
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCelecoxib attenuates hepatocellular proliferative capacity during hepatocarcinogenesis by modulating a PTEN/NF-κB/PRL-3 pathway†
Cong Zhang‡
a,
Junxuan Zhou‡a,
Junjie Hu‡ a,
Sheng Leia,
Ming Yuana,
Liang Chena,
Guihong Wangab and
Zhenpeng Qiu*ac
a,
Sheng Leia,
Ming Yuana,
Liang Chena,
Guihong Wangab and
Zhenpeng Qiu*ac
aCollege of Pharmacy, Hubei University of Chinese Medicine, No. 1, West Huangjiahu Road, Wuhan, People's Republic of China. E-mail: whuqq@hotmail.com
bKey Laboratory of Chinese Medicine Resource and Compound Prescription, Ministry of Education, Hubei University of Chinese Medicine, Wuhan, People's Republic of China
cKey Laboratory of Resources and Chemistry of Chinese Medicine, Hubei University of Chinese Medicine, Wuhan, People's Republic of China
First published on 2nd July 2019
Abstract
Although the efficacy of celecoxib on various cancer cell behaviors, including aberrant proliferation, in cultured hepatocellular carcinoma (HCC) cells has been demonstrated, whether celecoxib regulates cell proliferation by targeting PRL-3-associated signaling transduction during hepatocarcinogenesis in vivo has been incompletely studied. Here, we investigate the anti-proliferative efficacy of celecoxib in a rapid HCC mouse model established by hydrodynamic transfection of activated AKT and c-Met proto-oncogenes. The results show that celecoxib is effective at delaying the malignant transformation of hepatocytes by reducing the protein expression of Ki67, Cyclin D1 and c-Myc in the AKT/c-Met HCC-bearing mice. Mechanistically, celecoxib increases the protein expression of PTEN and suppresses the protein expression of NF-κB and PRL-3 in the liver of the HCC mice. Using PTEN-silenced and LPS-stimulated approaches in vitro, a mechanism by which celecoxib regulates a PTEN/NF-κB/PRL-3 pathway in HCC cells was illuminated. Altogether, our study demonstrates that celecoxib attenuates the hepatocellular proliferative capacity during hepatocarcinogenesis, which is probably attributable to its regulation of the PTEN/NF-κB/PRL-3 pathway.
Introduction
Hepatocellular carcinoma (HCC) is the 6th most prevalent cancer and the 3rd most frequent malignancy causing cancer-related death worldwide.1 Although therapeutic strategies against HCC, including surgical resection and systemic chemotherapy, have been used to improve the survival of HCC patients, the survival rate of these patients is still less than 14%.2 Excessive proliferation of hepatocytes is the primary indication of both hepatocarcinogenesis and poor prognosis in HCC patients.3 Thus, the discovery of therapeutic agents capable of inhibiting crucial oncogenic pathways fueling hepatocellular proliferation and malignant transformation is urgently needed for the management of HCC.Phosphatase and tensin homologue (PTEN) is a tumor suppressor that controls the intracellular levels of phosphatidylinositol-3,4,5-triphosphate and has an essential role in manipulating the phosphoinositide 3-kinase (PI3K) network to control cellular homeostasis.4 Mutations and loss of PTEN may occur during tumor initiation and progression that interferes with its enzymatic activity, subsequently leading to the activation of its downstream signaling pathways, and ultimately promoting aberrant proliferation, invasion and metastasis in human cancers.5 Hepatocytes lacking PTEN fails to harmonize the PI3K-linked program, which triggers a variety of cellular phenotypes that favor hepatocarcinogenesis.6 Furthermore, upon activation by systemic suppression of PTEN, AKT moves to the cytoplasm and nucleus, where it phosphorylates multiple downstream oncogenic targets,7,8 and further stimulates a pivotal transcriptional factor nuclear factor κB (NF-κB) DNA-binding activity for enhancing the transcription of pro-oncogenes, such as Cyclin D1 (ref. 9) and c-Myc.10
Mounting evidence shows that overexpression of phosphatases of regenerating liver (PRLs) accelerates tumorigenic and metastatic processes.11 Moreover, PRLs plays an important role in controlling cell cycles and accelerating tumor cell proliferation.12 Notably, PRL-3, one member of protein tyrosine phosphatase superfamily serving as a metastasis-associated phosphatase,13 is a potential molecular target for inhibition of proliferation through the AKT/mTORC1 signaling in glioblastoma.14 Furthermore, it has been reported that NF-κB is capable of binding the PRL-3 promoter and enhances gene transcription and expression of PRL-3 in breast cancer.15 However, whether chemicals could suppress hepatocellular proliferation during hepatocarcinogenesis by modulating the NF-κB/PRL-3 cascade remains unclear.
Recent studies have shown that selective cyclooxygenase type 2 (COX-2) inhibitors possess anti-tumor activity in various tumor types, including HCC.16 Celecoxib (Fig. 1), a kind of nonsteroidal anti-inflammatory drugs (NSAIDs), is effective at decelerating cell growth in multiple preclinical cancer models through suppression of proliferation.17 Although previous literatures showed the inhibitory efficacy of celecoxib in cultured HCC cells, the molecular mechanism by which celecoxib delays hepatocellular malignant transformation and tumorigenesis has not been well demonstrated. In this study, the anti-proliferative activity of celecoxib was investigated in an AKT/c-Met-driven HCC mouse model via hydrodynamic transfection that led to downregulation of PTEN expression and elevation of NF-κB and PRL-3 expression. Our data indicate that celecoxib impairs excessive proliferation during carcinogenesis of hepatocytes, potentially resulting from its regulation of a PTEN/NF-κB/PRL-3 pathway.
 | ||
| Fig. 1 The chemical structure of celecoxib. | ||
Methods and materials
Chemicals and reagents
Celecoxib powder and capsules were obtained from Sigma-Aldrich (St. Louis, MO, USA) and Pfizer Pharmaceuticals LLC (Celebrex, New York, USA), respectively. Lipopolysaccharide (LPS) was purchased from Invitrogen (San Diego, CA, USA). A hydrosoluble tetrazolium salt WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) was obtained from Dojindo Laboratories (Kumamoto, Japan) for the CCK-8 assay. The E.Z.N.A.® Plasmid Maxi Kit (Omega Bio-tek, Inc. Doraville, GA, USA) was used for plasmid extractions and purifications before hydrodynamic and transient transfection into mice and in vitro HCC cells, respectively.Hydrodynamic injection and animal administration
Female wild-type FVB/N mice were purchased from Charles River (Beijing, China) and housed under controlled temperature (23 °C) and lighting (12 h light/12 h dark cycle) with free access to water and standard mouse chow. FVB/N mice were divided into four groups (n = 6): (1) wild-type control mice (WT); (2) AKT/c-Met HCC mice (AKT/c-Met, co-expression of AKT/c-Met via hydrodynamic transfection); (3) AKT/c-Met HCC mice treated with low dose of celecoxib (AKT/c-Met + CELE-L, celecoxib 125 mg per kg per day); AKT/c-Met HCC mice treated with high dose of celecoxib (AKT/c-Met + CELE-H, celecoxib 250 mg per kg per day). Hydrodynamic transfection was performed as previously described.18 A volume of 2 mL normal saline solution containing 20 μg pT3-EF1α-HA-myr-AKT, 20 μg pT3-EF1α-V5-c-Met and 1.6 μg pCMV-sleeping beauty transposase (SB) was injected into the caudal vein of the FVB/N mice within 7 seconds. Since the AKT/c-Met-injected mice developed a lethal burden of liver tumor within 6 to 8 weeks post-injection,18 mice were sacrificed by decapitation 6 weeks after injection in the present study. This study was performed in accordance with the Animal Management Rules of the Ministry of Health of the People's Republic of China and were approved by the Animal Care and Use Committee of the Hubei University of Chinese Medicine (Wuhan, China).Cell culture, transient transfection and treatment
The human HCC cell lines (Huh-7 and SMMC-7721) were purchased from the American Type Culture Collection and cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (1 mM) and streptomycin (1 mM) at 37 °C in a humidified 5% CO2 atmosphere. The transient transfection of HCC cells with pT3-EF1α-HA-myr-AKT/c-Met or sgPTEN, a sgRNA-plasmid vector targeting human PTEN (Santa Cruz Biotechnology), was performed according to the manufacturer's protocol using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA). The transfected cells were then incubated with celecoxib (0–50 μM) for 24 h.Quantitative real-time polymerase chain reaction (qPCR)
The total cellular RNA was extracted using the TRIzol reagent (Invitrogen, USA). Oligo (dT) 18 primer was used to bind the poly-A tails of mRNA for generating complementary DNA (cDNA). The qPCR assay was performed on a Mini-Opticon (Bio-Rad, USA) using Fast Start Universal SYBR Green Master Mix (Roche, USA). The housekeeping gene β-actin was employed as an internal control. Primers for qPCR are listed in the ESI Table 1.†Western blotting and immunohistochemistry
The protein preparation and immunoblotting procedures for hepatic tissues and cultured cells were performed as previously described.19 The specific primary antibodies used in the immunoblotting assay are listed in ESI Table 2.†The nuclear antigen Ki67, NF-κB and E-cadherin were detected by immunohistochemistry in the liver tissues of mice. After deparaffinization of the liver tissue sections, antibody incubation and visualization of immunoreactivity were conducted as previously described.20 Specific primary antibodies used for immunohistochemistry are listed in ESI Table 2.†
Statistical analysis
Data analysis was performed with Prism 6 (GraphPad, USA). Data are shown as the mean ± S.D. from at least three independent experiments. Statistical significance was measured using a two-tailed unpaired t-test (between two groups) or ANOVA (between three or more groups) and a statistical cutoff of P < 0.05.Results
Celecoxib suppresses aberrant hepatocellular proliferation in AKT/c-Met-induced HCC-bearing mice
Immunohistochemical assessment of the proportion of cells positive for Ki67 is the most common approach for determining the proliferative index and evaluating the prognosis of cancer patients with HCC and other neoplasms.21 As expected, we found that hepatocellular tumors consisting mainly of lipid-rich cells (ESI Fig. 1†) and highly proliferative Ki67-positive liver tumor cells (Fig. 2A) were eradicated upon celecoxib administration in the CELE-H group. Moreover, compared to the control group, celecoxib treatment downregulated both the protein and mRNA expression of two kinds of G1-phase regulatory proteins, c-Myc and Cyclin D1 in the CELE-H group (Fig. 2B–D). Thus, the results indicate that the inhibition of cell proliferation might be a potential mechanism by which celecoxib alleviates AKT/c-Met-triggered hepatocarcinogenesis.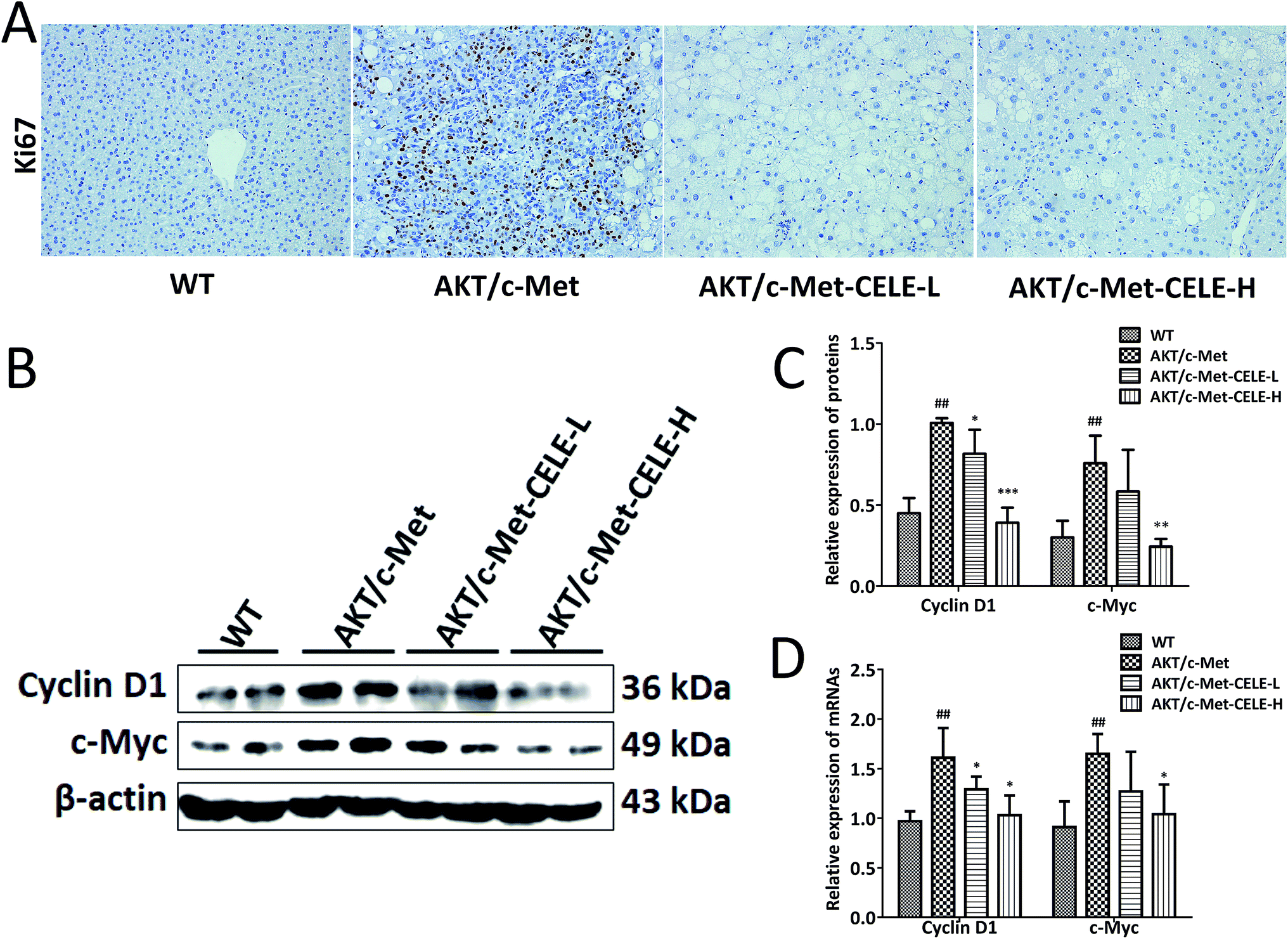 | ||
| Fig. 2 Celecoxib inhibits AKT/c-Met-driven hepatocellular proliferation during hepatocarcinogenesis. (A) Observation of nuclear Ki67 staining in the liver tissues. AKT/c-Met-induced high expression of Ki67 was suppressed by celecoxib treatment. Ki67 positive cells are recognized by their yellow-brown nuclear staining. Original magnification: 200×. (B) Celecoxib suppressed the protein expression of Cyclin D1 and c-Myc in the livers of AKT/c-Met HCC-bearing mice. Liver samples from each group were subjected to Western blot assay, and representative bands are shown. (C and D) Histograms show the relative protein and mRNA expression levels of Cyclin D1 and c-Myc quantified by densitometric analysis of Western blot and qPCR assay, respectively. The housekeeping gene β-actin was used as an internal control. Data are expressed as the mean ± S.D., n = 3 per group. ##P < 0.01 vs. the WT group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the AKT/c-Met group. CELE-L and CELE-H represent intragastric administration of celecoxib at low (125 mg) and high (250 mg) doses, respectively. | ||
Celecoxib elevates the protein expression of PTEN and inhibits the protein expression of NF-κB and PRL-3 in HCC-bearing mice
Using Western blotting, immunohistochemistry and qPCR analyses as described in the Methods, we investigated the molecular mechanisms underlying the inhibitory efficacy of celecoxib on hepatocellular proliferation in mice. The results revealed that celecoxib treatment elevated the level of PTEN expression in liver tissues relative to that in the AKT/c-Met group (Fig. 3A and B). Moreover, celecoxib effectively reduced the protein expression of NF-κB in mice of the CELE-H group (Fig. 3A–D). PRL-3 is commonly overexpressed to promote cell proliferation in tumor malignant transformation.22 Of note, our results indicated that the mRNA and protein expression of PRL-3 were suppressed upon celecoxib administration in mice of the CELE-L and CELE-H groups (Fig. 3A–C). Furthermore, celecoxib enhanced the immunohistochemical staining of E-cadherin, a known downstream negative effector of PRL-3, in mice of the CELE-L and CELE-H groups (Fig. 3D). Therefore, these data suggested that celecoxib obstructs hepatocellular proliferation in the AKT/c-Met-transfected mice, which is probably due to its regulation of PTEN, NF-κB and PRL-3 expression.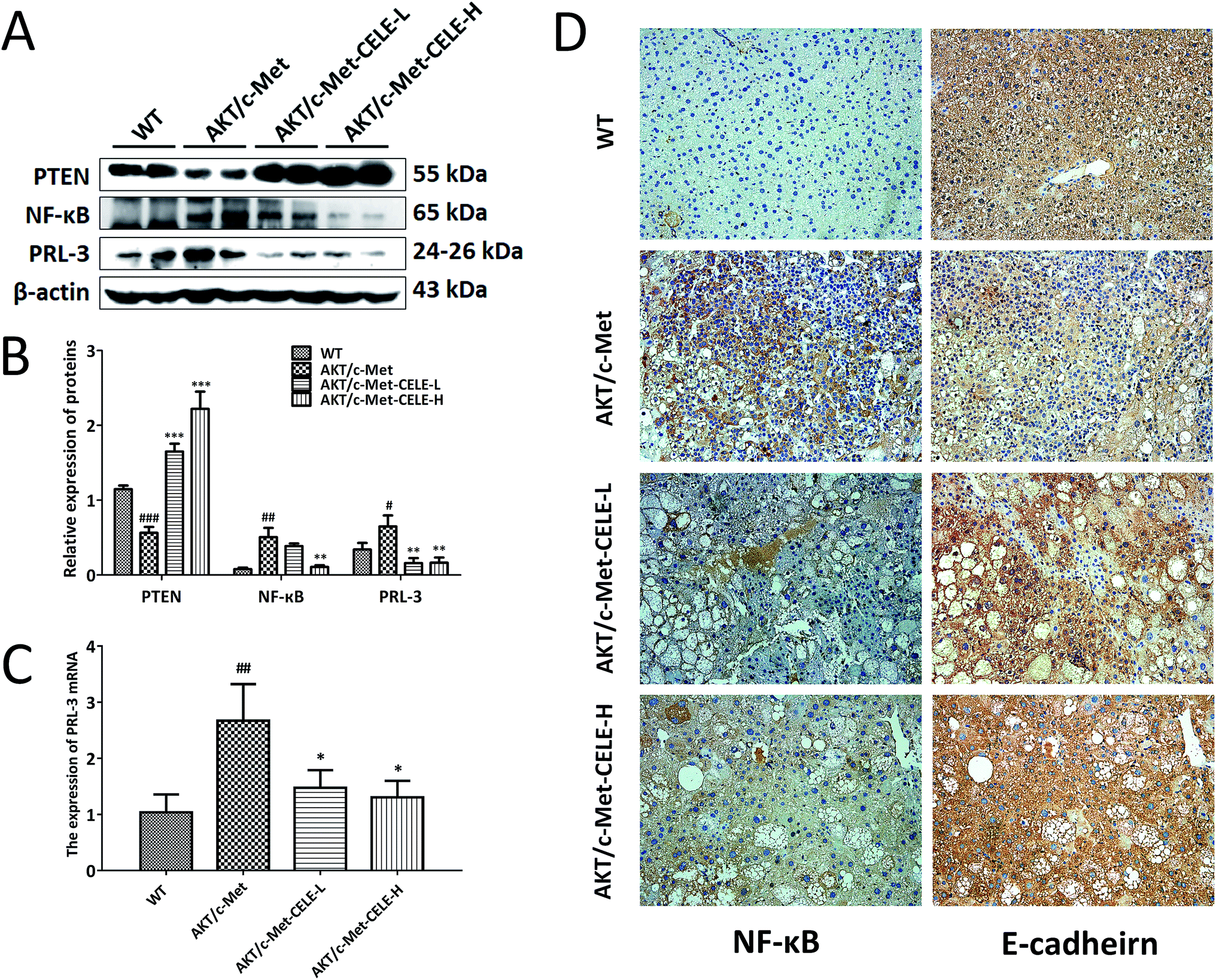 | ||
| Fig. 3 Celecoxib elevates the expression of PTEN and E-cadherin, and suppresses the expression of NF-κB and PRL-3. (A) Celecoxib enhances the protein expression of PTEN and downregulates the protein expression of NF-κB and PRL-3 in livers of AKT/c-Met HCC-bearing mice. Liver samples from each group were quantified by Western blot assay, and representative bands are shown. (B) Histograms represent the protein expression of PTEN, NF-κB and PRL-3 quantified by densitometric analysis of Western blots. (C) Histograms show the mRNA expression of PRL-3 quantified by qPCR. (D) The IHC staining of NF-κB and E-cadherin shows that the AKT/c-Met-induced high expression of NF-κB and low expression of E-cadherin were improved by celecoxib treatment. Original magnification: 200×. The housekeeping gene β-actin was used as an internal control. Data are expressed as the mean ± S.D., n = 3 per group. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the WT group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the AKT/c-Met group. CELE-L and CELE-H represent intragastric administration of celecoxib at low (125 mg) and high (250 mg) doses, respectively. | ||
Celecoxib upregulates the protein expression of PTEN and inhibits the protein expression of NF-κB and PRL-3 in HCC cells
To confirm whether celecoxib is efficacious in HCC-bearing mice by regulating a PTEN/NF-κB/PRL-3 pathway, we further evaluated the protein expression of the aforementioned targets at different concentrations (10–50 μM) of celecoxib in cultured HCC cells. The results revealed that celecoxib enhanced the expression of PTEN and repressed the expression of NF-κB and PRL-3 in a concentration-dependent manner in SMMC-7721 (Fig. 4A and B) and Huh-7 (Fig. 4C and D) cells. Similarly, celecoxib prevented the enhancing effect of AKT/c-Met transfection on the protein expression of NF-κB and PRL-3 in a dose-dependent manner (Fig. 4E and F). Furthermore, consistent with in vivo findings, the protein expression of Cyclin D1 and c-Myc was suppressed by celecoxib incubation in the AKT/c-Met overexpressing cells (Fig. 4E and F).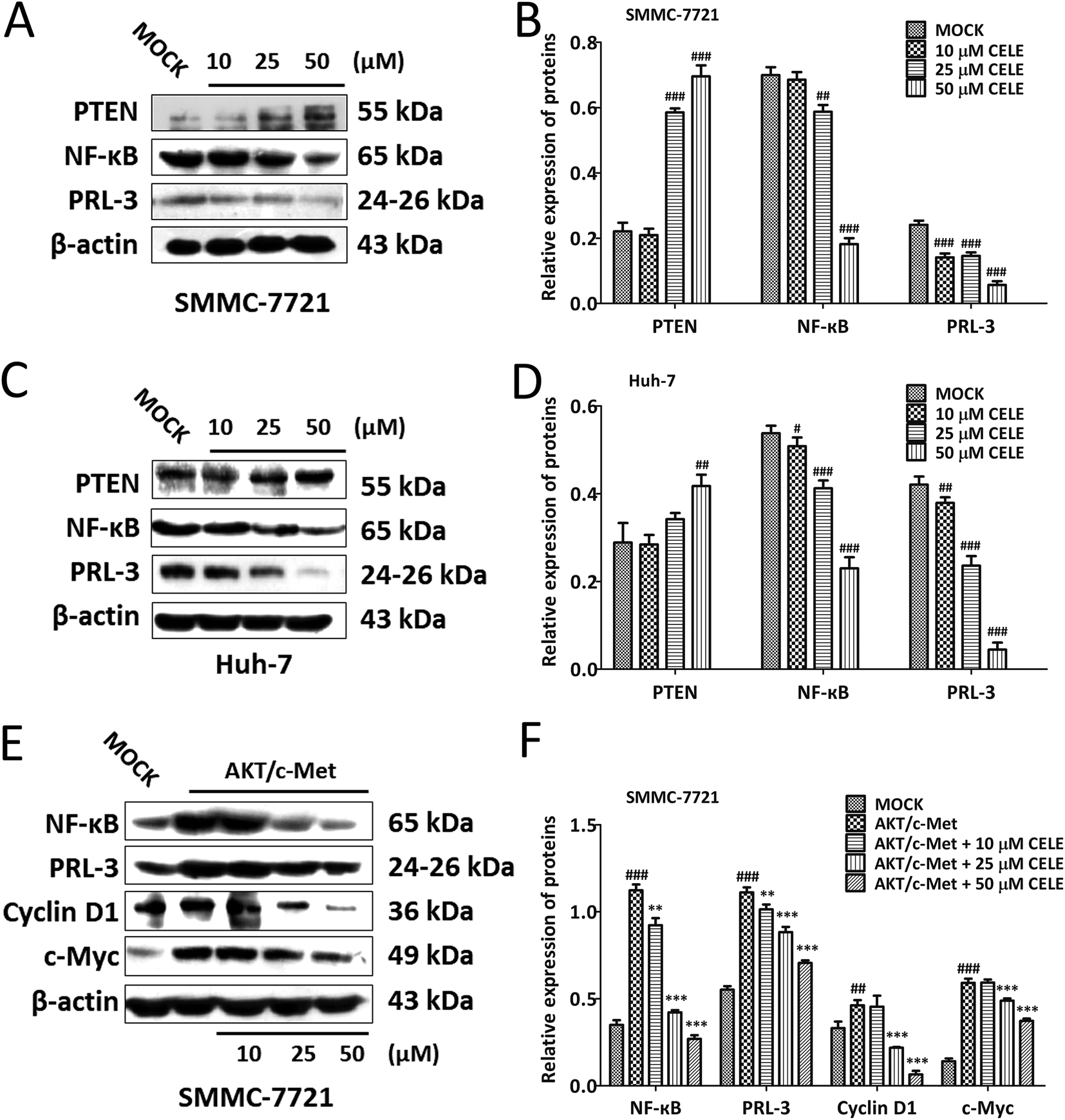 | ||
| Fig. 4 Celecoxib enhances the protein expression of PTEN and inhibits the protein expression of NF-κB and PRL-3 in HCC cells. Western blot analysis of the protein expression of PTEN, NF-κB and PRL-3 in (A) SMMC-7721 cells and (C) Huh-7 cells. Histograms in (B) and (D) represent the intensity of the immunoblot bands in (A) and (C), respectively. (E) Western blot analysis for the protein expression of PTEN, NF-κB and PRL-3 in AKT/c-Met-transfected SMMC-7721 cells. Histograms in (F) represent the intensity of the immunoblot bands in (E) quantified by densitometry. The housekeeping gene β-actin was used as an internal control. Data are expressed as the mean ± S.D., n = 3 per group. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the MOCK group. **P < 0.01, ***P < 0.001 vs. the AKT/c-Met group. | ||
Celecoxib regulates a PTEN/NF-κB/PRL-3 pathway in HCC cells
Next, genetic and LPS-induced approaches were used to expound the mechanism by which celecoxib alleviates the proliferative capacity of hepatocytes. To do so, SMMC-7721 cells were transiently transfected with the sgRNA-plasmid targeting PTEN were further incubated with celecoxib (10–50 μM) for 24 hours. The data suggested that PTEN silence led to the elevated expression of NF-κB and PRL-3, indicating that PTEN may act upstream of NF-κB and PRL-3 (Fig. 5A and B). Notably, celecoxib treatment restored the protein expression of PTEN, and repressed the sgPTEN-mediated elevated protein expression of NF-κB and PRL-3 (Fig. 5A and B). To determine whether celecoxib could regulate the NF-κB/PRL-3 signaling transduction in hepatocytes, we assessed the effect of celecoxib on PRL-3 expression in the presence of LPS, well known to trigger innate immune responses and enhance the expression and activity of NF-κB.23 As shown in Fig. 5C and D, the protein expression of NF-κB and PRL-3 was elevated by LPS stimulation in SMMC-7721 HCC cells. Further, the LPS-induced HCC cells in the presence of celecoxib (10–50 μM) for 24 h showed a dose-dependent reduction of NF-κB and PRL-3 expression at the protein level (Fig. 5C and D). Overall, our data indicate that celecoxib is capable of regulating the PTEN/NF-κB/PRL-3 pathway in HCC cells.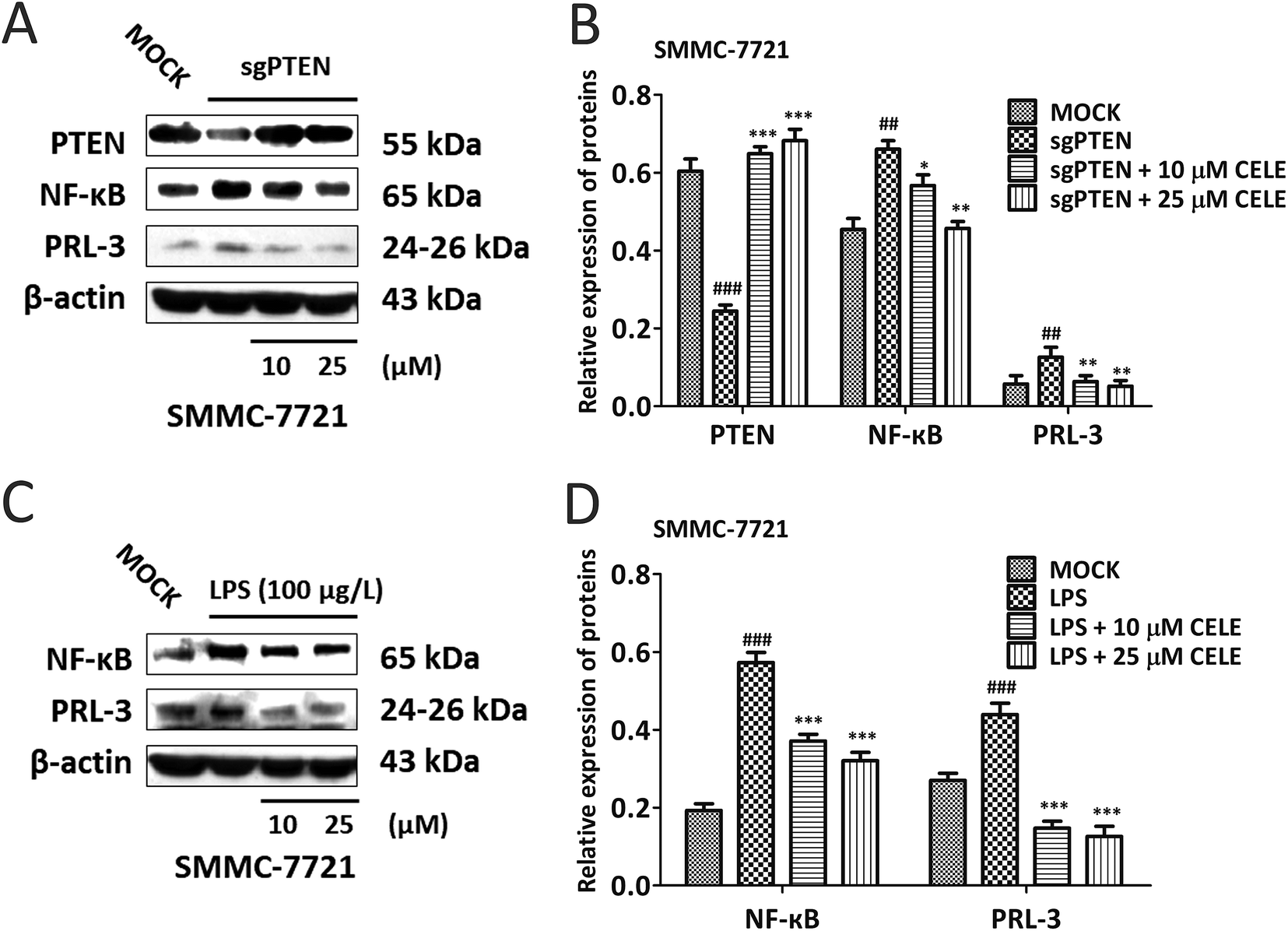 | ||
| Fig. 5 Celecoxib regulates a PTEN/NF-κB/PRL-3 pathway in HCC Cells. (A) Western blot analysis of the protein expression of PTEN, NF-κB and PRL-3 in PTEN-silenced SMMC-7721 cells. Histograms in (B) represent the intensity of the immunoblot bands in (A) quantified by densitometry. (C) Western blotting analysis for the protein expression of NF-κB and PRL-3 in LPS-induced SMMC-7721 cells. Histograms in (D) represent the intensity of the immunoblot bands in (C) quantified by densitometry. The housekeeping gene β-actin was used as an internal control. Data are expressed as the mean ± S.D., n = 3 per group. ##P < 0.01, ###P < 0.001 vs. the MOCK group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the sgPTEN-transfected group or the LPS-simulated group. | ||
Discussion
Although previous studies have reported that COX inhibitors suppress cell proliferation and tumor formation, whether COX-2 inhibitors can interfere with this process during hepatocarcinogenesis remains poorly defined. In the present study, we sought to determine the anti-proliferative efficacy of a selective COX-2 inhibitor, celecoxib, in a rapid HCC mouse model established by hydrodynamic injection. The present data suggest that celecoxib diminished aberrant hepatocellular proliferation during hepatocarcinogenesis via a PTEN/NF-κB/PRL-3 pathway.In this study, celecoxib could repress the expression of proliferation-associated regulators (Ki67, c-Myc and Cyclin D1) in AKT/c-Met-driven hepatocarcinogenesis, indicating that its efficacy delaying hepatic tumorigenesis is mainly due to the inhibition of proliferation. Mechanistically, the protein expression of PTEN was elevated by celecoxib treatment in the AKT/c-Met-transfected mice, supporting the important tumor suppressor role of PTEN for obstructing malignant transformation of hepatocytes in HCC, as observed in other chemotherapeutical processes in HCC cells.24,25 In the cytoplasm, activated/phosphorylated extracellular-signal-regulated kinases (ERKs) phosphorylate a separate set of substrates including kinases and cytoskeletal proteins in cellular signaling responses.26 Aberrant ERK1/2 signaling is in part responsible for oncogenesis by targeting PTEN. Indeed, blockade of ERK activation by the MAP kinase/ERK kinase (MEK) inhibitor rescues PTEN suppression at both the transcriptional and post-translated levels.27 Of note, our group28 and others29 have previously reported that celecoxib was effective at precluding the phosphorylation of ERK1/2 at Thr202/Tyr204 residues in hepatocytes. Thus, we believe that celecoxib-induced elevation of PTEN expression is highly relevant to its effect on ERK phosphorylation, especially in the context of hepatocarcinogenesis with overexpression of c-Met that serves as an upstream modulator of ERK/MEK signaling for stimulating ERK activation. In addition, COX-2 is the inducible isoform of cyclooxygenases (COXs), the rate-limiting enzymes converting arachidonic acid to prostaglandins (PGEs), expressed constitutively during hepatocarcinogenesis, potentially fueling the excessive inflammatory response required for malignant transformation.30 Selective COX-2 inhibitors (including celecoxib) bind the active center of COX-2 and further block its enzymatic function, thereby alleviating the production of PGEs (especially PGE2). Indeed, previous reports suggests that induction of PTEN expression is a typical feature for the efficacy of celecoxib and another selective COX-2 inhibitor NS398 on cancer progression,31,32 indicating that suppression of PGE synthesis by celecoxib treatment contributes to the elevation of PTEN expression in the present investigation. Furthermore, accumulating evidence demonstrates that microRNAs (miRNAs) with pro-oncogenic properties may represent available targets for the anti-proliferative actions of NSAIDs in a wide variety of cancer subtypes.33 For instance, aberrant expression of miR-21 is involved in both the initiation and progression of human cancers and plays its oncogenic roles in tumor malignant transformation by modulating PTEN expression directly at the 3′ untranslated regions of PTEN.34 Notably, upon administration of aspirin and NS398 repressed the expression of miR-21 in colon gastric carcinoma cells.31,33 Thus, it would be important to further test whether celecoxib influences a potential COX-2/PGEs/miR-21 axis that sequentially regulates the expression of PTEN in HCC initiation.
The deficiency or loss of PTEN contributes to the high proliferation index in HCC by acting through its downstream AKT/mTOR pathway involved in modulating phenotypic characteristics of cancer cells, including cell proliferation.34 Inversely, elevation of PTEN expression inhibits colon cancer progression by repressing AKT phosphorylation.35 Although the previous evidence demonstrates that celecoxib induced apoptosis through its regulation of PDK1/PTEN/AKT pathway in HCC cells in vitro,36 the PTEN-associated mechanism by which celecoxib inhibits cell proliferation in hepatocarcinogenesis remains unclear. In fact, NF-κB is a dimer of Rel proteins that is sequestered in the cytoplasm as an inactive form through interaction with an inhibitory κB (IκB) protein. The PTEN/AKT/mTORC1 cascade activates NF-κB predominantly through mechanisms that regulate nuclear translocation or transactivation potential. Moreover, the degradation of IκB is responsible for constitutive activation of the NF-κB signaling in HCC.37 Furthermore, ubiquitination regulates the degradation of IκB, processing of NF-κB precursors, and activation of IκB kinase (IKK) in the NF-κB pathway.38 Inhibition of NF-κB by proteasome inhibitors or prevention of IκB phosphorylation induces apoptosis in myeloma cell lines.39 In accordance with previous data in human gastric cancer cell lines,40 our results revealed that the efficacy of celecoxib in AKT/c-Met HCC-bearing mice was associated with a marked reduction in the protein expression of NF-κB subunit p65. Furthermore, our in vitro genetic studies demonstrated that PTEN is required for celecoxib-induced repression of NF-κB expression. Therefore, it is possible that celecoxib decreases the elevated protein expression of NF-κB during AKT/c-Met-driven hepatocarcinogenesis due to its suppressing action on IκB ubiquitination and degradation, which is associated with the celecoxib-induced elevation of PTEN expression. Obviously, it would be important to further test whether celecoxib influences the post-translational modification of IκB that regulates NF-κB activity in HCC.
Accumulating evidence indicates that PRL-3 could promote tumor cell proliferation and survival in various kinds of neoplasms, including lung, breast, colorectal and gastric carcinoma.41–44 The aberrant PRL-3 expression fuels cell cycle progression by elevating the expression of Cyclin D1 and cyclin A-cyclin-dependent kinase 2 (CDK2), activating the signal transducer and activator of transcription 5 (STAT5)/AKT pathways and suppressing the expression of p21 WAF1/CIP1, a CDK inhibitory protein that serves as an effector of the p53-dependent G1 arrest.45 However, whether PRL-3 is a potential target for the anti-proliferative efficacy of celecoxib during HCC initiation is still unclear. For the first time, we found that celecoxib suppressed the expression of PRL-3 at the transcriptional and post-transcriptional levels during hepatocarcinogenesis. Importantly, NF-κB, the ubiquitous transcription factor which binds DNA as a heterodimeric complex composed of members of the Rel/NF-κB family of polypeptides, is responsible for transcriptionally activating the PRL-3 expression by binding the PRL-3 promoter.15 Our data suggests that celecoxib is capable of downregulating the expression of NF-κB, which might impair its nuclear translocation and obstruct transcriptional activity, subsequently resulting in the suppression of PRL-3 mRNA transcription. Intriguingly, PRLs can also enhance the phosphorylation of ERK1/2, which could promote tumor cell proliferation by transcriptionally controlling the expression of c-Myc and Cyclin D1 in multiple types of cancer.46–48 Furthermore, as a metastasis associated tyrosine phosphatase, PRL-3 post-transcriptionally decreases the expression of PTEN by affecting multiple effectors (e.g., vinculin and RhoA).49 Thus, further investigations need to be performed to elucidate whether celecoxib could affect the PRL-3/ERK1/2 pathway or a potential PRL-3/PTEN feedback loop in excessively proliferating hepatocytes during hepatocarcinogenesis with AKT and c-Met co-activation.
Conclusion
Altogether, our in vitro and in vivo data demonstrate that celecoxib could suppress hepatocellular proliferation by interfering with a PTEN/NF-κB/PRL-3 pathway in hepatocarcinogenesis. Importantly, our study introduces PRL-3 as a novel therapeutic target of celecoxib in hepatocarcinogenesis. The present study also warrants further investigation of celecoxib and other COX-2 inhibitors for their anti-proliferative effect against HCC initiation. Furthermore, combined therapy of celecoxib with other drugs such as multikinase inhibitors should be evaluated to establish the clinical utility of NSAIDs for the prevention and treatment of HCCs.Conflicts of interest
The authors declare that they have no competing interests.Abbreviations
| cDNA | complementary DNA |
| CDK2 | cyclin-dependent kinase 2 |
| COX-2 | cyclooxygenase-2 |
| COXs | cyclooxygenases |
| ERK1/2 | extracellular signal-regulated kinases 1/2 |
| FASN | fatty acid synthase |
| HCC | hepatocellular carcinoma |
| IκB | inhibitory κB |
| IKK | IκB kinase |
| Ki-67 | antigen Ki-67 |
| LPS | lipopolysaccharide |
| MEK | MAPK/ERK kinase |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NF-κB | nuclear factor κB |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| PGs | prostaglandins |
| PI3K | phosphoinositide 3-kinase |
| PRL | phosphatase of regenerating liver |
| PTP4A | class IVa of protein tyrosine phosphatase |
| PTEN | phosphatase and tensin homologue |
| qPCR | quantitative real-time polymerase chain reaction |
| STAT5 | signal transducer and activator of transcription 5 |
Acknowledgements
This study was supported by a grant from the National Natural Science Foundation of China (81603338) Dr Zhenpeng Qiu. We thank Dr Xin Chen of the University of California, San Francisco for providing the materials for the hydrodynamic transfection.References
- J. Wang, T. Yang, H. Chen, H. Li and S. Zheng, Biochem. Biophys. Res. Commun., 2018, 498, 424–430 CrossRef CAS PubMed.
- G. Deng, S. Zeng, Y. Qu, Q. Luo, C. Guo, L. Yin, Y. Han, Y. Li, C. Cai, Y. Fu and H. Shen, J. Exp. Clin. Cancer Res., 2018, 37, 156 CrossRef PubMed.
- Y. Xiong, S. Wu, H. Yu, J. Wu, Y. Wang, H. Li, H. Huang and H. Zhang, Exp. Cell Res., 2018, 371, 185–195 CrossRef CAS PubMed.
- M. L. Sulis and R. Parsons, Trends Cell Biol., 2003, 13, 478–483 CrossRef CAS PubMed.
- L. Liu, H. Long, Y. Wu, H. Li, L. Dong, J. L. Zhong, Z. Liu, X. Yang, X. Dai, L. Shi, M. Ren and Z. Lin, Cell. Signalling, 2018, 50, 90–99 CrossRef CAS PubMed.
- J. J. Li, H. K. Yin, D. X. Guan, J. S. Zhao, Y. X. Feng, Y. Z. Deng, X. Wang, N. Li, X. F. Wang, S. Q. Cheng, Y. Bao and D. Xie, Br. J. Cancer, 2018, 118, 1337–1348 CrossRef CAS PubMed.
- C. Y. Chen, J. Chen, L. He and B. L. Stiles, Front. Endocrinol., 2018, 9, 338 CrossRef PubMed.
- R. L. Dillon, D. E. White and W. J. Muller, Oncogene, 2007, 26, 1338–1345 CrossRef CAS PubMed.
- A. S. Baldwin, J. Clin. Invest., 2001, 107, 241–246 CrossRef CAS PubMed.
- F. A. La Rosa, J. W. Pierce and G. E. Sonenshein, Mol. Cell. Biol., 1994, 14, 1039–1044 CrossRef CAS PubMed.
- M. D. Lin, H. T. Lee, S. C. Wang, H. R. Li, H. L. Hsien, K. W. Cheng, Y. D. Chang, M. L. Huang, J. K. Yu and Y. H. Chen, BMC Dev. Biol., 2013, 13, 18 CrossRef.
- J. M. Salamoun, K. E. McQueeney, K. Patil, S. J. Geib, E. R. Sharlow, J. S. Lazo and P. Wipf, Org. Biomol. Chem., 2016, 14, 6398–6402 RSC.
- W. B. Zhao, Y. Li, X. Liu, L. Y. Zhang and X. Wang, Int. J. Mol. Med., 2008, 22, 187–192 CAS.
- L. Wang, J. Liu, Z. Zhong, X. Gong, W. Liu, L. Shi and X. Li, Brain Res., 2016, 1646, 441–450 CrossRef CAS PubMed.
- H. H. Gari, G. D. DeGala, M. S. Lucia and J. R. Lambert, Oncogenesis, 2016, 5, e255 CrossRef CAS PubMed.
- C. Y. Wu, C. J. Wang, C. C. Tseng, H. P. Chen, M. S. Wu, J. T. Lin, H. Inoue and G. H. Chen, World J. Gastroenterol., 2005, 11, 3197–3203 CrossRef CAS PubMed.
- B. Vijayakumar, V. Kannappan and V. Sathyanarayanamoorthi, J. Mol. Struct., 2016, 1121, 16–25 CrossRef CAS.
- J. J. Hu, L. Che, L. Li, M. G. Pilo, A. Cigliano, S. Ribback, X. L. Li, G. Latte, M. Mela, M. Evert, F. Dombrowski, G. H. Zheng, X. Chen and D. F. Calvisi, Sci. Rep., 2016, 6, 20484 CrossRef CAS PubMed.
- J. J. Hu, X. Li, J. X. Zhou, C. Zhang, G. H. Zheng and Z. P. Qiu, RSC Adv., 2018, 8, 13976–13983 RSC.
- S. S. Zhang, X. H. Song, D. Cao, Z. Xu, B. A. Fan, L. Che, J. J. Hu, B. Chen, M. J. Dong, M. G. Pilo, A. Cigliano, K. Evert, S. Ribback, F. Dombrowski, R. M. Pascale, A. Cossu, G. Vidili, A. Porcu, M. M. Simile, G. M. Pes, G. Giannelli, J. Gordan, L. X. Wei, M. Evert, W. M. Cong, D. F. Calvisi and X. Chen, J. Hepatol., 2017, 67, 1194–1203 CrossRef CAS PubMed.
- C. Stroescu, A. Dragnea, B. Ivanov, C. Pechianu, V. Herlea, O. Sgarbura, A. Popescu and I. Popescu, J. Gastrointestin. Liver Dis., 2008, 17, 411–417 Search PubMed.
- F. Liang, J. Liang, W. Q. Wang, J. P. Sun, E. Udho and Z. Y. Zhang, J. Biol. Chem., 2007, 282, 5413–5419 CrossRef CAS.
- T. Luedde and R. F. Schwabe, Nat. Rev. Gastroenterol. Hepatol., 2011, 8, 108–118 CrossRef CAS PubMed.
- C. He, X. Dong, B. Zhai, X. Jiang, D. Dong, B. Li, H. Jiang, S. Xu and X. Sun, Oncotarget, 2015, 6, 28867–28881 Search PubMed.
- X. Zhang, S. Jia, S. Yang, Y. Yang, T. Yang and Y. Yang, J. Cell. Biochem., 2012, 113, 3528–3535 CrossRef CAS.
- J. W. Ramos, Int. J. Biochem. Cell Biol., 2008, 40, 2707–2719 CrossRef CAS PubMed.
- S. E. Beck and J. M. Carethers, Cancer Biol. Ther., 2007, 6, 1313–1317 CAS.
- Z. P. Qiu, C. Zhang, J. X. Zhou, J. J. Hu, L. Sheng, X. Li, L. Chen, X. Li, X. K. Deng and G. H. Zheng, Mol. Carcinog., 2019, 58, 31–41 CrossRef CAS PubMed.
- Y. H. Paik, J. K. Kim, J. I. Lee, S. H. Kang, D. Y. Kim, S. H. An, S. J. Lee, D. K. Lee, K. H. Han, C. Y. Chon, S. I. Lee, K. S. Lee and D. A. Brenner, Gut, 2009, 58, 1517–1527 CrossRef CAS PubMed.
- H. Koga, S. Sakisaka, M. Ohishi, T. Kawaguchi, E. Taniguchi, K. Sasatomi, M. Harada, T. Kusaba, M. Tanaka, R. Kimura, Y. Nakashima, O. Nakashima, M. Kojiro, T. Kurohiji and M. Sata, Hepatology, 1999, 29, 688–696 CrossRef CAS PubMed.
- H. Li, J. Cheng, Y. Mao, M. Jiang and X. Fan, OncoTargets Ther., 2015, 8, 3245–3253 CrossRef CAS PubMed.
- T. D. Kabir, R. J. Leigh, H. Tasena, M. Mellone, R. D. Coletta, E. K. Parkinson, S. S. Prime, G. J. Thomas, I. C. Paterson, D. Zhou, J. McCall, P. M. Speight and D. W. Lambert, Aging, 2016, 8, 1608–1635 CAS.
- E. Yiannakopoulou, Cell. Oncol., 2014, 37, 167–178 CrossRef CAS PubMed.
- F. Meng, R. Henson, H. Wehbe-Janek, K. Ghoshal, S. T. Jacob and T. Patel, Gastroenterology, 2007, 133, 647–658 CrossRef CAS PubMed.
- L. L. Zhang, G. G. Mu, Q. S. Ding, Y. X. Li, Y. B. Shi, J. F. Dai and H. G. Yu, J. Biol. Chem., 2015, 290, 15018–15029 CrossRef CAS PubMed.
- J. Leng, C. Han, A. J. Demetris, G. K. Michalopoulos and T. Wu, Hepatology, 2003, 38, 756–768 CrossRef CAS PubMed.
- G. He and M. Karin, Cell Res., 2011, 21, 159–168 CrossRef CAS PubMed.
- Z. J. Chen, Nat. Cell Biol., 2005, 7, 758–765 CrossRef CAS PubMed.
- H. Ni, M. Ergin, Q. Huang, J. Z. Qin, H. M. Amin, R. L. Martinez, S. Saeed, K. Barton and S. Alkan, Br. J. Haematol., 2001, 115, 279–286 CrossRef CAS PubMed.
- Z. Chen, M. Liu, X. Liu, S. Huang, L. Li, B. Song, H. Li, Q. Ren, Z. Hu, Y. Zhou and L. Qiao, Int. J. Mol. Med., 2013, 32, 93–100 CrossRef PubMed.
- Y. X. Lian, R. Chen, Y. H. Xu, C. L. Peng and H. C. Hu, Gene, 2012, 511, 169–176 CrossRef CAS PubMed.
- P. den Hollander, K. Rawls, A. Tsimelzon, J. Shepherd, A. Mazumdar, J. Hill, S. A. Fuqua, J. C. Chang, C. K. Osborne, S. G. Hilsenbeck, G. B. Mills and P. H. Brown, Cancer Res., 2016, 76, 1942–1953 CrossRef CAS PubMed.
- J. Zhang, Z. Xiao, D. Lai, J. Sun, C. He, Z. Chu, H. Ye, S. Chen and J. Wang, Br. J. Cancer, 2012, 107, 352–359 CrossRef CAS PubMed.
- E. R. Sharlow, P. Wipf, K. E. McQueeney, A. Bakan and J. S. Lazo, Expert Opin. Invest. Drugs, 2014, 23, 661–673 CrossRef CAS PubMed.
- S. Qu, B. Liu, X. L. Guo, H. S. Shi, M. F. Zhou, L. Li, S. L. Yang, X. Z. Tong and H. H. Wang, Cancer, 2014, 120, 2130–2141 CrossRef CAS PubMed.
- G. Pintus, B. Tadolini, A. M. Posadino, B. Sanna, M. Debidda, F. Bennardini, G. Sava and C. Ventura, Eur. J. Biochem., 2002, 269, 5861–5870 CrossRef CAS PubMed.
- E. Halilovic, Q. B. She, Q. Ye, R. Pagliarini, W. R. Sellers, D. B. Solit and N. Rosen, Cancer Res., 2010, 70, 6804–6814 CrossRef CAS PubMed.
- Y. Cao, Y. Tu, J. Mei, Z. Li, Z. Jie, S. Xu, L. Xu, S. Wang and Y. Xiong, Mol. Med. Rep., 2013, 7, 1805–1811 CrossRef CAS PubMed.
- H. Wang, S. Y. Quah, J. M. Dong, E. Manser, J. P. Tang and Q. Zeng, Cancer Res., 2007, 67, 2922–2926 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00429g |
| ‡ Cong Zhang, Junxuan Zhou and Junjie Hu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |