
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA versatile strategy for the synthesis and mechanical property manipulation of networked biodegradable polymeric materials composed of well-defined alternating hard and soft domains†
Hideto Tsuji *,
Ken-ichi Tamura and
Yuki Arakawa
*,
Ken-ichi Tamura and
Yuki Arakawa
Department of Environmental and Life Sciences, Graduate School of Engineering, Toyohashi University of Technology, Tempaku-cho, Toyohashi, Aichi 441-8580, Japan. E-mail: tsuji@ens.tut.ac.jp
First published on 1st March 2019
Abstract
The present paper proposes a versatile strategy for the synthesis and mechanical property manipulation of networked biodegradable polymeric materials composed of well-defined alternating soft and hard domains. As an example of the strategy, we selected biodegradable poly(L-lactide) (PLLA) and poly(ε-caprolactone) (PCL) as the hard and soft components, respectively, and synthesized networked biodegradable polymeric materials composed of well-defined alternating PLLA and PCL domains with different L-lactide (LLA) unit contents via crosslinking of well-defined four-armed diblock copolymers of PLLA and PCL (4-C-L). The strategy reported here, which is also applicable to non-biodegradable polymeric materials, successfully facilitated the synthesis of the networked biodegradable materials composed of alternating hard and soft domains and their mechanical properties of the synthesized materials were largely manipulated by the LLA unit contents of the precursor four-armed diblock 4-C-L copolymers. Moreover, the crystallization behavior and thermal properties of 4-C-L copolymers before and after crosslinking were investigated and discussed.
1. Introduction
Poly(L-lactide) or poly(L-lactic acid) (PLLA) is a glassy biodegradable polyester produced from renewable resources such as a variety of polysaccharides, whereas poly(ε-caprolactone) (PCL) is a rubbery biodegradable polyester with a high microbial biodegradation rate in the environment.1–10 The block11–47 and random48–58 copolymers of hard L-lactide (LLA) and soft ε-caprolactone (CL) units, which are abbreviated as PLLA-b-PCL or PCL-b-PLLA and P(LLA-co-CL) or P(CL-co-LLA), respectively, have been extensively synthesized and their crystallization behavior, mechanical and thermal properties, hydrolytic degradation, and drug release properties were studied.11–59 Compared to homopolymers PLLA and PCL, block or random copolymers of LLA and CL are soft.51,58 The hydrolytic degradation rate11,16,17,20,48,50,58 of the copolymers of LLA and CL increases with increasing LLA unit content, except for PLLA homopolymers and copolymers with a very low CL unit content, whereas the randomness of LLA and CL accelerated the hydrolytic degradation rate.24 The drug release rate14,15 of the copolymers of LLA and CL decreases with an increase in glassy LLA unit content.To manipulate the mechanical properties, hydrolytic degradation and drug release rate of LLA- and CL-based materials, numerous methods have been proposed in addition to copolymerization of LLA and CL. Blending of PLLA with PCL or copolymers of LLA and CL is versatile and facile method to prepare LLA and CL-based biodegradable materials with a wide variety of mechanical properties and hydrolytic degradation behavior.60–77 However, due to the phase-separation between PLLA-rich and PCL-rich domains, mechanical properties change dramatically at the critical blending ratio where continuous PLLA-rich domains become dispersed PLLA-rich domains and vice versa for PCL-rich domains. Compatibilizers such as P(LLA-co-CL) and PLLA-b-PCL have been added to avoid phase-separation between PLLA and PCL.61,66,68,70,73,74
Recently, to increase the toughness of LLA- or DL-lactide (DLLA)- and CL-based materials, crosslinked or networked materials are prepared by urethane linkage formation between multi-armed P(DLLA-co-CL)78 or three or more-armed PLLA and two or more-armed PCL and vice versa79,80 and by synthesis of vinyl- or vinylene-functionalized P(DLLA-co-CL),81 multi-block copolymers of PLLA and PCL,82,83 and three-armed or four-armed P(DLLA-co-CL)83–86 and their reaction. Although this strategy has been effectively utilized to prepare tough LLA- or DLLA- and CL-based materials, LLA or DLLA and CL units are randomly arranged or the array of PLLA and PCL domains is not controlled. Since high molecular weights of PLLA and PCL will induce phase-separation between PLLA and PCL to form respective domains with low interfacial interaction, resulting in low mechanical properties after crosslinking. Therefore, the molecular weights of PLLA and PCL before crosslinking should be kept low to avoid the phase separation.79,80 In the present study, to overcome this issue and to show the versatile strategy for the synthesis and mechanical property manipulation of the networked biodegradable polymeric materials with well-defined alternating hard and soft domains, as an example of our strategy, we selected biodegradable PLLA and PCL as the hard and soft components, respectively, and synthesized the biodegradable polymeric materials composed of well-defined alternating PLLA and PCL domains with different LLA unit contents via crosslinking of well-defined four-armed diblock PCL-b-PLLA (4-C-L) copolymers (Fig. 1). The crosslinking or network formation was attained by diisocyanate. The mechanical properties as well as crystallization behavior and thermal properties of the prepared materials were investigated by wide-angle X-ray diffractometry (WAXD), differential scanning calorimetry (DSC), and tensile testing, in comparison with those from a four-armed PLLA (4-L)/four-armed PCL (4-C) blend. The strategy proposed in the present study is applicable to the preparation of networked materials composed of well-defined alternating hard and soft domains of non-biodegradable polymers as well as biodegradable polymers with a wide variety of mechanical properties.
 | ||
| Fig. 1 Schematic representation for synthesis of 4-C-L (a), crosslinking of 4-C-L (b), and structures of crosslinked 4-C-L (c). In panel (b), the quadrants are quarter parts of 4-C-L and R in OCN-R-NCO is –C6H4–CH2–C6H4–. In panel (c), the ratio of each domain is determined by the content of LLA unit contents or PLLA block length in 4-C-L. | ||
2. Experimental section
2.1. Materials
The 4-C-L copolymers were synthesized by two-step process, i.e., the synthesis of four-armed PCL (4-C) using pentaerythritol as coinitiator and the synthesis of 4-C-L using 4-C as coinitiator.87–90 4-C polymers with different molecular weights and four-armed PLLA (4-L) were synthesized by bulk ring-opening polymerization of CL (Guaranteed grade, Nacalai Tesque, Inc., Kyoto, Japan) and LLA (PURASORB L, Purac Biochem BV, Gorinchem, The Netherlands), respectively, initiated with 0.03 wt% of tin(II) 2-ethylhexanoate (Nacalai Tesque, Inc.) in the presence of different amounts of pentaerythritol (Sigma-Aldrich Japan, K.K., Tokyo, Japan) as the coinitiator (Table 1) at 150 °C for 4-C synthesis and 140 °C for 4-L synthesis for 10 h.89,90 Synthesized 4-C and 4-L polymers were purified by reprecipitation using chloroform and methanol (both Guaranteed grades, Nacalai Tesque, Inc.) as the solvent and non-solvent, respectively, and then dried in vacuo for at least 6 days. 4-C-L copolymers with different LLA or CL unit contents were synthesized by ring-opening polymerization of LLA (0.2–1.0 g) in toluene (2 mL) initiated with 0.3 wt% of tin(II) 2-ethylhexanoate in the presence of the purified and dried 4-C polymers as the coinitiator at 120 °C for 36 h.89,90 The weight ratios of precursor (4-C polymer) to LLA are shown in Table 1. Synthesized 4-C-L polymers were purified by precipitation using a mixed solvent of chloroform and methanol as the solvent and non-solvent, respectively, and then dried in vacuo for at least 6 days. CL and tin(II)2-ethylhexanoate were purified by distillation under reduced pressure before use, whereas LLA was purified by repeated recrystallization using ethyl acetate (Guaranteed grade, Nacalai Tesque, Inc.) as a solvent. The crosslinked materials were prepared as follows. 4-C-L copolymers, 4-C homopolymer (4-C93), 4-L homopolymer (4-L90), or 4-C93/4-L90 (w/w = 1/1) blend (0.3–0.8 g) and 4,4′-methylenebis(phenyl isocyanate) (MBPI) or methylenediphenyl-4,4′-diisocyanate (98%, Sigma-Aldrich Japan, K.K.) [4-C-L, 4-C93, 4-L90, or 4-C93/4-L90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MBPI = 1:2 (mol:mol)] were dissolved in dichloromethane (Guaranteed grade, Nacalai Tesque, Inc.). The solutions of 4-C-L copolymers or 4-C homopolymer (4-C93) and MBPI were poured into a Petri dish and then dried at ambient temperature under atmospheric pressure for 2 h. Crosslinking of the dried films was carried out at 150 °C under reduced pressure for 3 h. In the case of 4-L homopolymer (4-L90), imperfect crosslinking of its solution containing MBPI poured into a Petri dish was performed at a lower temperature of 110 °C under reduced pressure for 30 min to avoid the crack formation. The imperfectly crosslinked film was dissolved by addition of dichloromethane (5 mL g−1) in the Petri dish, dried at ambient temperature under atmospheric pressure, and then complete crosslinking was performed at 150 °C under reduced pressure for 3 h. The crosslinked films were dried under reduced pressure for at least 6 days to remove residual solvent.
:MBPI = 1:2 (mol:mol)] were dissolved in dichloromethane (Guaranteed grade, Nacalai Tesque, Inc.). The solutions of 4-C-L copolymers or 4-C homopolymer (4-C93) and MBPI were poured into a Petri dish and then dried at ambient temperature under atmospheric pressure for 2 h. Crosslinking of the dried films was carried out at 150 °C under reduced pressure for 3 h. In the case of 4-L homopolymer (4-L90), imperfect crosslinking of its solution containing MBPI poured into a Petri dish was performed at a lower temperature of 110 °C under reduced pressure for 30 min to avoid the crack formation. The imperfectly crosslinked film was dissolved by addition of dichloromethane (5 mL g−1) in the Petri dish, dried at ambient temperature under atmospheric pressure, and then complete crosslinking was performed at 150 °C under reduced pressure for 3 h. The crosslinked films were dried under reduced pressure for at least 6 days to remove residual solvent.
| Codea | Synthesis | Polymer | ||||||
|---|---|---|---|---|---|---|---|---|
| Coinitiator | Monomer | Monomer/coinitiator mol/mol or (w/w) | Mn(NMR)a (g mol−1) | Mw(GPC)b (g mol−1) | Mw(GPC)/Mn(GPC)b | L-Lactyl unit contenta (mol%) | LLA unit content (wt%) | |
| a Number-average molecular weight [Mn(NMR)] and L-lactyl unit content were estimated by 1H NMR spectroscopy.b Weight- and number-average molecular weights [Mw(GPC) and Mn(GPC), respectively] were estimated by GPC. | ||||||||
| 4-L90 | Pentaerythritol | LLA | 143 | 9.00 × 103 | 2.27 × 104 | 1.21 | ||
| 4-C28 | Pentaerythritol | CL | 70 | 2.82 × 103 | 4.64 × 103 | 1.80 | ||
| 4-C49 | 122 | 4.94 × 103 | 8.92 × 103 | 1.51 | ||||
| 4-C69 | 165 | 6.85 × 103 | 1.90 × 104 | 1.94 | ||||
| 4-C93 | 254 | 9.27 × 103 | 2.59 × 104 | 1.74 | ||||
| 4-C28-L62 | 4-C28 | LLA | (1.0/0.3) | 9.01 × 103 | 2.18 × 104 | 1.14 | 74.9 | 66.2 |
| 4-C49-L38 | 4-C49 | (0.5/0.4) | 8.74 × 103 | 2.15 × 104 | 1.20 | 53.2 | 42.7 | |
| 4-C69-L28 | 4-C69 | (0.2/0.6) | 9.77 × 103 | 2.29 × 104 | 1.45 | 17.3 | 12.1 | |
2.2. Measurements and observation
The number-average molecular weight [Mn(NMR)] values of the synthesized polymers were determined from the 400 MHz 1H NMR spectra obtained in deuterated chloroform (50 mg mL−1) by a Bruker BioSpin (Kanagawa, Japan) AVANCE III 400 using tetramethylsilane as the internal standard. In the present study, the polymer codes of 4-LX, 4-CY and 4-CX-LY are used for abbreviations of 4-L, 4-C, and 4-C-L polymers, respectively, wherein X and Y indicate the total Mn/100 (not Mn of each arm/100) of PLLA and PCL blocks, respectively. Fig. S1† shows the typical 1H NMR spectrum of 4-C-L copolymer (4-C49-L38). As seen in this figure, four-armed diblock copolymers were successfully synthesized. The Mn values of 4-C [Mn(4-C)] and of 4-C-L and 4-L [Mn(4-C-L and 4-L)] were estimated according to the following equations using the peak intensities of pentaerythritol (I1) observed at around 4.1 ppm, methylene protons of CL units neighboring ester carbonyl carbon (I2) observed at around 2.3 ppm, and methine protons of LLA units inside the chain (I3) and at the hydroxyl terminal (I4), observed at around 5.2 and 4.4 ppm, respectively,87–90| Mn(NMR) (4-C) = 136.2 + 4 × 114.1 × (I2/I1), |
| Mn(NMR) (4-C-L and 4-L) = 136.2 + 4 × [114.1 × I2/(2 × I4) + (144.1/2) × (I3 + I4)/I4], |
The highly crosslinked polymers become insoluble to the solvents of a polymer before crosslinking. Therefore, the fraction of insoluble materials after crosslinking can be used as an index for the degree of crosslinking. The insoluble fractions of the crosslinked polymers were estimated as follows. Crosslinked materials (100 mg) were extracted three times with fresh chloroform (10 mL, Guaranteed grade, Nacalai Tesque, Inc.) for 8 h. After the extraction, the crosslinked materials were dried under reduced pressure for at least 7 days. The degree of crosslinking (DC) was evaluated by the following equation:
| DC (%) = 100 × Wafter/Wbefore |
The thermal properties of samples (3 mg) were measured with a Shimadzu (Kyoto, Japan) DSC-60 differential scanning calorimeter under a nitrogen gas flow at a rate of 50 mL min−1. For DSC measurements, the sample was heated at a rate of 10 °C min−1 from −10 °C to 180 °C, and then cooled at a rate of −3 °C min−1 to −10 °C. From heating measurements, glass transition and cold crystallization temperatures (Tg and Tcc, respectively), and melting temperatures of PCL and PLLA [Tm(PCL) and Tm(PLLA), respectively], and enthalpies of cold crystallization and melting of PCL and PLLA [ΔHcc and ΔHm(PCL) and ΔHm(PLLA), respectively] were evaluated. From cooling measurements, crystallization temperatures and enthalpy (Tc and ΔHc, respectively) were estimated. The transition temperatures and enthalpies were calibrated using tin, indium, and benzophenone as standards. The crystalline species and crystallinity (Xc) values of the polymers before and after crosslinking were estimated by the use of WAXD. The WAXD measurements were performed at 25 °C using a Rigaku (Tokyo, Japan) RINT-2500 equipped with a Cu-Kα source (λ = 1.5418 Å), which was operated at 40 kV and 200 mA. In a 2θ range of 5–30°, the crystalline diffraction peak areas of respective crystalline species relative to the total area between a diffraction profile and a baseline were used to estimate the Xc values.91 The crystallization behavior of the samples was observed using an Olympus (Tokyo, Japan) polarized optical microscope (BX50) equipped with a heating–cooling stage and a temperature controller (LK-600PM, Linkam Scientific Instruments, Surrey, UK) under a constant nitrogen gas flow. The samples were heated from ambient temperature to 180 °C at 100 °C min−1, held at this temperature for 1 min, then cooled at a rate of 3 °C min−1 to −10 °C. The mechanical properties of the crosslinked samples (3 mm × 30 mm × 100 μm) were measured at 25 °C and 50% relative humidity using a Shimadzu tensile tester (EZ Test) at a cross-head speed of 100% min−1 (20 mm min−1). The initial length between the two gauges was always kept at 20 mm.92
3. Results and discussion
3.1. Crystallizability and crystallinity
To determine the crystallizability and crystallinity of PLLA and PCL chains of the samples before and after crosslinking, WAXD measurements were performed (Fig. 2). The samples before crosslinking are the purified (or precipitated) polymers and cast (or solvent evaporated) blend. As seen in Fig. 2(a), all samples were crystallizable before crosslinking. 4-L90, 4-C28-L62, and 4-C49-L38 had α- or δ-form PLLA crystalline peaks at 15, 17, and 19°, whereas 4-L90 and 4-C28-L62 had the α-form PLLA crystalline peaks at 22.5° but this peak cannot be recognized for 4-C49-L38 probably due to a low peak height.93–95 On the other hand, the 4-L90/4-C93 blend had a very small α- or δ-form PLLA crystalline peak at 17°. This result indicates that PLLA chains in 4-L90 and 4-C28-L62 crystallized in α-form, whereas those in 4-C49-L38 and 4-L90/4-C93 blend crystallized in α- or δ-form crystallites.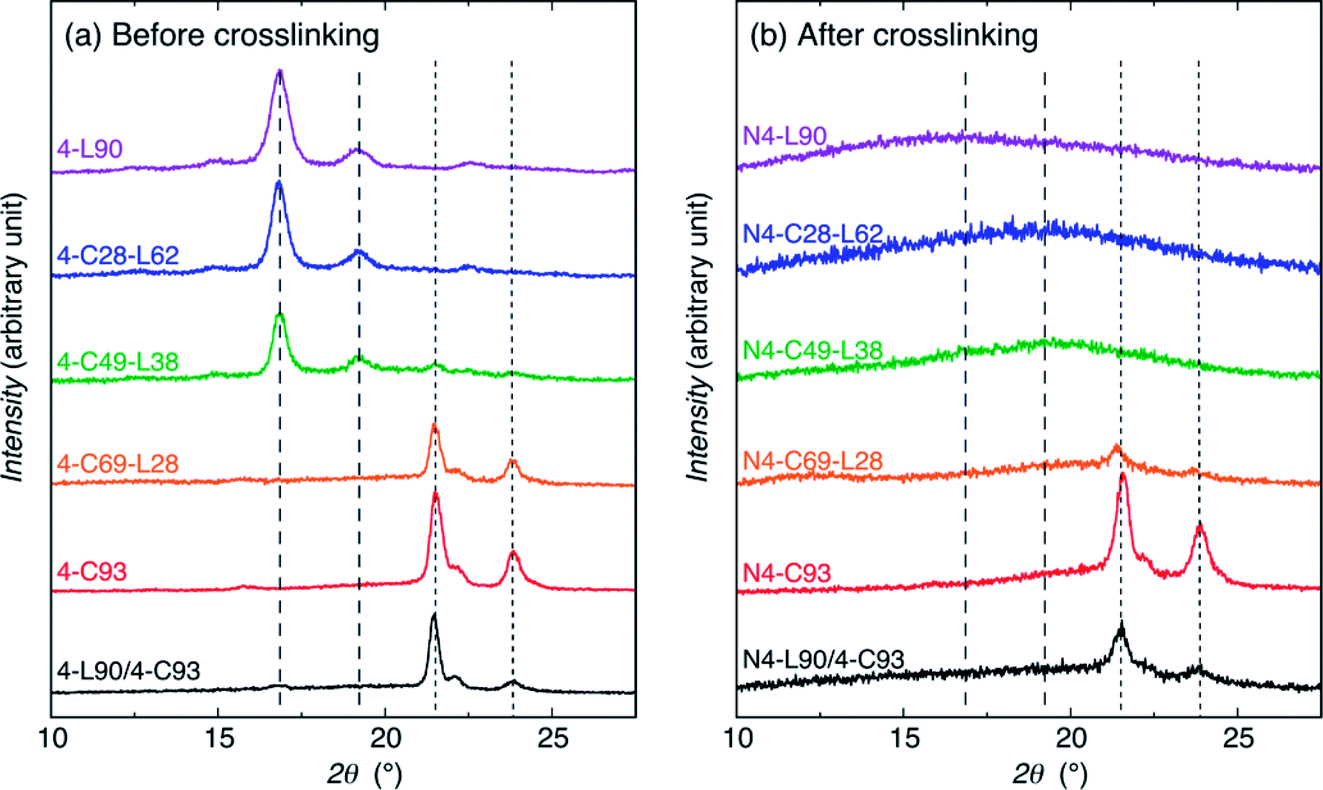 | ||
| Fig. 2 WAXD profiles of samples before (a) and after (b) crosslinking. The dashed and dotted lines indicate the main crystalline peak angles of PLLA α-(or δ-)form and PCL crystallites, respectively. | ||
4-C93, 4-C69-L28, and 4-L90/4-C93 blend had PCL crystalline peak at around 21.5, 22, and 24°, in agreement with those reported for orthorhombic lattice with the cell parameters of a = 7.47 Å, b = 4.98 Å, c = 17.05 Å,96–98 whereas in the case of 4-C49-L38 has only one PCL crystalline peak at around 21.5° was observed. In summary, 4-C28-L62 and 4-C69-L28 contained only PLLA and PCL crystallites, respectively, whereas 4-C49-L38 and 4-L90/4-C93 blend contained both PLLA and PCL crystallites. After crosslinking, N4-C93, N4-C69-L28, and N4-L90/4-C93 blend were crystallizable to have only PCL crystallites [Fig. 2(b)].
The Xc values of PLLA and PCL crystallites [Xc(PLLA) and Xc(PCL), respectively] were estimated from WAXD profiles in Fig. 2 and summarized in Table S1† and are plotted in Fig. 3 as a function of LLA unit content (wt%). As expected, the Xc(PLLA) and Xc(PCL) values correspondingly increased and decreased with an increase in LLA unit content and all the Xc values were largely decreased by crosslinking, indicating the crosslinking disturbed the crystallization of polymer chains. 4-C49-L38 before crosslinking had higher Xc(PLLA) (28.5%) compared to Xc(PCL) (4.4%), despite the fact that its LLA unit content (42.7 wt%) was higher than CL unit content (57.3 wt%) (Table 1). This result indicates that hard PLLA chains at a shell has high crystallizability compared to soft PCL chains at the core part, due to the facile interaction between the PLLA chains at a shell. The nil values of Xc(PLLA) after crosslinking shows the complete suppression of crystallization of PLLA chains by crosslinking, whereas the decreased but non-zero values of Xc(PCL) for N4-C69-L28 and N4-C93 (7.5 and 35.7%, respectively) after crosslinking reflect that the crystallizability of PCL chains were reduced by crosslinking but remained after crosslinking. The higher crystallizability of PCL chains after crosslinking compared to those of PLLA chains should be ascribed to higher segmental mobility of PCL, which is supported by low Tg of soft PCL (ca. −60 °C) compared to that of hard PLLA (ca. 60 °C).64 On the other hand, 4-L90/4-C93 blend before crosslinking had lower Xc(PLLA) and higher Xc(PCL) values (1.6 and 35.1%, respectively) compared those of the 4-C-L copolymer having similar LLA unit content (4-C49-L38) (4.4 and 28.5% respectively). The value difference between the 4-C-L copolymer and blend samples can be mainly ascribed to the molecular architectural difference between the 4-C-L copolymer and blend samples and the contribution of the difference in sample preparation, i.e., precipitation for 4-C-L copolymers and solvent evaporation for the blend should be very small.
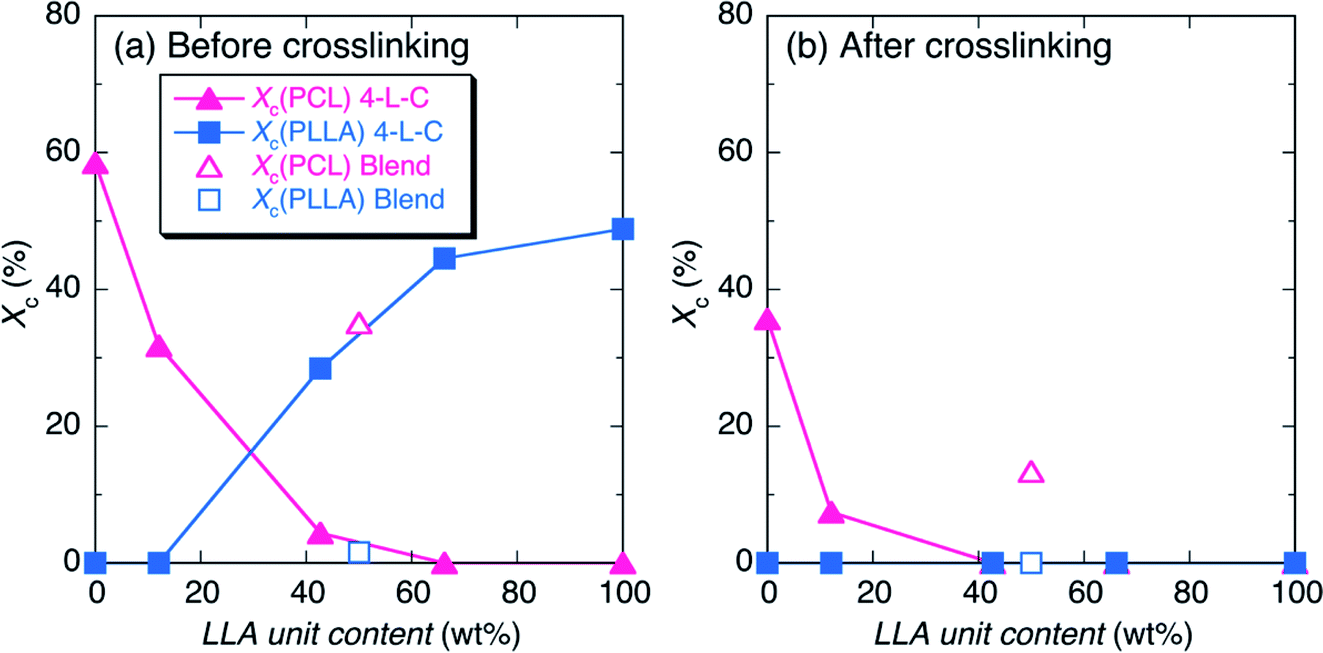 | ||
| Fig. 3 Crystallinity (Xc) of samples before (a) and after (b) crosslinking. | ||
To confirm this assumption, we prepared the typical 4-C-L copolymer (4-C49-L38) sample by solvent evaporation with the same procedure used to prepare the blend sample and its WAXD measurement was performed (Fig. S3†). The solvent evaporated 4-C49-L38 had a WAXD profile very similar to that of the precipitated 4-C49-L38. The Xc(PLLA) and Xc(PCL) values (30.1 and 5.8%, respectively) of the solvent evaporated 4-C49-L38 estimated from Fig. S3† were very similar to those of the precipitated 4-C49-L38 (28.5 and 4.4%, respectively) but completely different from those of the solvent evaporated blend (1.6 and 35.1%, respectively). This result confirms that the difference in Xc(PLLA) and Xc(PCL) values between the 4-C-L copolymer and blend samples is attributed to the molecular architectural difference between the 4-C-L copolymer and blend samples not the difference in preparation method.
3.2. Thermal properties
To assess the thermal properties and crystallization during cooling from the melt of the sample before and after crosslinking, DSC measurements by heating were carried out (Fig. 4 and 5). For reference, DSC measurements of 4-C49-L38 prepared by solvent evaporation were carried out (Fig. S4†). Evidently, for both heating and cooling, the solvent evaporated 4-C49-L38 exhibited the DSC thermograms very similar to those of the precipitated 4-C49-L38. This result supported that the molecular architectural difference played the main role in determining the thermal properties and crystallization behavior during cooling from the melt, but sample preparation method (precipitation or solvent evaporation) had an insignificant effect on them. Also, it is well known that crystallization during cooling after melting is not normally affected by the sample preparation method. The exothermic peaks with arrows in Fig. 5 and S4(b)† were formed due to replenishment of liquid nitrogen to the DSC apparatus. The thermal properties [obtained from Fig. 4, 5, and S4†] are summarized in Table S2.†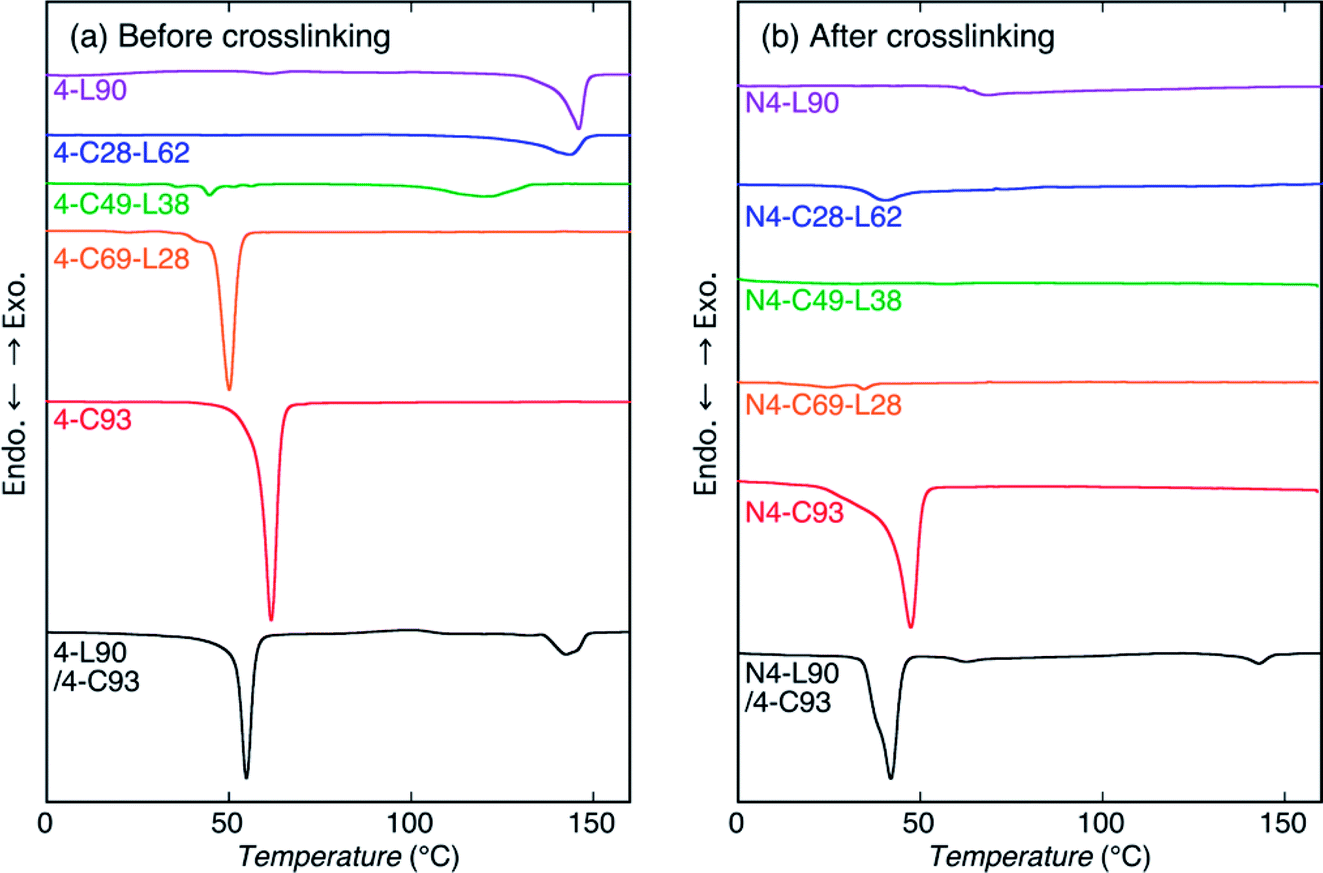 | ||
| Fig. 4 DSC heating thermograms of samples before (a) and after (b) crosslinking. | ||
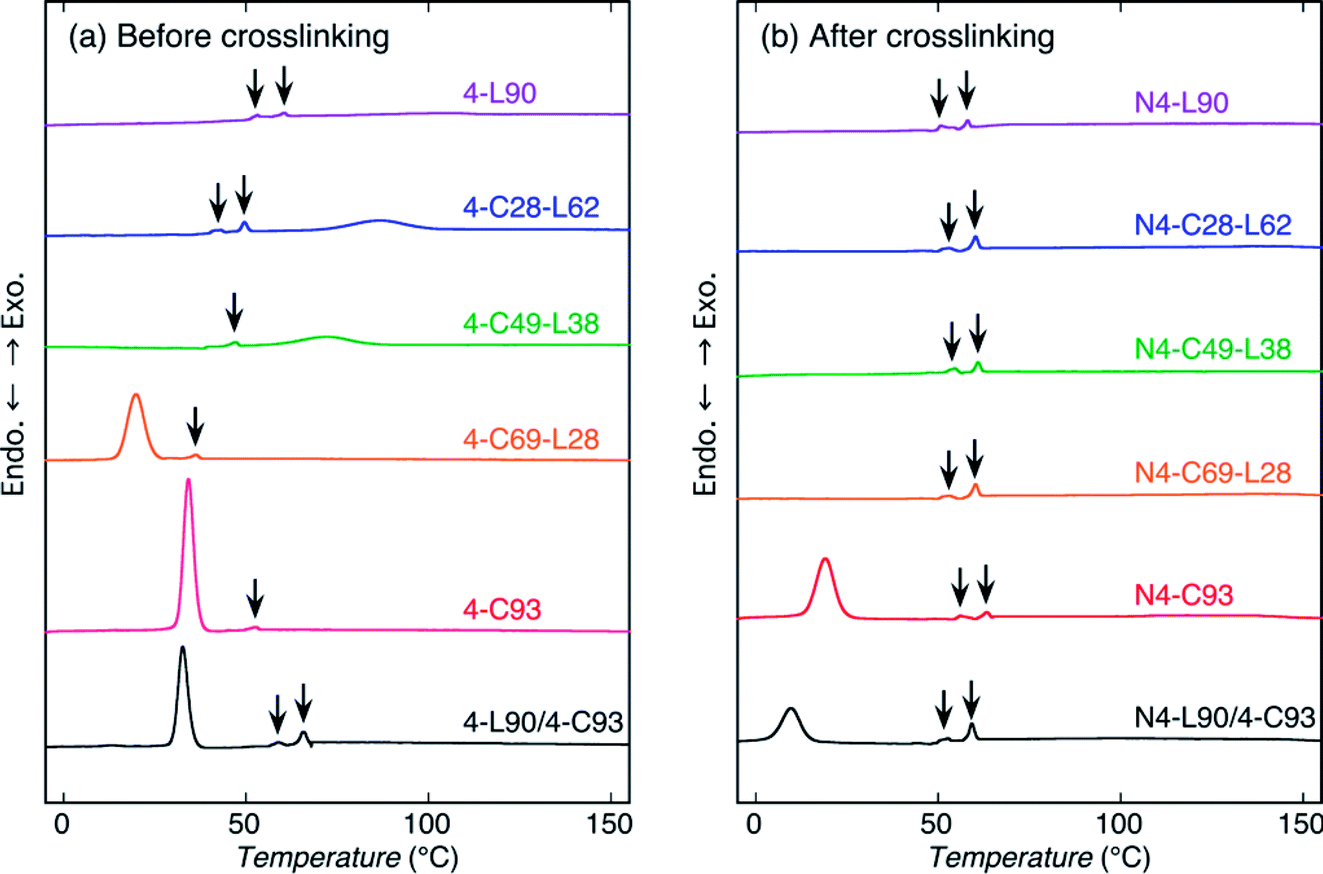 | ||
| Fig. 5 DSC cooling thermograms from the melt of samples before (a) and after (b) crosslinking. The exothermic peaks with arrows were formed due to replenishment of liquid nitrogen to the DSC apparatus. | ||
For heating of the samples before crosslinking [Fig. 4(a)], all the samples showed a melting peak. The melting peaks of PLLA chains were observed at 146.0, 143.6, and 120.0 °C for 4-L90, 4-C28-L63, and 4-C49-L28, respectively, whereas the melting peaks of PCL chains were seen at 61.6, 50.1, and 44.6 °C for 4-C93, 4-C69-L28, and 4-L49-L38, respectively. As expected, the Tm values of PLLA and PCL blocks in the polymer samples decreased with decreasing molecular weights of PLLA and PCL blocks, respectively, indicating the molecular weight of each block determine the crystalline thickness. On the other hand, 4-L90/4-C93 blend had both melting peaks of 4-armed PLLA and PCL polymers at 142.6 and 54.8 °C, which were lower than those of nonblended 4-L90 (146.0 °C) and 4-L93 (61.6 °C). This reflects that although PLLA and PCL polymers were phase-separated in the blends, the PLLA molecules dissolved in the PCL rich-phase and vice versa, and the dissolved PCL molecules in PLLA-rich domains and PLLA molecules in PCL-rich domains correspondingly disturbed the crystallization of PLLA and PCL chains during solvent evaporation compared to that of nonblended 4-L90 and 4-C93 during precipitation. For heating of the samples after crosslinking [Fig. 4(b)], N4-C93, N4-C69-L28, and N4-L90/4-C93 blend exhibit a melting peak of PCL chains at 47.4, 34.5, and 41.9 °C, respectively. Similar to the samples before crosslinking, the crystalline thickening was limited by the lower molecular weight of PCL chains in N4-C69-L28 compared to N4-C93 and the presence of PLLA chains dissolved in the PCL-rich domain in N4-L90/4-C93.
For cooling from the melt of the samples before crosslinking [Fig. 5(a)], all the samples except for 4-L90 were crystallized. Considering the Tm values of PLLA and PCL chains (120.0–146.0 °C and 44.6–61.6 °C, respectively), the exothermic peaks of 4-C28-L62 and 4-C49-L38 observed correspondingly at 86.6 and 72.0 °C were attributed to crystallization of PLLA chains, whereas the exothermic peaks of 4-C93 and 4-C69-L28, and 4-L90/4-C93 blend monitored at 34.2, 19.8, and 32.6 °C were ascribed to crystallization of PCL chains. The 4-L90/4-C93 blend had only crystallization peak of PCL chains, as can be expected from non-crystallizability of PLLA chains in 4-L90 during cooling. After crosslinking, only N4-C93 and N4-L90/4-C93 blend had a crystallization peak at 19.1 and 9.6 °C, indicating the disturbance of crystallization by crosslinking, in agreement with the WAXD results, and the higher crystallizability of PCL chains only in 4-C homopolymers in nonblended and blend samples compared to that of PLLA chains. The latter result can be ascribed to the high chain mobility of soft PCL chains compared to that of hard PLLA chains, as stated before.
The ΔHcc and ΔHm values for heating and ΔHc values for cooling are tabulated in Table S2† and are plotted in Fig. S5 and S6† as a function of LLA unit content. The ΔHm(PCL) and ΔHm(PLLA) values of the polymer samples for heating scanning in Fig. S5 and Table S2† had the similar trend with Xc(PCL) and Xc(PLLA) in Fig. 3. That is, the ΔHm(PCL) and ΔHm(PLLA) values of the samples correspondingly decreased and increased with increasing with LLA unit contents and the ΔHm(PCL) and ΔHm(PLLA) values of the samples before crosslinking were higher than those after crosslinking. Also, the trend observed for the samples in Fig. S6† for cooling scanning was similar to that in Fig. S5.† That is, the −ΔHc(PCL) and −ΔHc(PLLA) values of the polymer samples for cooling scanning respectively decreased and increased with an increase in LLA unit content although 4-L90 before and after crosslinking did not crystallized during cooling [Fig. S6(a)†] [i.e., −ΔHc(PLLA) = 0 J g−1], and the −ΔHc(PCL) and −ΔHc(PLLA) values were higher for of the polymer samples before crosslinking than for those after crosslinking. The higher absolute enthalpy values of both non-crosslinked and crosslinked samples during heating scanning compared to those during cooling scan confirmed the WAXD results, i.e., facile crystallization of the samples by precipitation and solvent evaporation compared to that by cooling from the melt. In summary, the trend of enthalpy values of the blend sample in Fig. S5 and S6† was similar to the crystallinity values in Fig. 3.
3.3. Overall crystallization behavior
The relative crystallinity (Xr) of the samples during cooling from the melt was estimated from the DSC thermograms shown in Fig. 5 using the following equation. The thus obtained Xr is plotted as a function of temperature in Fig. S7:†
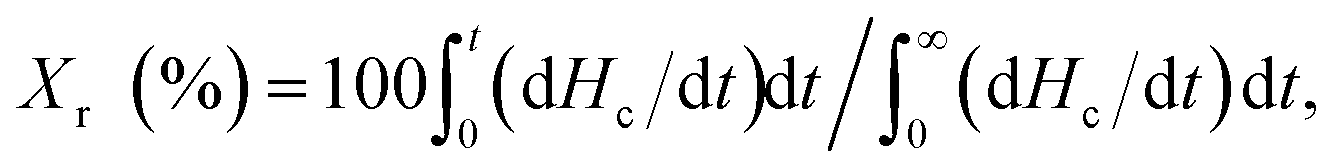 | (1) |
| 1 − Xr (%)/100 = exp(−ktcn), | (2) |
|
log[−ln(1 − Xr/100)] = logk + nlogtc.
| (3) |
To avoid deviation from the theoretical curves, which is suggested by Mandelkern et al. and Lorenzo et al.,102,103 we used Xr in the range of 3–20% for estimating n and k. The plots with eqn (3) are shown in Fig. 6. The plots in Fig. 6 give n as slope and lnk as intercept. The thus obtained n and k values are summarized in Table 2. Before crosslinking, the n values of 4-C-L copolymers (4-C28-L62, 4-C49-L38, and 4-C69-L28) are approximately 3, whereas those of 4-C93 and 4-L90/4-C93 blend were 5.6 and 5.0, respectively. After crosslinking, the n values of N4-C93 and N4-L90/4-C93 blend were approximately 4.0, which were lower than those before crosslinking. Assuming the thermal nucleation,104 before crosslinking, n ≈ 3 for 4-C-L copolymers reflects circular growth geometry, whereas the 3 < n < 6 for 4-C93 and 4-L90/4-C93 blend means growth geometry between spherical and solid sheaf. Considering the fact that PLLA blocks crystallized in 4-C28-L62 and 4-C49-L38 and PCL blocks crystallized in 4-C69-L28, 4-C93, and 4-L90/4-C93 blend (in PCL-rich domain), absence of PLLA blocks is concluded to increase the n values or changed the growth geometry. After crosslinking, the n values around 4 for N4-C93 and N4-L90/4-C93 blend, which were lower than those before crosslinking (5.0 and 5.6), reflect spherical growth geometry and indicates that crosslinking decreased the n values or altered the growth geometry. The values of tc(1/2)(cal) thus obtained are listed in Table 2, which were very similar to the experimental tc(1/2) [tc(1/2)(exp)].
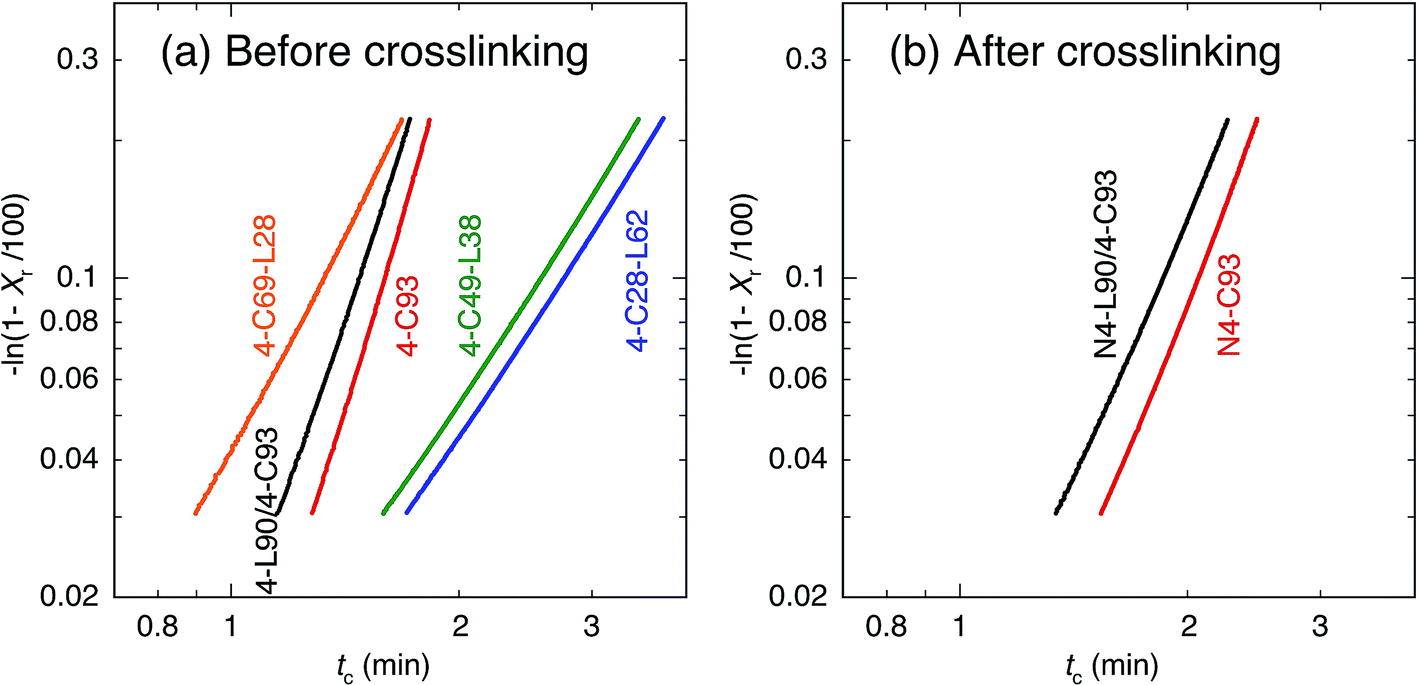 | ||
| Fig. 6 −ln(1 − Xr/100) of before (a) and after (b) crosslinking for cooling scanning as a function of crystallization time (tc). | ||
| Code | Temperature range for evaluationa (°C) | Temperature range for Avrami plot (°C) | Tca (°C) | n | k (min−n) | tc(1/2)(exp)b (min) | tc(1/2)(cal)b (min) |
|---|---|---|---|---|---|---|---|
| a Temperature range for evaluation is that used for relative crystallinity (Xr) estimation and Tc is crystallization temperature.b tc(1/2)(exp) and tc(1/2)(cal) are experimental and calculated crystallization half times, respectively. | |||||||
| 4-C28-L62 | 65.1–102.7 | 91.5–97.6 | 86.6 | 2.6 | 7.6 × 10−3 | 5.58 | 5.67 |
| 4-C49-L38 | 52.6–86.8 | 76.5–82.1 | 72.0 | 2.6 | 9.0 × 10−3 | 5.15 | 5.32 |
| 4-C69-L28 | 8.3–26.8 | 21.7–24.1 | 19.8 | 3.2 | 4.2 × 10−2 | 2.31 | 2.40 |
| 4-C93 | 23.3–41.0 | 35.5–37.2 | 34.2 | 5.6 | 7.5 × 10−3 | 2.22 | 2.24 |
| 4-L90/4-C93 | 25.6–39.1 | 34.0–35.7 | 32.6 | 5.0 | 1.5 × 10−2 | 2.28 | 2.15 |
| N4-C93 | 5.6–28.5 | 21.1–23.9 | 19.1 | 4.2 | 4.8 × 10−3 | 3.18 | 3.27 |
| N4-L90/4-C93 | −1.9 to 18.4 | 11.6–14.4 | 9.6 | 3.8 | 9.6 × 10−3 | 2.98 | 3.08 |
3.4. Polarized optical microscopy
To further inquire the crystallization of the samples during cooling from the melt, POM observation was performed. Fig. 7 shows the polarized optical photomicrographs of the samples after cooling from the melt. The photos of samples which did not crystallize are not shown in Fig. 7. Before crosslinking, the polymer samples except for 4-L90 which did not crystallize [Fig. 7(a)–(d)] were filled with a large number of spherulites, whereas 4-C93/4-C93 blend was composed of two types of relatively bright and dark parts. Considering the phase-separation between PLLA-rich and PCL-rich domains and non-crystallizability of 4-L90 during cooling from the melt, the brightness difference depending on the place is attributable to the difference in thickness of PCL-rich layers whose thickness should be reduced by the presence of overlapping PLLA-rich layers and relative layer thicknesses of PCL- and PLLA-rich domains should vary depending on the place. That is, the bright and dark domains can be assigned to the domains with thick and thin PCL-rich layers, respectively. After crosslinking, only N4-C93 and N4-L90/C93 [Fig. 7(f) and (g)] were crystallizable during cooling from the melt, in agreement with WAXD and DSC results, and had vague crystalline assemblies. | ||
| Fig. 7 Polarized optical photomicrographs of samples after cooling from the melt (a) 4-C28-L62, (b) 4-C49-L38, (c) 4-C69-L28, (d) 4-C93, (e) 4-L90/4-C93, (f) N4-C93, (g) N4-L90/4-C93. Photos were obtained at 0 °C, excluding −10 °C for panel (g) N4-L90/4-C93. | ||
3.5. Mechanical properties
Since all the samples before crosslinking and N4-L90/4-C93 blend sample after crosslinking were fragile and their mechanical properties could not be measured, only the mechanical properties of samples after crosslinking, except for the blend sample, are summarized in Fig. 8. It should be noted that for facile recognition of the dependence of mechanical properties on LLA unit content, most mechanical properties in Fig. 8(b)–(d) were plotted logarithmically, whereas tensile strength in Fig. 8(a) is plotted linearly. The tensile strength decreased with a decrease in hard LLA unit content (or an increase in soft CL unit content) from 29.8 MPa at LLA unit content = 100 wt% to 7.1 MPa at LLA unit content = 12.1 wt%, but then increased slightly to 15.8 MPa at LLA unit content = 0 wt% [Fig. 8(a)]. The exceptional low tensile strength value at LLA unit content = 12.1 wt% (7.1 MPa) compared to that at LLA unit content = 0 wt% (15.8 MPa) can be ascribed to the lower Xc(PCL) value at 12.1 wt% (7.5%) compared to that at 0 wt% (35.7%) (Fig. 3 and Table S1†). The higher Xc(PCL) at LA unit content = 0 wt% should have increased the density of physical crosslinking of crystalline regions. The values of tensile strength for LLA unit contents at 100% (29.8 MPa) and at 0% (15.8 MPa) with initial degrees of polymerization (DP) of 123 and 80 before crosslinking were correspondingly comparable to and higher than those reported for crosslinked low molecular weight four-armed PLLA (19.4–38.9 MPa) and PCL (2.1–8.3 MPa) with initial DP before crosslinking from 3 to 10.79 Also, the tensile strength of crosslinked 4-L, i.e., N4-L90 (29.8 MPa) was comparable with 31.6 MPa reported for linear one-armed PLLA.92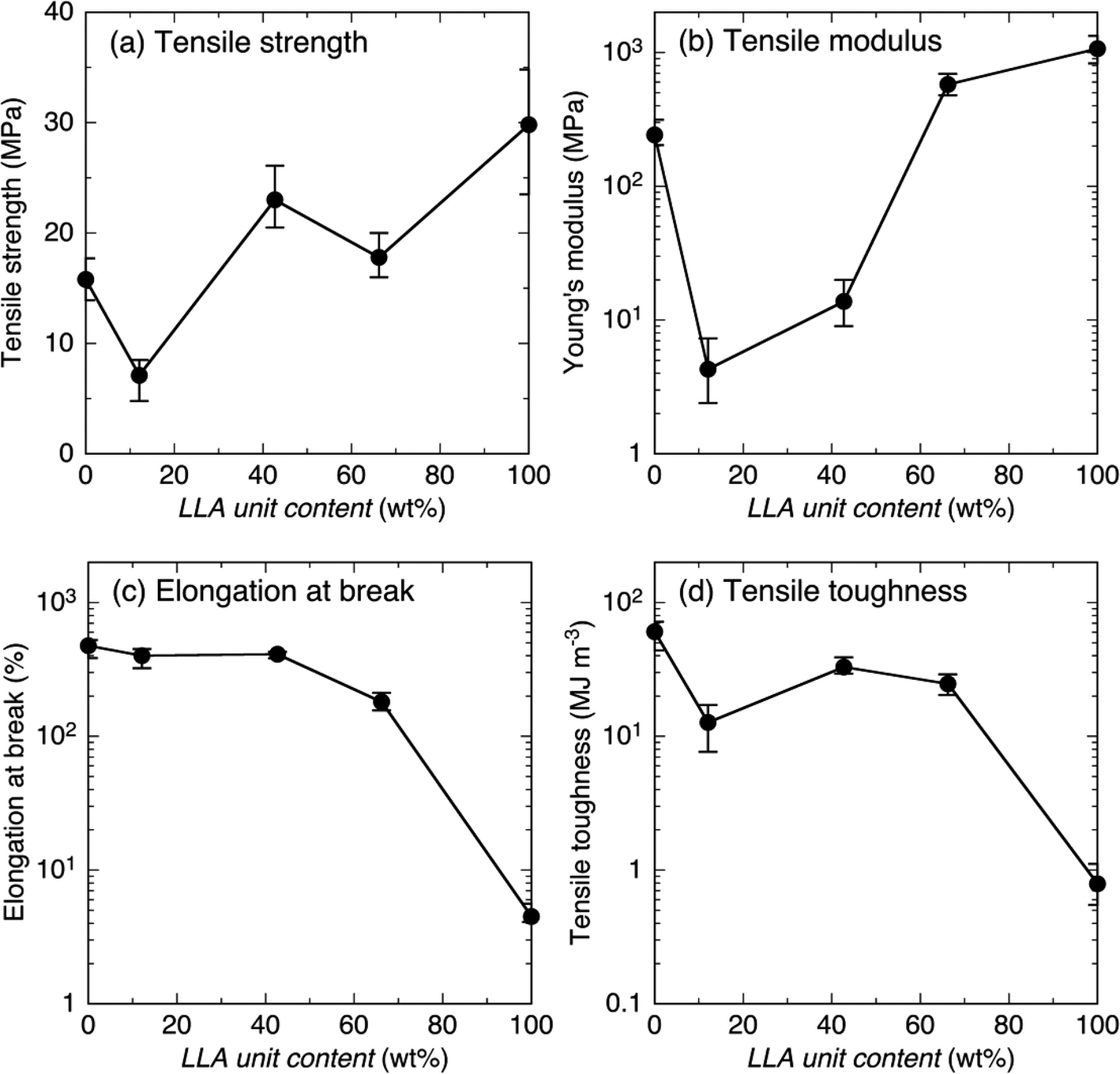 | ||
| Fig. 8 Tensile strength (a), tensile modulus (b), elongation at break (c) and tensile toughness (d) of crosslinked samples as a function of LLA content. | ||
Similar to tensile strength, tensile modulus decreased dramatically with decreasing hard LLA unit content (or increasing soft CL unit content) from 1.07 × 103 MPa at LLA unit content = 100 wt% to 5.77 × 102 MPa at LLA unit content = 66.2 wt%, and then to 4.30 MPa at LLA unit content = 12.1 wt%, but increased dramatically to 2.42 × 102 MPa at LLA unit content = 0 wt% [Fig. 8(b)]. The values of tensile modulus of for LLA unit contents at 100% (1.07 × 103 MPa) and at 0% (2.42 × 102 MPa) were respectively comparable to and higher than those reported for crosslinked low molecular weight four-armed PLLA (0.73–1.19 × 103 MPa) and PCL (0.04–1.12 × 102 MPa) with initial DP before crosslinking from 3 to 10.79 Also, the tensile modulus of crosslinked 4-L, i.e., N4-L90 (1.07 × 103 MPa) was comparable with 9.3 × 102 MPa reported for linear one-armed PLLA.92 The final increase in tensile strength and modulus at LA unit content = 0 wt% compared to that at LLA unit content = 12.1 wt% is also attributable to the higher Xc(PCL) value at LA unit content = 0 wt% (35.7%) compared to that at LA unit content = 12.1 wt% (7.5%), as stated above at the result of tensile strength.
Elongation at break increased with a decrease in hard LLA unit content (or an increase in soft CL unit content) from 4.5% at LLA unit content = 100 wt% to 180.8% at LLA unit content = 100 wt%, and finally reached 475.7% at LLA unit content = 0 wt% [Fig. 8(c)]. Elongation at break dramatically increased in corporation of a small amount of CL units such as 33.8 wt%. The values of elongation at break for LLA unit contents at 100% (4.5%) and at 0% (475.7%) were correspondingly comparable to and higher than those reported for crosslinked low molecular weight four-armed PLLA (4.3–10.9%) and PCL (47.7–282%) with initial DP before crosslinking from 3 to 10.79 However, the elongation at break of crosslinked 4-L, i.e., N4-L90 (4.5%) was lower than 13.6% reported for linear one-armed PLLA.92
Tensile toughness increased dramatically with decreasing in hard LLA unit content (or increasing soft CL unit content) from 0.79 MJ m−3 at LLA unit content = 100 wt% to 32.9 MJ m−3 at LLA unit content = 42.7 wt%, slightly decreased to 12.7 MJ m−3 at LLA unit content = 12.1 wt%, and then increased again and reached 60.6 MJ m−3 at LLA unit content = 0 wt% [Fig. 8(d)]. Probably, incorporated soft CL chains facilitated the absorption of applied energy by their elongation, resulting in elevated tensile toughness. The values of tensile toughness for LLA unit contents at 100% (0.79 MJ m−3) and at 0% (60.6 MJ m−3) were respectively comparable to and higher than those reported for crosslinked low molecular weight 4-armed PLLA (0.29–2.11 MJ m−3) and PCL (0.60–20.5 MJ m−3) with DP before crosslinking from 3 to 10.79 From the comparison of the tensile properties between the present and the previous studies, it was found that the tensile properties of rather soft 4-armed PCL-based materials were susceptible to the initial DP before crosslinking, whereas those of rather rigid 4-armed PLLA-based materials were independent of initial DP.
The present study revealed that networked biodegradable polymeric materials having well-defined alternating soft and hard domains were successfully synthesized utilizing the proposed strategy. For crosslinked materials composed of alternating soft PCL and hard PLLA domains had a high tensile toughness higher than 10 MJ m−3, a high elongation at break over 100%, and a wide variety of tensile modulus from 4.30 to 1.07 × 103 MPa by varying LLA unit content from 12.1 to 100 wt% can be obtained by crosslinking four-armed PCL-PLLA diblock copolymers. The method proposed in the present study is applicable for the preparation of well-defined networked materials composed of alternating domains of two types of biodegradable polymers with a wide variety of mechanical properties. The strategy proposed in the present study can be applied to the preparation of networked materials composed of well-defined alternating soft and hard domains of non-biodegradable polymers as well as biodegradable polymers having a wide variety of mechanical properties.
4. Conclusions
The networked biodegradable polymeric materials having well-defined alternating hard and soft domains of two types of biodegradable polymers (hard PLLA and soft PCL) with different LLA unit contents (N4-C-L) were successfully synthesized by the proposed strategy of crosslinking of well-defined four-armed diblock copolymers of PLLA and PCL (4-C-L) and their crystallization and mechanical behavior was investigated in comparison with those from a four-armed PLLA (4-L)/four-armed PCL (4-C) blend. All 4-C-L copolymers and the blend were crystallizable before crosslinking, whereas only four-armed PCL and 4-L/4-C blend were crystallizable after crosslinking. The presence of rigid PLLA blocks and crosslinking affected Avrami exponent values or growth geometry. The tensile strength and modulus of crosslinked networked N4-C-L samples decreased with decreasing LLA unit content from correspondingly 29.8 and 1.07 × 103 MPa at LLA unit content = 100 wt% to the minimum values 7.1 and 4.3 MPa at LLA unit content = 12.1 wt% and then reached 15.8 and 2.42 × 102 MPa at LLA unit content = 0 wt%. On the other hand, the elongation at break and tensile toughness of N4-C-L samples increased dramatically with decreasing LLA unit content from correspondingly 4.5% and 0.79 MJ m−3 at LLA unit content = 100 mol% to 180.8% and 32.9 MJ m−3 at LLA unit content = 66.2 and 42.7 wt%, respectively, and finally reached 475.7% and 60.6 MJ m−3 at LLA unit content = 0 wt% LLA unit content. In contrast, the mechanical properties of networked 4-L/4-C blend sample could not be measured due to the fragility caused by phase-separation before crosslinking or network formation. The present study revealed that the networked biodegradable materials with a high tensile toughness higher than 10 MJ m−3, a high elongation at break over 100%, a tensile strength up to 29.8 MPa, and a wide variety tensile modulus from 4.30 to 1.07 × 103 MPa by varying LLA unit content from 12.1 to 100 wt%. The strategy proposed in the present study is applicable to the preparation of networked materials composed of well-defined alternating hard and soft domains of non-biodegradable polymers as well as biodegradable polymers having a wide variety of mechanical properties.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by JSPS KAKENHI Grant Number 16K05912.References
- Biodegradable polymers and plastics, ed. M. Vert, J. Feijen, A.-C. Albertsson, G. Scott and E. Chiellini, Royal Society of Chemistry,Cambridge, 1992 Search PubMed.
- Plastics from microbes, ed. D. P. Mobley, Hanser Publishers, New York, 1994 Search PubMed.
- M. Vert, G. Schwarch and J. Coudane, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32, 787–796 CrossRef.
- Handbook of biodegradable polymers (Drug Targeting and Delivery), ed. A. J. Domb, J. Kos and D. M. Wieseman, Harwood Academic Publishers, Amsterdam (The Netherlands), 1997, vol. 7 Search PubMed.
- Biopolymers from renewable resources, ed. D. L. Kaplan, Springer, Berlin (Germany), 1998 Search PubMed.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- A. Södergård and M. Stolt, Prog. Polym. Sci., 2002, 27, 1123–1163 CrossRef.
- Degradable aliphatic polyesters (Advances in Polymer Science), ed. A. C. Albertsson, Springer, Berlin (Germany), 2002, vol. 157 Search PubMed.
- Polyesters I, II, III (Biopolymers), ed. Y. Doi and A. Steinbüchel, Wiley-VCH, Weinheim (Germany), 2002, vol. 3a, 3b, 4 Search PubMed.
- Poly(lactic acid): Synthesis, structures, properties, processing, and applications (Wiley Series on Polymer Engineering and Technology), ed. R. Auras, L. T. Lim, S. E. M. Selke and H. Tsuji, John Wiley & Sons, Inc., New Jersey, 2010 Search PubMed.
- X. D. Feng, S. T. Voong, C. X. Song and W. Y. Chen, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 593–600 CrossRef CAS.
- C. X. Song and X. D. Feng, Macromolecules, 1984, 17, 2764–2767 CrossRef CAS.
- D. Tian, Ph. Dubois, R. Jerome and Ph. Teyssie, Macromolecules, 1994, 27, 4134–4144 CrossRef CAS.
- W.-P. Ye and Y. W. Chien, Pharm. Dev. Technol., 1996, 1, 1–9 CrossRef CAS PubMed.
- W.-P. Ye and Y. W. Chien, J. Controlled Release, 1996, 41, 259–269 CrossRef CAS.
- W.-P. Ye, F. S. Du, W. H. Jin, J. Y. Yang and Y. Xu, React. Funct. Polym., 1997, 32, 161–168 CrossRef CAS.
- M. R. Lostocco, C. A. Murphy, J. A. Cameron and S. J. Huang, Polym. Degrad. Stab., 1998, 59, 303–307 CrossRef CAS.
- M. Trollsås, M. A. Kelly, H. Claesson, R. Siemens and J. L. Hedrick, Macromolecules, 1999, 32, 4917–4924 CrossRef.
- S. Pensec, M. Leroy, H. Akkouche and N. Spassky, Polym. Bull., 2000, 45, 373–380 CrossRef CAS.
- H. Qian, J. Bei and S. Wang, Polym. Degrad. Stab., 2000, 68, 423–429 CrossRef CAS.
- J. K. Kim, D.-J. Park, M.-S. Lee and K. J. Ihn, Polymer, 2001, 42, 7429–7441 CrossRef CAS.
- O. Jeon, S.-H. Lee, S. H. Kim, Y. M. Lee and Y. H. Kim, Macromolecules, 2003, 36, 5585–5592 CrossRef CAS.
- Y. Nakayama, S. Okuda, H. Yasuda and T. Shiono, React. Funct. Polym., 2007, 67, 798–806 CrossRef CAS.
- J. Fernández, A. Larrañaga, A. Etxeberría and J. R. Sarasua, Polym. Degrad. Stab., 2003, 98, 481–489 CrossRef.
- G. Maglio, A. Migliozzi and R. Palumbo, Polymer, 2003, 44, 369–375 CrossRef CAS.
- C. Teng, K. Yang, P. Ji and M. Yu, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5045–5053 CrossRef CAS.
- M.-H. Huang, S. Li and M. Vert, Polymer, 2004, 45, 8675–8681 CrossRef CAS.
- I. W. Hamley, V. Castelletto, R. V. Castillo, A. J. Müller, C. M. Martin, E. Pollet and P. Dubois, Macromolecules, 2005, 38, 463–472 CrossRef CAS.
- H. R. Kricheldorf, S. Rost, C. Wutz and A. Domb, Macromolecules, 2005, 38, 7018–7025 CrossRef CAS.
- D. Cohn and A. H. Salomon, Biomaterials, 2005, 26, 2297–2305 CrossRef CAS PubMed.
- W. Yuan, J. Yuan, F. Zhang and X. Xie, Biomacromolecules, 2007, 8, 1101–1108 CrossRef CAS PubMed.
- M. Florczak, J. Libiszowski, J. Mosnacek, A. Duda and S. Penczek, Macromol. Rapid Commun., 2007, 28, 1385–1391 CrossRef CAS.
- W. Zhang, S. Zheng and Q. Guo, J. Appl. Polym. Sci., 2007, 106, 417–424 CrossRef CAS.
- F. Liu, Z. Zhao, J. Yang, J. Wei and S. M. Li, Polym. Degrad. Stab., 2009, 94, 227–233 CrossRef CAS.
- J. Zhang, J. Xu, H. Wang, W. Jin and J. Li, Mater. Sci. Eng., C, 2009, 29, 889–893 CrossRef CAS.
- R. V. Castillo, A. J. Müller, J.-M. Raquez and P. Dubois, Macromolecules, 2010, 43, 4149–4160 CrossRef CAS.
- J. Ren, Z. Zhang, Y. Feng, J. Li and W. Yuan, J. Appl. Polym. Sci., 2010, 118, 2650–2658 CrossRef CAS.
- M. T. Casas, J. Puiggalí, J.-M. Raquez, P. Dubois, M. E. Córdova and A. J. Müller, Polymer, 2011, 52, 5166–5177 CrossRef CAS.
- J. F. Kong, V. Lipik, M. J. M. Abadie, G. Roshan Deen and S. S. Venkatraman, J. Biomed. Mater. Res., Part A, 2012, 100A, 3436–3445 CrossRef CAS PubMed.
- M. Jiao, K. Yang, J. Cao, H. Liu, W. Pan and P. Gao, J. Macromol. Sci., Part B: Phys., 2014, 53, 191–204 CrossRef CAS.
- Z. Ning, N. Jiang and Z. Gan, Polym. Degrad. Stab., 2014, 107, 120–128 CrossRef CAS.
- R. Liénard, N. Zaldua, T. Josse, J. De Winter, M. Zubitur, A. Mugica, A. Iturrospe, A. Arbe, O. Coulembier and A. J. Müller, Macromol. Rapid Commun., 2016, 37, 1676–1681 CrossRef PubMed.
- K. Matsubara, K. Eda, Y. Ikutake, M. Dan, N. Tanizaki, Y. Koga and M. Yasuniwa, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2536–2544 CrossRef CAS.
- D. B. Konwar, J. Jacob and B. K. Satapathy, Polym. Int., 2016, 65, 1107–1117 CrossRef CAS.
- X. Len, Y. Ren, Z. Wei, Y. Bian and Y. Li, Macromol. Chem. Phys., 2017, 218, 1700178 CrossRef.
- M. Jikei, Y. Yamadoi, T. Suga and K. Matsumoto, Polymer, 2017, 123, 73–80 CrossRef CAS.
- T. Rosen, I. Goldberg, W. Navarra, V. Venditto and M. Kol, Angew. Chem., Int. Ed., 2018, 57, 7191–7195 CrossRef CAS PubMed.
- C. G. Pitt, M. M. Gratzl, G. L. Kimmel, J. Surles and A. Schindler, Biomaterials, 1981, 2, 215–220 CrossRef CAS PubMed.
- J. M. Vion, R. Jerome, P. Teyssie, M. Aubin and R. E. Prudhomme, Macromolecules, 1986, 19, 1828–1838 CrossRef CAS.
- S. M. Li, J. L. Espartero, P. Foch and M. Vert, J. Biomater. Sci., Polym. Ed., 1997, 8, 165–187 CrossRef.
- J. Fernández, A. Etxeberria and J.-R. Sarasua, J. Mech. Behav. Biomed. Mater., 2012, 9, 100–112 CrossRef PubMed.
- J. Fernández, E. Meaurio, A. Chaos, A. Etxeberria, A. Alonso-Varona and J. R. Sarasua, Polymer, 2013, 54, 2621–2631 CrossRef.
- H. R. Kricheldorf, K. Ahrensdorf and S. Rost, Macromol. Chem. Phys., 2004, 205, 1602–1610 CrossRef CAS.
- Z. Wei, L. Liu, C. Qu and M. Qi, Polymer, 2009, 50, 1423–1429 CrossRef CAS.
- D. J. Darensbourg and O. Karroonnirun, Macromolecules, 2010, 43, 8880–8886 CrossRef CAS.
- N. Nomura, A. Akita, R. Ishii and M. Mizuno, J. Am. Chem. Soc., 2010, 132, 1750–1751 CrossRef CAS PubMed.
- G. Li, M. Lamberti, D. Pappalardo and C. Pellecchia, Macromolecules, 2012, 45, 8614–8620 CrossRef CAS.
- J. Odent, P. Leclère, J.-M. Raquez and P. Dubois, Eur. Polym. J., 2013, 49, 914–922 CrossRef CAS.
- A. Dalmoro, A. A. Barba, M. Lamberti, M. Mazze, V. Venditto and G. Lamberti, J. Mater. Sci., 2014, 49, 5986–5996 CrossRef CAS.
- A. J. Domb, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1973–1981 CrossRef CAS.
- M. Hiljanen-Vainio, P. Varpomaa, J. Seppälä and P. Törmälä, Macromol. Chem. Phys., 1996, 197, 1503–1523 CrossRef CAS.
- H. Tsuji and Y. Ikada, J. Appl. Polym. Sci., 1996, 60, 2367–2375 CrossRef CAS.
- J.-M. Yang, H.-L. Chen, J.-W. You and J. C. Hwang, Polym. J., 1997, 29, 657–662 CrossRef CAS.
- H. Tsuji and Y. Ikada, J. Appl. Polym. Sci., 1998, 67, 405–415 CrossRef CAS.
- H. Tsuji, A. Mizuno and Y. Ikada, J. Appl. Polym. Sci., 1998, 70, 2259–2268 CrossRef CAS.
- G. Maglio, A. Migliozzi, R. Palumbo, B. Immirzi and M. G. Volpe, Macromol. Rapid Commun., 1999, 20, 236–238 CrossRef CAS.
- Z. Gan, D. Yu, Z. Zhong, Q. Liang and X. Jing, Polymer, 1999, 40, 2859–2862 CrossRef CAS.
- S. Aslan, L. Calandrelli, P. Laurienzo, M. Malinconico and C. Migliaresi, J. Mater. Sci., 2000, 35, 1615–1622 CrossRef CAS.
- L. Liu, S. Li, H. Garreau and M. Vert, Biomacromolecules, 2000, 1, 350–359 CrossRef CAS PubMed.
- R. Dell'Erba, G. Groeninckx, G. Maglio, M. Malinconico and A. Migliozzi, Polymer, 2001, 42, 7831–7840 CrossRef.
- N.-S. Choi, C.-H. Kim, K. Y. Cho and J.-K. Park, J. Appl. Polym. Sci., 2002, 86, 1892–1898 CrossRef CAS.
- M. E. Broz and D. L. VanderHart, Biomaterials, 2003, 24, 4181–4190 CrossRef CAS PubMed.
- H. Tsuji, T. Yamada, M. Suzuki and S. Itsuno, Polym. Int., 2003, 52, 269–275 CrossRef CAS.
- H. Tsuji and T. Yamada, J. Appl. Polym. Sci., 2003, 87, 412–419 CrossRef CAS.
- M. Todo, S.-D. Park, T. Takayama and K. Arakawa, Eng. Fract. Mech., 2007, 74, 1872–1883 CrossRef.
- H. Tsuji, G. Horikawa and S. Itsuno, J. Appl. Polym. Sci., 2007, 104, 831–841 CrossRef CAS.
- H. Bai, C. Huang, H. Xiu, Y. Gao, Q. Zhang and Q. Fu, Polymer, 2013, 54, 5257–5266 CrossRef CAS.
- R. F. Sorey, J. S. Wiggins and A. D. Puckett, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2345–2363 CrossRef.
- A. Shibita, T. Shimasaki, N. Teramoto and M. Shibata, Polymer, 2015, 74, 54–62 CrossRef CAS.
- M. Shibata, M. Katoh, H. Takasea and A. Shibita, Polym. Chem., 2015, 6, 4123–4132 RSC.
- A. O. Helminen, H. Korhonen and J. V. Seppälä, Macromol. Chem. Phys., 2002, 203, 2630–2639 CrossRef CAS.
- M. Nagata and Y. Sato, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2426–2439 CrossRef CAS.
- M. Nagata and Y. Yamamoto, Macromol. Chem. Phys., 2010, 211, 1826–1835 CrossRef CAS.
- H. Miyasako, K. Yamamoto, A. Nakao and T. Aoyagi, Macromol. Biosci., 2007, 7, 76–83 CrossRef CAS PubMed.
- T. Muroya, K. Yamamoto and T. Aoyagi, Polym. Degrad. Stab., 2009, 94, 285–290 CrossRef CAS.
- R. Chapanian, M. Y. Tse, S. C. Pang and B. G. Amsden, J. Biomed. Mater. Res., Part A, 2010, 92, 830–842 Search PubMed.
- T. Cai, M. Li, K.-G. Neoh and E.-T. Kang, J. Mater. Chem. B, 2012, 22, 16248–16258 RSC.
- H. Tsuji, Y. Sakamoto and Y. Arakawa, Macromol. Chem. Phys., 2017, 218, 1700286 CrossRef.
- H. Tsuji, N. Matsumura and Y. Arakawa, J. Phys. Chem. B, 2016, 120, 1183–1193 CrossRef CAS PubMed.
- H. Tsuji, M. Ogawa and Y. Arakawa, J. Phys. Chem. B, 2016, 120, 11052–11063 CrossRef CAS PubMed.
- H. Tsuji, N. Masaki, Y. Arakawa, K. Iguchi and T. Sobue, Cryst. Growth Des., 2018, 18, 521–530 CrossRef CAS.
- H. Tsuji, T. Aratani and H. Takikawa, Macromol. Mater. Eng., 2013, 298, 1149–1159 CrossRef CAS.
- P. Pan, W. Kai, B. Zhu, T. Dong and Y. Inoue, Macromolecules, 2007, 40, 6898–6905 CrossRef CAS.
- P. Pan and Y. Inoue, Prog. Polym. Sci., 2009, 34, 605–640 CrossRef CAS.
- H. Tsuji, K. Tashiro, L. Bouapao and M. Hanesaka, Macromol. Chem. Phys., 2012, 213, 2099–2112 CrossRef CAS.
- H. Hu and D. L. Dorset, Macromolecules, 1990, 23, 4604–4607 CrossRef CAS.
- Y. Liu, X. Yang, W. Zhang and S. Zheng, Polymer, 2006, 47, 6814–6825 CrossRef CAS.
- S. Nojima, K. Hashizume, A. Rohadi and S. Sasaki, Polymer, 1997, 38, 2711–2718 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103–1112 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1940, 8, 212–224 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1941, 9, 177–184 CrossRef CAS.
- L. Mandelkern, Crystallization of Polymers, McGraw-Hill, New York, 1964 Search PubMed.
- A. T. Lorenzo, M. L. Arnal, J. Albuerne and A. J. Müller, Polym. Test., 2007, 26, 222–231 CrossRef CAS.
- See for example, U. W. Gedde, Polymer Physics, Chapman & Hall, London, 1995, ch. 8, pp. 170−198 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Typical 1H NMR spectrum of 4-C-L copolymer (4-C49-L38) (Fig. S1), typical GPC curves of 4-C and 4-C-L polymers (before and after 2nd step polymerization, 4-C49 and 4-C49-L38, respectively) (Fig. S2), degree of crosslinking and crystallinity of samples (Table S1), WAXD profile of 4-C49-L38 sample prepared by solvent evaporation (Fig. S3), DSC heating and cooling thermograms of 4-C49-L38 sample prepared by solvent evaporation (before crosslinking) (Fig. S4), thermal properties of samples (Table S2), enthalpies of cold crystallization and melting of samples before and after crosslinking for heating scanning (Fig. S5), enthalpies of crystallization of samples before and after crosslinking for cooling scanning (Fig. S6), relative crystallinity (Xr) of samples before and after crosslinking for cooling scanning (Fig. S7). See DOI: 10.1039/c9ra00255c |
| This journal is © The Royal Society of Chemistry 2019 |