., 2014, 99, 131 CrossRef CAS.
- C. D. Wagner, D. E. Passoja, H. F. Hillery, T. G. Kinisky, H. A. Six and W. T. Jansen, et al., Auger and photoelectron line energy relationships in aluminum–oxygen and silicon–oxygen compounds, J. Vac. Sci. Technol., 1982, 21(4), 933 CrossRef CAS.
- V. Georgakilas, J. N. Tiwari, K. Christian Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Noncovalent Functionalization of Graphene and Graphene Oxide for Energy Materials, Biosensing, Catalytic, and Biomedical Applications, Chem. Rev., 2016, 116(9), 5464 CrossRef CAS PubMed.
- H.-S. Tsai, C.-H. Hsiao, C.-W. Chen, H. Ouyang and J.-H. Liang, Synthesis of nonepitaxial multilayer silicene assisted by ion implantation, Nanoscale, 2016, 8(18), 9488 RSC.
- Z. Lei, G. Zhang, Y. Deng and C. Wang, Thermoresponsive Melamine Sponges with Switchable Wettability by Interface-Initiated Atom Transfer Radical Polymerization for Oil/Water Separation, ACS Appl. Mater. Interfaces, 2017, 9(10), 8967 CrossRef CAS PubMed.
- N. Liu, X. Huang and J. J. Dubowski, Selective area in situ conversion of Si (0 0 1) hydrophobic to hydrophilic surface by excimer laser irradiation in hydrogen peroxide, J. Phys. D: Appl. Phys., 2014, 47(38), 385106 CrossRef.
- G. Beamson and D. Briggs, Wiley, Chichester, Hantsche H. High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database (Beamson, G.; Briggs, D.),, J. Chem. Educ., 1993, 70(1), A25 Search PubMed.
- H. Yifan, X. Zhao, X. Song, H. Li, X. Zhu and C. Hao, The Interaction between Graphene and Oxygen Atom, Open Phys., 2016, 14, 690 Search PubMed.
- X. Qi, X. Guo and C. Zheng, Density functional study the interaction of oxygen molecule with defect sites of graphene, Appl. Surf. Sci., 2012, 259, 195 CrossRef CAS.
- F. R. Bagsican, A. Winchester, S. Ghosh, X. Zhang, L. Ma and M. Wang, et al., Adsorption energy of oxygen molecules on graphene and two-dimensional tungsten disulfide, Sci. Rep., 2017, 7(1), 1774 CrossRef PubMed.
- K. Elersic, M. Picman, N. Hauptman, U. Cvelbar and M. Mozetic, Interaction of Oxygen Species With Graphene and Pyrolytic-Graphite Surfaces, IEEE Trans. Plasma Sci., 2011, 39(11), 2812 CAS.
- G. Zu, T. Shimizu, K. Kanamori, Y. Zhu, A. Maeno and H. Kaji, et al., Transparent, Superflexible Doubly Cross-Linked Polyvinylpolymethylsiloxane Aerogel Superinsulators via Ambient Pressure Drying, ACS Nano, 2018, 12(1), 521 CrossRef CAS PubMed.
- H. Tamon, H. Ishizaka, T. Yamamoto and T. Suzuki, Influence of freeze-drying conditions on the mesoporosity of organic gels as carbon precursors, Carbon, 2000, 38(7), 1099 CrossRef CAS.
- J. D. Rimer, O. Trofymluk, R. F. Lobo, A. Navrotsky and D. G. Vlachos, Thermodynamics of Silica Nanoparticle Self-Assembly in Basic Solutions of Monovalent Cations, J. Phys. Chem. C, 2008, 112(38), 14754 CrossRef CAS.
- A. Pattammattel and C. V. Kumar, Kitchen Chemistry 101: Multigram Production of High Quality Biographene in a Blender with Edible Proteins, Adv. Funct. Mater., 2015, 25(45), 7088 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso and J. Rouquerol, et al., Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87(9), 1051 CAS.
- P. I. Ravikovitch and A. V. Neimark, Experimental Confirmation of Different Mechanisms of Evaporation from Ink-Bottle Type Pores: Equilibrium, Pore Blocking, and Cavitation, Langmuir, 2002, 18(25), 9830 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00994a |
| This journal is © The Royal Society of Chemistry 2019 |