
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDonor–acceptor polymers containing thiazole-fused benzothiadiazole acceptor units for organic solar cells†
Tomoya Nakamuraa,
Yasuhisa Ishikuraa,
Noriko Arakawaa,
Megumi Horia,
Motoi Satoua,
Masaru Endoa,
Hisashi Masuib,
Shinichiro Fusec,
Takashi Takahashib,
Yasujiro Murata a,
Richard Murdeya and
Atsushi Wakamiya*a
a,
Richard Murdeya and
Atsushi Wakamiya*a
aInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: wakamiya@scl.kyoto-u.ac.jp
bDepartment of Pharmaceutical Sciences, Yokohama University of Pharmacy, 601, Matano-cho, Totsuka-ku, Yokohama 245-0066, Japan
cLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8503, Japan
First published on 1st March 2019
Abstract
Two p-type semiconducting donor–acceptor polymers were designed and synthesized for use in organic solar cells. The polymers combine a benzodithiophene (BDT) donor and a thiazole-fused benzothiadiazole (TzBT) acceptor. Two TzBT acceptor units are compared, one with an alkylthio group (P1) and the other with a more strongly electron-withdrawing alkylsulfonyl group (P2) at the fused thiazole ring. The strongly electron-accepting nature of the TzBT unit lowers the lowest unoccupied molecular orbital (LUMO) energy of P1 and P2 relative to that of the BT analog (PBDT-BT), without altering the energy of the highest occupied molecular orbital (HOMO). Despite the smaller optical band gaps, bulk heterojunction organic solar cells fabricated using these polymers in a PC71BM blend showed high open-circuit voltages. The power conversion efficiency (PCE) of the P1-based device reached 6.13%. Though efficiency of the P2-based device was lower, photoelectric conversion extended into the near-IR region up to 950 nm.
Introduction
Organic solar cells (OSCs) have attracted considerable attention as low cost, flexible, and light-weight sources of electrical power.1–4 In bulk-heterojunction OSC devices, p-type materials, typically semiconducting polymers, are blended with n-type molecular materials, typically fullerene derivatives such as [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM).5,6 The p-type semiconductor is often a conjugated donor–acceptor (D–A) polymer consisting of alternating electron-rich donor units and electron-deficient acceptor units.7,8 A promising approach to improve the power conversion efficiency (PCE) of OSCs is using p-type materials with a narrow highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) band gap (Eg) to increase short-circuit current density (JSC).9 The open circuit voltage, VOC, scales closely with the energy difference between the HOMO of the p-type polymer and LUMO of the n-type fullerene.10 Hence, lowering the LUMO energy level of the p-type polymer without changing the HOMO energy level should lead to a narrow band gap without sacrificing the VOC. The development of strong acceptor units is therefore a key element for fine-tuning the electronic structure. The difference in energy between the optical band gap of the absorber and the open-circuit voltage of the device is defined as the photon energy loss (Eloss = Eg − eVOC).11–13 For high PCE, the photon energy loss should be minimized.2,1,3-Benzothiadiazole (BT) is a widely used acceptor unit in D–A polymers.14–16 The strong electron-accepting ability of benzothiadiazole stems from the strong butadiene character and the presence of the two electron-withdrawing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds. A common approach to enhance the electron-accepting ability is the introduction of electron-withdrawing substituents such as fluoro,17–19 chloro,20 and cyano21 groups. As a unique electronic modification, we have demonstrated the utility of the formation of intramolecular B–N bonds, which makes it possible to lower the LUMO energy level without changing the orbital distribution.22–29 Another way for modification of BT unit is the heteroannulation at the 5- and 6-positions to give acceptors such as thiadiazoloquinoxaline (TDQ) and benzobisthiadiazole (BBT).30,31 We have recently developed a thiazole-fused BT skeleton (TzBT) as a new type of electron acceptor unit with further enhancement of the electron-accepting ability of BT skeleton.32–34 Additional tuning of the electron-accepting character is realized by changing the alkylthio group to alkylsulfonyl (Fig. 1a).34 We anticipated that this simple substitution of the alkylthio group in the TzBT derivative can fine-tune the electronic structure of a D–A polymer when TzBT is used as the acceptor unit.
N double bonds. A common approach to enhance the electron-accepting ability is the introduction of electron-withdrawing substituents such as fluoro,17–19 chloro,20 and cyano21 groups. As a unique electronic modification, we have demonstrated the utility of the formation of intramolecular B–N bonds, which makes it possible to lower the LUMO energy level without changing the orbital distribution.22–29 Another way for modification of BT unit is the heteroannulation at the 5- and 6-positions to give acceptors such as thiadiazoloquinoxaline (TDQ) and benzobisthiadiazole (BBT).30,31 We have recently developed a thiazole-fused BT skeleton (TzBT) as a new type of electron acceptor unit with further enhancement of the electron-accepting ability of BT skeleton.32–34 Additional tuning of the electron-accepting character is realized by changing the alkylthio group to alkylsulfonyl (Fig. 1a).34 We anticipated that this simple substitution of the alkylthio group in the TzBT derivative can fine-tune the electronic structure of a D–A polymer when TzBT is used as the acceptor unit.
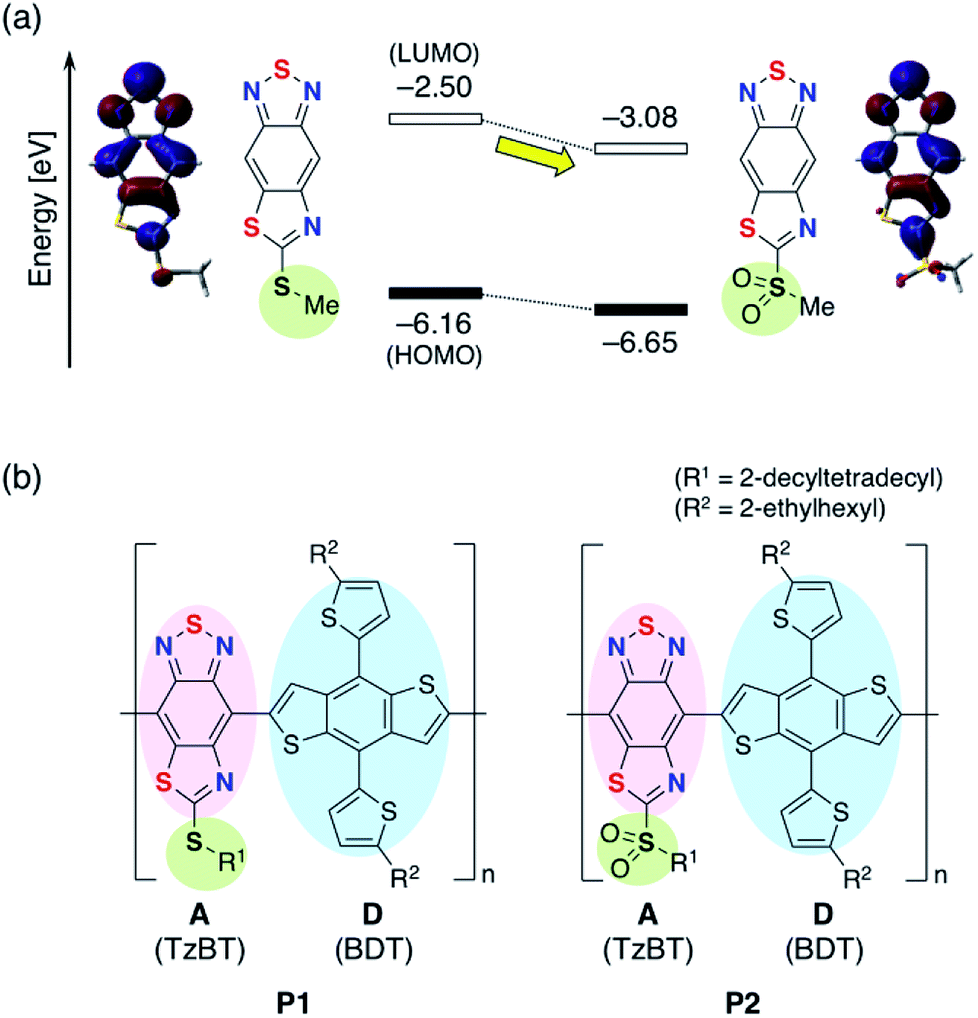 | ||
| Fig. 1 (a) Fine-tuning of electron-accepting ability in TzBT bearing alkylthio group. Calculated energy level diagram with depictions of the LUMO at B3LYP/6-31G(d) level of theory.37 (b) Chemical structures of TzBT-based D–A polymers P1 and P2. | ||
Wong, Jones, and co-workers reported a D–A polymer composed of a benzodithiophene (BDT) donor and a BT acceptor (PBDT-BT) for the use as a p-type semiconductor in OSC devices with a blend with PC71BM.35,36 Although a high PCE of 9.4% is reported, the device shows a relatively large photon energy loss of 0.83 eV as a result of the high-lying LUMO energy level of PBDT-BT. The PCE might therefore be further improved by reducing the band gap of the polymer while maintaining the VOC. To examine the potential of the TzBT unit in OSC devices, we designed donor–acceptor polymers containing two types of TzBT acceptor units, P1 with alkylthio groups and P2 with alkylsulfonyl groups at the fused thiazole rings, respectively (Fig. 1b). The strong electron-accepting nature of TzBT unit, especially in the unit with alkylsulfonyl substituent, is expected to lower the LUMO energy levels in these polymers compared to the BT analog, resulting in a smaller photon energy loss (Eloss).
In the present work, the synthesis of the D–A polymers is reported, followed by a summary of the photophysical properties evaluated by ultraviolet-visible-NIR (UV-Vis-NIR) spectroscopy and photoemission yield spectroscopy (PYS). Smaller optical band gaps compared to BT-based polymers were observed, and PYS confirmed that the HOMO energy levels are relatively unchanged. Bulk heterojunction organic solar cell devices were fabricated using the blend of these polymers with PC71BM as the light-absorbing layer. P1 and P2-based devices exhibited smaller photon energy losses than that for PBDT-BT based device.
Results and discussion
Synthesis and characterization of polymers
For the synthesis of TzBT acceptor unit, we followed the same method we previously developed for the synthesis of the unit with a methylthio group substituent (Scheme 1).34 The reaction of commercially available 2-chloro-5-nitrobenzene-1,4-diamine (1) with sodium ethylxanthate gave the intermediate 2, which was treated with 2-decyltetradecyl bromide to give benzothiazole 3 in 68% yield in two steps. Reduction of the nitro group by tin(II) chloride gave diamine 4, and the following condensation with thionyl chloride gave thiazole-fused BT 5 in 81% yield in two steps. The reaction of 5 with bromine in the presence of iron(III) chloride yielded 6 with an alkylthio group in 63% yield. Furthermore, 7 with an alkylsulfonyl group was synthesized by the reaction with two equivalents of mCPBA in 89% yield.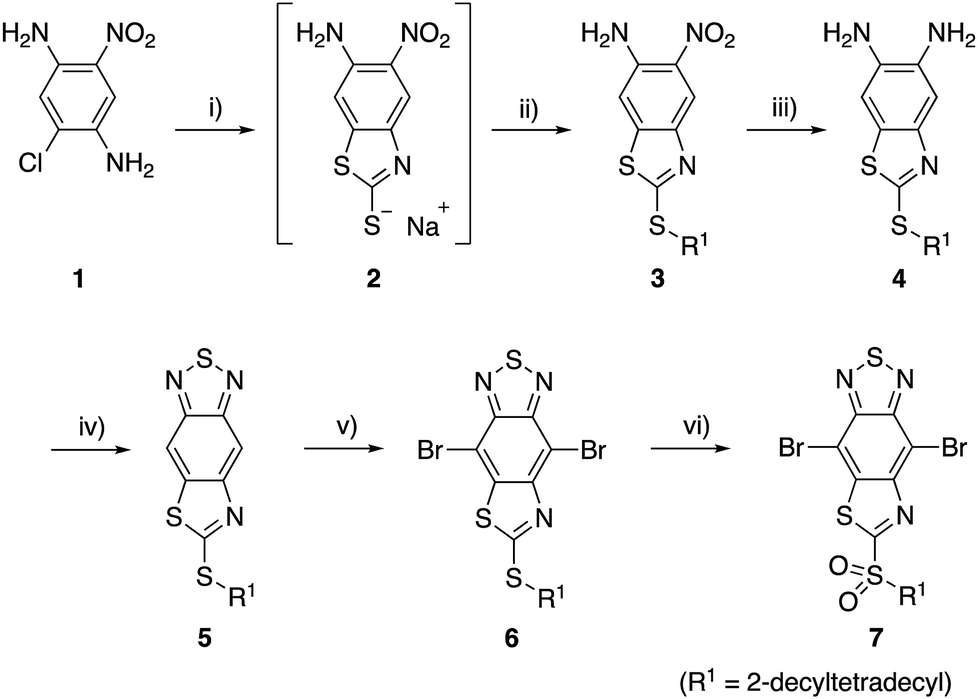 | ||
| Scheme 1 Synthesis of thiazole-fused BT units. Reagents and conditions: (i) sodium ethylxanthate (2.0 equiv.), DMF, 120 °C, 3 h; (ii) R1Br (1.5 equiv.), DMF, 0 °C to rt, 16 h, 68% in two steps; (iii) SnCl2·2H2O (5.0 equiv.), MeOH/HCl aq., 70 °C, 22 h; (iv) SOCl2 (3.0 equiv.), Et3N (6.0 equiv.), CH2Cl2, 0 °C to rt, 2 h, 81% in two steps; (v) Br2 (48 equiv.), FeCl3·6H2O (0.6 equiv.), 50 °C, 19 h, 63%; (vi) mCPBA (2.0 equiv.), CH2Cl2, rt, 28 h, 89%. | ||
The monomers 6 and 7 were polymerized with the benzodithiophene (BDT) unit via the Stille coupling reaction assisted by microwave heating to give P1 and P2, respectively (Scheme 2). The number-average molecular weight (Mn) and weight-average molecular weight (Mw) were estimated to be 16 kDa and 35 kDa with a polydispersity index (PDI) of 2.15 for P1 by high-temperature gel permeation chromatography (GPC) using o-dichlorobenzene as the eluent at 140 °C. Mn and Mw were 22 kDa and 66 kDa with a PDI of 3.04 for P2 (Table 1).
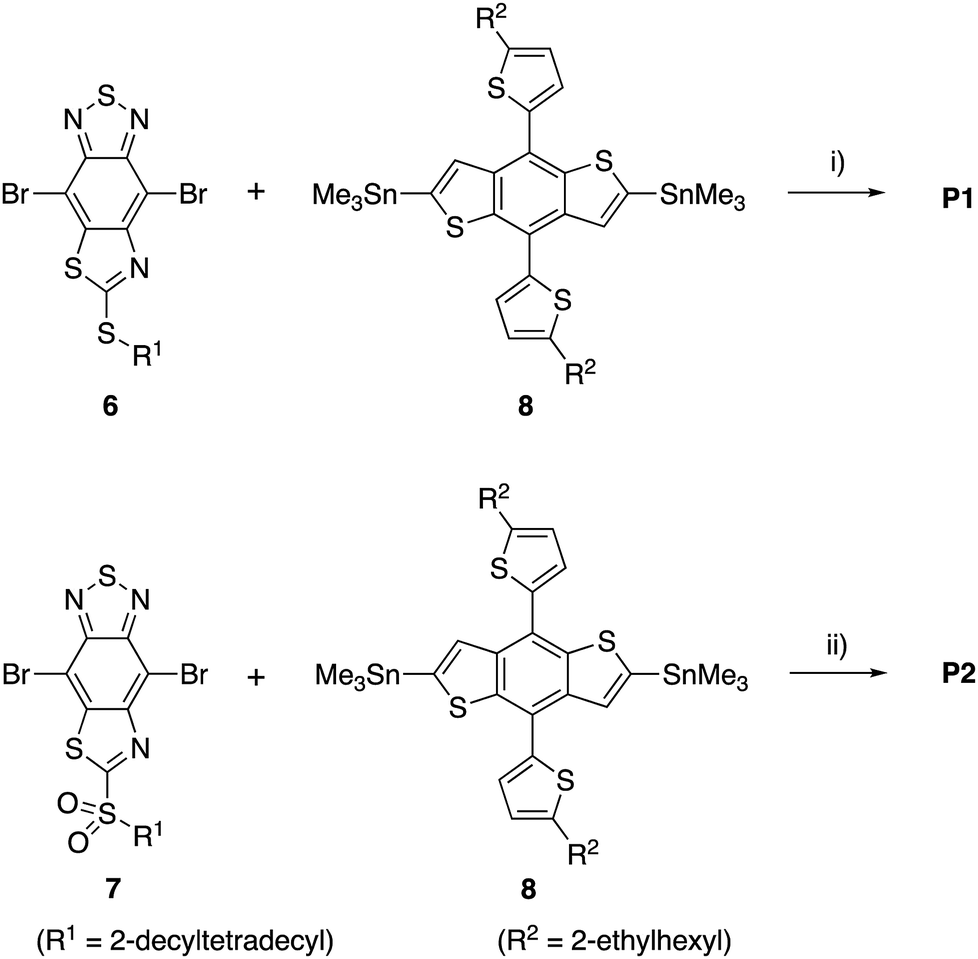 | ||
| Scheme 2 Synthesis of D–A polymers. Reagents and conditions: (i) Pd2(dba)3·CHCl3 (2 mol%), P(o-tolyl)3 (8 mol%), toluene, microwave, 160 °C (ca. 3.4 atm), 1 h, 86%; (ii) Pd2(dba)3·CHCl3 (2 mol%), P(o-tolyl)3 (8 mol%), toluene, microwave, 160 °C (ca. 3.4 atm), 1 h, 69%. | ||
The thermal stability of the polymers was examined by thermogravimetric analysis (TGA) under nitrogen atmosphere with a heating rate of 10 °C min−1 (Fig. S1†). The 5% weight loss temperatures (Td5) were 332 °C and 340 °C for P1 and P2, respectively, confirming that these polymers have good thermal stability.
Photophysical properties
The UV-Vis-NIR absorption spectra of the polymers were measured in dichloromethane solution and in the solid state (Fig. 2). In solution, P1 with alkylthio substituents shows an absorption maximum (λmax) at 746 nm. This value is red-shifted compared to that of the BT analog, PBDT-BT (λmax = 650 nm),35,36 by ca. 100 nm, as a result of the enhanced electron-accepting nature of the TzBT unit. P2 with alkylsulfonyl substituents exhibits λmax of 829 nm in the near-IR region, which is further red-shifted by ca. 80 nm compared to that of P1. In the thin films, the absorption is broadened and red-shifted to 756 nm for P1 and 848 nm for P2, respectively, suggesting some degree of intermolecular interactions in the solid state.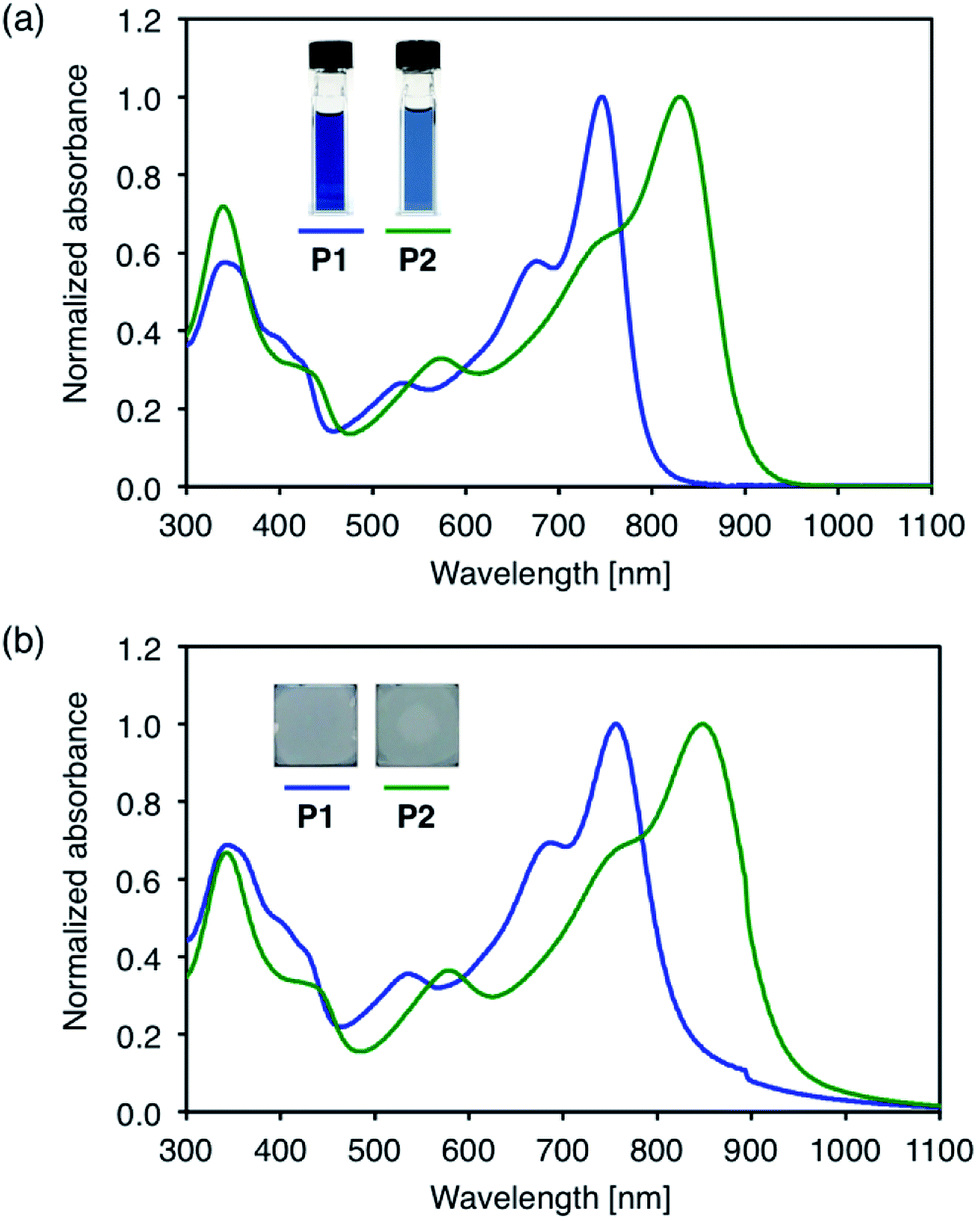 | ||
| Fig. 2 UV-Vis absorption spectra of D–A polymers; (a) in CH2Cl2 solution and (b) in the solid state. | ||
The optical band gaps (Eg) were estimated using Tauc plots of the solid state absorption spectra (Fig. S2†). Eg of P1 and P2 are 1.54 eV and 1.37 eV, respectively, which are smaller than that of PBDT-BT (1.75 eV).
Electronic properties
In this section, the solid state energy levels of these polymers as well as n-type PC71BM are determined by experimental and theoretical methods. The HOMO energy level (EHOMO) of the polymer thin films on ITO substrates was estimated using photoemission yield spectroscopy (PYS) in air. EHOMO of n-type PC71BM was also investigated. The values of −5.26 eV, −5.35 eV, and −6.08 eV were obtained for P1, P2, and PC71BM, respectively (Fig. 3a). The EHOMO values of P1 and P2 do not significantly differ from the value of PBDT-BT (−5.27 eV) estimated similarly by PYS.35 The LUMO energy levels (ELUMO) were determined as the sum of EHOMO and Eg. The values were −3.72 eV, −3.98 eV, and −4.19 eV for P1, P2, and PC71BM, respectively. Compared to the ELUMO of PBDT-BT (−3.52 eV),35 the ELUMO of P1 and P2 were found to be deeper by 0.20 eV and 0.46 eV, respectively. The lowering of LUMO energy levels in TzBT-based polymers was also confirmed by cyclic voltammetry (CV) measurements (Fig. S3†). As the energetic parameters determined by CV measurements can be affected by the solvent, supporting electrolyte, and electrodes,38 the parameters determined by PYS and Eg are considered to be more reliable. Fig. 3b shows the energy level diagram of P1, P2, and PC71BM, together with the energy levels of PBDT-BT35 estimated by PYS and the optical band gap. Table 2 summarizes the physicochemical properties. As a result of the effective lowering of the LUMO energy levels in P1 and P2, the energy differences in the LUMO energy of p-type polymers and PC71BM decrease from 0.67 eV for PBDT-BT to 0.47 eV for P1 and 0.21 V for P2, respectively. Consequently, smaller photon energy losses are anticipated in organic solar cells using these TzBT-based polymers, especially in the cells using P2 with alkylsulfonyl groups.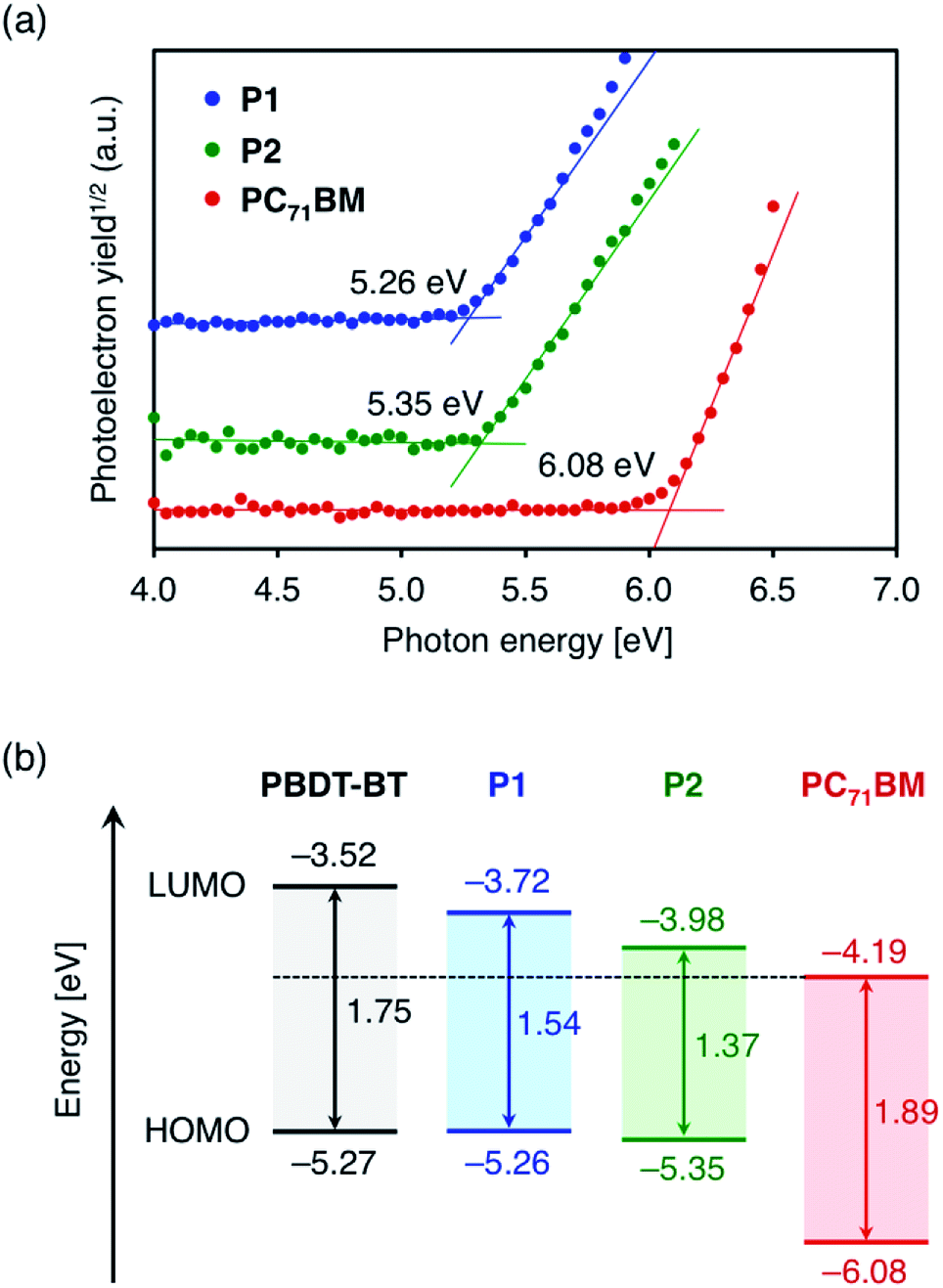 | ||
| Fig. 3 (a) Photoemission yield spectra of the polymers as well as PC71BM in the solid state and (b) energy level diagram of P1, P2, and PC71BM, estimated from photoemission yield spectroscopy and optical band gap in the solid state. Energy levels of PBDT-BT35 are also shown for comparison. | ||
| Polymer | λmaxa [nm] | EHOMOb [eV] | Egc [eV] | ELUMOd [eV] | |
|---|---|---|---|---|---|
| Solution | Film | ||||
| a Absorption maximum.b The HOMO energy levels estimated by photoemission yield spectroscopy (PYS) in air.c Optical band gap estimated from the edge of the Tauc plot.d The LUMO energy levels calculated from the HOMO energy level and optical band gap (ELUMO = EHOMO + Eg). | |||||
| P1 | 746 | 756 | −5.26 | 1.54 | −3.72 |
| P2 | 829 | 848 | −5.35 | 1.37 | −3.98 |
The DFT calculations on the tetramer model compounds clarifies the selective reduction in the LUMO energy levels in these polymers. The calculated electron density of the LUMO is located on the TzBT moieties, whereas the HOMO is mainly distributed at the BDT donor units (Fig. S4†). The use of a strong acceptor unit should therefore have a greater impact on the LUMO energy than the HOMO energy.
Device fabrication and photovoltaic properties
To evaluate the photovoltaic properties of these TzBT-based polymers, bulk heterojunction solar cells using the two polymers were prepared with the following structure; indium tin oxide (ITO)/zinc oxide (ZnO)/polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC71BM/molybdenum oxide (MoOx)/Ag. The light-absorbing layer was spin-coated from the polymer:PC71BM blend solution (optimized ratio, 1:1.2 wt/wt for P1, 1:1 wt/wt for P2) in chlorobenzene with 3 vol% 1,8-diiodooctane (DIO) as an additive to control the bulk heterojunction morphology.39 The optimal thickness of the light-absorbing layer was 80 nm, similar to the BT analog PBDT-BT device (90 nm).35
:PC71BM/molybdenum oxide (MoOx)/Ag. The light-absorbing layer was spin-coated from the polymer:PC71BM blend solution (optimized ratio, 1:1.2 wt/wt for P1, 1:1 wt/wt for P2) in chlorobenzene with 3 vol% 1,8-diiodooctane (DIO) as an additive to control the bulk heterojunction morphology.39 The optimal thickness of the light-absorbing layer was 80 nm, similar to the BT analog PBDT-BT device (90 nm).35
Fig. 4a shows the current density–voltage (J–V) characteristics of the representative polymer:PC71BM cells under AM 1.5 G irradiation (100 mW cm−2). The solar cell devices using P1 and P2 showed high open-circuit voltages (VOC) of 0.78 V and 0.79 V, respectively (Table 3). The high VOC of the cell using P2 with alkylsulfonyl substituents is especially striking given the small band gap of P2. From the optical band gaps of these polymers (1.54 eV for P1 and 1.37 eV for P2), the photon energy losses (Eloss = Eg − eVOC) were estimated to be 0.76 eV and 0.58 eV for P1 and P2-based cells, respectively. The energy losses are lower compared to that for the BT analog PBDT-BT (0.83 eV) by 0.06 eV and 0.25 eV for P1 and P2, respectively.35 The reduced energy loss is attributed to the enhanced electron-accepting ability of the TzBT units, which lowers the LUMO energy levels of D–A polymers.
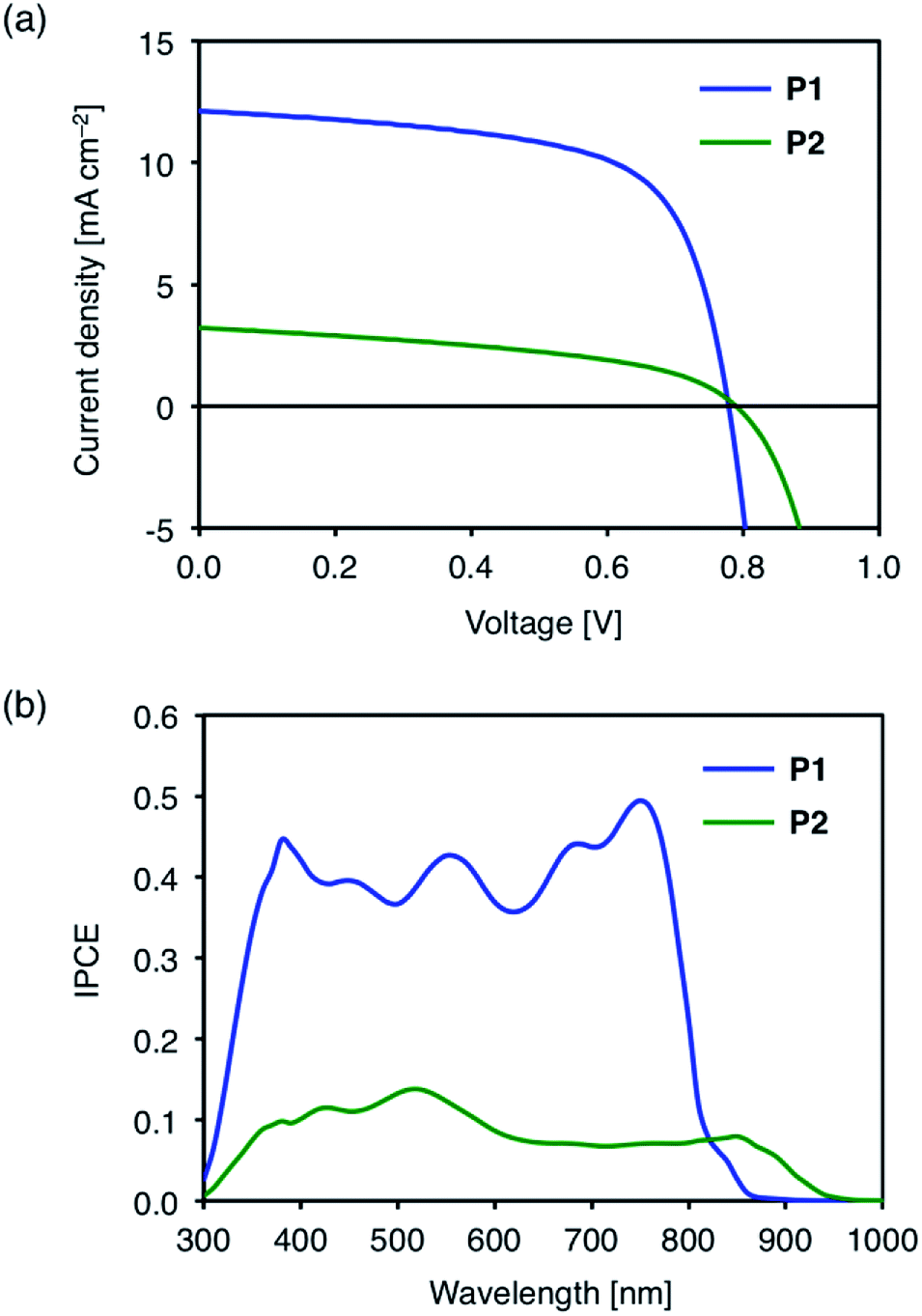 | ||
| Fig. 4 (a) J–V characteristics, and (b) incident photon to current conversion efficiency (IPCE) spectra of polymer/PC71BM cells. | ||
| Polymer | JSC [mA cm−2] | VOC [V] | FF | PCE [%] | Elossa [eV] |
|---|---|---|---|---|---|
| a Photon energy loss defined by Eloss = Eg − eVOC.b Average PCE with standard deviation from 15 independent devices.c Average PCE with standard deviation from 6 independent devices. | |||||
| P1 | 12.1 | 0.78 | 0.65 | 6.13 (5.97 ± 0.21)b | 0.76 |
| P2 | 3.2 | 0.79 | 0.45 | 1.15 (1.12 ± 0.03)c | 0.58 |
The PCEs of the P1 and P2-based devices were 6.13% and 1.15%, respectively (Table 3). The lower PCEs than the reported value for the PBDT-BT based cell (9.4%) are mainly due to the lower short-circuit current densities (JSC) of 12.1 mA cm−2 and 3.2 mA cm−2 for the P1 and P2-based devices, respectively, compared to 15.4 mA cm−2 for the PBDT-BT based device. The reduced current output correlates with the lower incident photon to current conversion efficiencies (IPCE) of P1 (0.4–0.5) and P2 (ca. 0.1) based devices (Fig. 4b) compared to the PBDT-BT based device (0.6–0.8).35 The IPCE spectra closely resemble the thin film absorption spectra of the polymer:PC71BM blends (Fig. S5†). Though the IPCE of P2-based device was low, it should be noted that the photon to current conversion in the near-IR region extends up to 950 nm.
The different IPCE and JSC values could be caused by at least three different factors: (i) the morphology of polymer:PC71BM heterojunction, (ii) the charge carrier mobility of the blends, or (iii) the energy offset of the LUMOs between the polymer and PC71BM. These possibilities were examined as follows:
The film morphology of polymer and PC71BM blends was imaged by atomic force microscopy (AFM). Without 1,8-diiodooctane (DIO) additive, the films of P1:PC71BM exhibits large-size phase separation with isolated PC71BM domains of ca. 300 nm (Fig. S6a†). The addition of 3 wt% DIO significantly decreased phase separation size to ca. 100 nm (Fig. S6b†). This observation is in good agreement with the increased JSC in the P1-based device with increased amount of DIO additive (Fig. S7 and Table S1†). The phase separation size is, however, still larger than the ideal value of ∼20 nm, which is likely the reason of the moderate IPCE and JSC of P1-based device. Though the P2:PC71BM blends have slightly smaller domain size of ca. 80 nm (Fig. S6d†), the overall morphology is similar, making it difficult to assign any causal relationship between morphology and performance.
The hole mobility of the polymer:PC71BM blends was measured using the space-charge-limited current (SCLC) method. Hole only devices were fabricated with the structure of ITO/poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS)/polymer:PC71BM/Au. Both polymers exhibit similar hole mobility, which was (1.06 ± 0.09) × 10−5 cm2 V−1 s−1 for P1 and (1.21 ± 0.08) × 10−5 cm2 V−1 s−1 for P2 (Fig. S8 and Table S2†). It seems then that hole mobility is also unlikely a limiting factor in the present instance.
As no significant differences were observed in P1 and P2 in regard to film morphology and hole mobility, it would be reasonable to ascribe the different JSC values to the energy offsets in the LUMOs of the polymer and PC71BM. Whereas the energy offset is 0.47 eV for P1, it is only 0.21 eV for P2, the latter being smaller than the empirical value of 0.3 eV considered necessary for efficient charge separation.10 This would explain why the P2-based device shows low JSC.
Conclusions
Two donor–acceptor polymers combining a thiazole-fused benzothiadiazole (TzBT) acceptor unit with the benzodithiophene (BDT) donor unit were designed and synthesized. In polymer P1, the TzBT acceptor unit had an alkylthio substituent. In polymer P2, this was replaced with a more electron-withdrawing alkylsulfonyl group. The strong electron-accepting nature of the TzBT units lowered the LUMO energy levels of P1 and P2 compared to that of the BT analog (PBDT-BT) by 0.20 and 0.46 eV, respectively. Since the HOMO energies were remained largely unchanged, the band gaps were reduced. Bulk heterojunction organic solar cells using a polymer/PC71BM blend showed high VOC of 0.78 V and 0.79 V for P1 and P2-based devices, despite their small optical band gaps of 1.54 eV and 1.37 eV. The photon energy loss was determined to be 0.76 eV and 0.58 eV, respectively, smaller than for the PBDT-BT based device (0.83 eV). The overall power conversion efficiencies of these devices were found to be 6.13% for P1 and 1.15% for P2. NIR photon-to-current conversion was found to extend to 860 nm for P1 and 950 nm for P2.Experimental section
General
Melting points (mp) were measured on a Yanaco Micro Melting Point Apparatus or Stanford Research Systems Opti Melt. 1H and 13C NMR spectra were recorded with JEOL JNM ECA500 (500 MHz for 1H and 126 MHz for 13C). Chemical shifts are reported in δ ppm using residual protons in the deuterated solvents for 1H NMR and using solvent peaks for 13C NMR as internal standards. UV-Vis-NIR absorption spectra were recorded with a Shimadzu UV-3150 spectrometer. Mass spectra were measured on a Bruker micrOTOF-Q II (APCI). Ionization potentials in the solid states were determined by an ambient photoelectron spectroscopy method with a Riken-Keiki AC-3 spectrometer. Thermogravimetric analysis (TGA) was performed on a SHIMADZU TGA-50 apparatus. Thin layer chromatography (TLC) was performed on plates coated with 0.25 mm thick silica gel 60F-254 (Merck). Column chromatography was performed using silica gel PSQ 60B (Fuji Silysia) or PSQ 100B (Fuji Silysia). The microwave reaction was performed using Anton Paar Monowave 300. All reactions were carried out under an argon atmosphere except as otherwise noted.Analytical GPC was performed on a HLC 8120 GPC system with a TOSOH TSKgel GMHHR-H(S)HT column. o-Dichlorobenzene was used as the mobile phase with a flow rate of 1.0 mL min−1 at 140 °C. The columns were calibrated against nine standard polystyrene samples (Mn = 1200–1410000).
Photocurrent-voltage measurements for organic solar cells were measured in air with an OTENTO-SUNIII (BUNKOUKEIKI Co., Ltd.) and a Keithley 2400. The light intensity of the illumination source was adjusted by using standard silicon photodiodes: BS520 for J–V characteristics and SiPD S1337-1010BQ for EQE measurements.
All calculations were conducted using the Gaussian 09 program.37 The geometries were optimized at the B3LYP/6-31G(d) level of theory. The fact that these geometries are at the energy minimum was confirmed by frequency calculations at the same level of theory.
Synthesis
:1, Rf = 0.25) to give 3 (18.3 g, 68% in two steps) as dark red oil.1H NMR (500 MHz, CDCl3): δ 8.55 (s, 1H), 7.08 (s, 1H), 5.97 (brs, 2H), 3.36 (d, J = 5.7 Hz, 2H), 1.77–1.82 (m, 1H), 1.25–1.43 (m, 40H), 0.86–0.89 (m, 6H); 13C NMR (126 MHz, CDCl3): δ 166.7, 145.5, 145.0, 141.2, 132.2, 117.7, 108.2, 38.2, 37.6, 33.2 (2C), 31.9 (2C), 29.8 (2C), 29.6 (8C), 29.3 (2C), 26.5 (2C), 22.7 (2C), 14.1 (2C); HRMS (APCI) (m/z): [M − H]− calcd. for C31H52N3O2S2, 562.3506; found, 562.3506.
:1, Rf = 0.25) to give 5 (14.0 g, 81% in two steps) as brown oil.1H NMR (500 MHz, CDCl3): δ 8.32 (s, 1H), 8.29 (s, 1H), 3.47 (d, J = 6.3 Hz, 2H), 1.83–1.87 (m, 1H), 1.24–1.46 (m, 40H), 0.85–0.89 (m, 6H); 13C NMR (126 MHz, CDCl3): δ 172.9, 155.8, 153.9, 152.0, 140.6, 111.5, 110.3, 38.1, 37.7, 33.3 (2C), 31.9 (2C), 29.8 (2C), 29.6 (8C), 29.3 (2C), 26.6 (2C), 22.7 (2C), 14.1 (2C); HRMS (APCI) (m/z): [M]− calcd for C31H51N3S3, 561.3245; found, 561.3240; elemental analysis calcd (%) for C31H51N3S3: C 66.26, H 9.15, N 7.48; found: C 66.05, H 9.26, N 7.50.
:1, Rf = 0.6) to give 6 (243 mg, 63%) as yellow oil.1H NMR (500 MHz, CDCl3): δ 3.50 (d, J = 6.3 Hz, 2H), 1.87–1.91 (m, 1H), 1.24–1.46 (m, 40H), 0.85–0.89 (m, 6H); 13C NMR (126 MHz, CDCl3): δ 173.5, 152.6, 152.1, 149.8, 142.9, 102.7, 102.2, 38.6, 38.0, 33.4 (2C), 31.9 (2C), 29.9 (2C), 29.6 (8C), 29.3 (2C), 26.6 (2C), 22.7 (2C), 14.1 (2C); HRMS (APCI) (m/z): [M]− calcd for C31H49Br2N3S3, 717.1455; found, 717.1471.
:1, Rf = 0.6) to give 7 (201 mg, 89%) as a pale-yellow solid.Mp (decomp.): 280.6 °C; 1H NMR (500 MHz, CDCl3): δ 3.61 (d, 2H), 2.18–2.21 (m, 1H), 1.21–1.57 (m, 40H), 0.86–0.89 (t, overlapped, 12H); 13C NMR (126 MHz, CDCl3): δ 171.7, 152.3, 151.2, 151.1, 141.8, 110.3, 104.8, 57.8, 33.2 (2C), 32.9, 31.9 (2C), 29.7–29.5 (12C), 26.0 (2C), 22.7 (2C), 14.1 (2C); HRMS (APCI) (m/z): [M]− calcd for C31H49Br2N3O2S3, 749.1354; found, 749.1372.
Solar cell fabrication
The ITO-coated glass substrate (5 Ω cm−2, 2.5 cm × 2.5 cm, GEOMATEC) was washed carefully under ultrasonic irradiation using water (15 min), acetone (15 min), detergent solution (Semico Clean 56, Furuuchi chemical, 15 min), water (15 min) and ethanol (15 min). The substrate was further cleaned with a Filgen UV230 UV/ozone cleaner.0.2 M Zn(OAc)2 solution was prepared by dissolving Zn(OAc)2·2H2O (110 mg, 0.50 mmol) in ethanolamine (30 μL) and anhydrous ethanol (2.5 mL). A thin layer of ZnO was prepared onto the ITO substrate by spin-coating of precursor solution at 1200 rpm for 50 s under relative humidity of 30%. The resulting substrate was heated at 150 °C for 30 min under ambient conditions.
The photoactive layers were deposited in a glove box filled with an inert gas. For P1-based device, chlorobenzene solution containing 10 mg mL−1 of P1 with PC71BM (P1:PC71BM = 1:1.2 wt/wt) and 3 vol% 1,8-diiodooctane was deposited by spin-coating at 900 rpm for 40 s. For P2-based device, chlorobenzene solution containing 5 mg mL−1 of P2 with PC71BM (P2:PC71BM = 1:1 wt/wt) and 3 vol% 1,8-diiodooctane was deposited by spin-coating at 900 rpm for 40 s. Finally, a layer of MoOx (10 nm) and silver (90 nm) were vacuum deposited.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JST ALCA (JPMJAL 1603), ERATO (JPMJER 1302), COI, and NEDO. T. N. thanks the JSPS for a Research Fellowship for Young Scientists. The elemental analysis and HRMS measurements were supported by the Joint Usage/Research Center (JURC) at the ICR, Kyoto University.Notes and references
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- M. Kaltenbrunner, M. S. White, E. D. Głowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 770 CrossRef PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- T. Kumari, S. M. Lee, S. Kang, S. Chen and C. Yang, Energy Environ. Sci., 2017, 10, 258–265 RSC.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef CAS PubMed.
- B. D. Mühlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef.
- P. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- W. Cai, X. Gong and Y. Cao, Sol. Energy Mater. Sol. Cells, 2010, 94, 114–127 CrossRef CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, D. Patrick, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed.
- K. Kawashima, T. Fukuhara, Y. Suda, Y. Suzuki, T. Koganezawa, H. Yoshida, H. Ohkita, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 10265–10275 CrossRef CAS PubMed.
- I. Osaka and K. Takimiya, Adv. Mater., 2017, 29, 1605218 CrossRef PubMed.
- J. Peet, A. J. Heeger and G. C. Bazan, Acc. Chem. Res., 2009, 42, 1700–1708 CrossRef CAS PubMed.
- J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS PubMed.
- J. Du, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2016, 4, 15771–15787 RSC.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang and L. Lan, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang and H. Lin, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 CrossRef CAS PubMed.
- W. Chen and F. He, Polym. Chem., 2018, 9, 940–947 RSC.
- Z. Fei and M. Heeney, J. Mater. Chem. C, 2015, 3, 265–275 RSC.
- A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170–3173 CrossRef CAS PubMed.
- H. Nishimura, N. Ishida, A. Shimazaki, A. Wakamiya, A. Saeki, L. T. Scott and Y. Murata, J. Am. Chem. Soc., 2015, 137, 15656–15659 CrossRef CAS PubMed.
- C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648–3652 CrossRef CAS PubMed.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS PubMed.
- H. Shimogawa, O. Yoshikawa, Y. Aramaki, M. Murata, A. Wakamiya and Y. Murata, Chem.–Eur. J., 2017, 23, 3784–3791 CrossRef CAS PubMed.
- H. Shimogawa, M. Endo, T. Taniguchi, Y. Nakaike, M. Kawaraya, H. Segawa, Y. Murata and A. Wakamiya, Bull. Chem. Soc. Jpn., 2017, 90, 441–450 CrossRef CAS.
- H. Shimogawa, M. Endo, Y. Nakaike, Y. Murata and A. Wakamiya, Chem. Lett., 2017, 46, 715–718 CrossRef CAS.
- H. Shimogawa, Y. Murata and A. Wakamiya, Org. Lett., 2018, 20, 5135–5138 CrossRef CAS PubMed.
- T. C. Parker, D. G. Patel, K. Moudgil, S. Barlow, C. Risko, J. L. Brédas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2015, 2, 22–36 RSC.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- M. Satou, K. Uchinaga, A. Wakamiya and Y. Murata, Chem. Lett., 2014, 43, 1386–1388 CrossRef CAS.
- M. Satou, T. Nakamura, Y. Aramaki, S. Okazaki, M. Murata, A. Wakamiya and Y. Murata, Chem. Lett., 2016, 45, 892–894 CrossRef CAS.
- T. Nakamura, S. Okazaki, N. Arakawa, M. Satou and M. Endo, J. Photopolym. Sci. Technol., 2017, 30, 561–568 CrossRef CAS.
- J. Subbiah, B. Purushothaman, M. Chen, T. Qin, M. Gao, D. Vak, F. H. Scholes, X. Chen, S. E. Watkins, G. J. Wilson, A. B. Holmes, W. W. H. Wong and D. J. Jones, Adv. Mater., 2015, 27, 702–705 CrossRef CAS PubMed.
- M. Gao, J. Subbiah, P. B. Geraghty, M. Chen, B. Purushothaman, X. Chen, T. Qin, D. Vak, F. H. Scholes, S. E. Watkins, M. Skidmore, G. J. Wilson, A. B. Holmes, D. J. Jones and W. W. H. Wong, Chem. Mater., 2016, 28, 3481–3487 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- B. W. D'Andrade, S. Datta, S. R. Forrest, P. Djurovich, E. Polikarpov and M. E. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef.
- C. V. Hoven, X. D. Dang, R. C. Coffin, J. Peet, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2010, 22, 63–66 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Thermogravimetric analysis, Tauc plots, cyclic voltammetry, theoretical calculations, thin film absorption spectra, atomic force microscopy images, photovoltaic characterization, space-charge limited current measurements, and NMR spectra. See DOI: 10.1039/c9ra00229d |
| This journal is © The Royal Society of Chemistry 2019 |