DOI:
10.1039/C9RA00167K
(Paper)
RSC Adv., 2019,
9, 7086-7093
The mechanism and activity of oxygen reduction reaction on single atom doped graphene: a DFT method†
Received
8th January 2019
, Accepted 4th February 2019
First published on 1st March 2019
Abstract
Heteroatom doped graphene as a single-atom catalyst for oxygen reduction reaction (ORR) has received extensive attention in recent years. In this paper, the ORR activity of defective graphene anchoring single heteroatom (IIIA, IVA, VA, VIA and VIIA) was systematically investigated using a dispersion-corrected density functional theory method. For all of the 34 catalysts, 14 of which were further analyzed, and the Gibbs free energy of each elementary reaction was calculated. According to the scaling relationship between ΔGOOH* and ΔGOH*, we further analyzed the rate-determining step of the remaining 20 catalysts. The results show that when the ORR reaction proceeds in the path O2 → OOH → O → OH → H2O, the reaction energy barriers are lower than 0.8 eV for Te-SV, Sb-DV, Pb-SV, Pb-DV, As-SV, As-DV, B-SV, Sn-SV and N-SV. Our result provides a theoretical basis for further exploration of carbon-based single-atom catalysts for ORR.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) are one of the most promising clean energy technology candidates with simplicity, low working temperature, high power density, and quick start-up characteristics. These advantages promote their widespread commercial applications in the areas of transportation, stationary and portable power generation. However, to achieve a desirable fuel cell performance, the sluggish reaction kinetics of the oxygen reduction reaction (ORR) requires much higher Pt loading (∼0.4 mg cm−2), even on the best Pt-based catalyst. Due to the scarcity and high price of Pt metal, reducing the cathode Pt loading or even completely replacing it with abundant and cheap catalysts would be advantageous. In recent years, a large number of non-precious metal catalysts (NPMCs) have been explored to substitute Pt in ORR, such as metal oxide, metal carbide, metal nitride and carbon based catalysts.1–3 Among all of the NPMCs, graphene and carbon nanotube (CNT) based materials receive widespread attention due to their low price, fuel tolerance, good electrical and mechanical properties, high surface area, and noticeable electrochemical durability under actual working environments.4,5
As an effective method, doping heteroatoms into a graphene matrix could adjust their electronic properties and enhance the ORR performance. Tremendous efforts have been undertaken to this filed, both on the experimental and theoretical level.6–11 Dai's group6,12 is one of the pioneers in the study of nitrogen-doped carbon material for ORR. They synthesized nitrogen-doped CNT (N-CNT) and graphene (N-graphene) by pyrolyzing and chemical vapor deposition of methane in the presence of NH3. The resultant N-CNT and N-graphene were demonstrated to act as metal-free electrode with a much better electrocatalytic activity, long-term operation stability, and tolerance to crossover effect than platinum for oxygen reduction via a four-electron pathway in alkaline fuel cells. Sheng et al.13 prepared boron doped graphene (B-graphene) on a large scale using boron oxide as the boron source. Compared to pristine graphene, the B-graphene exhibits excellent electrocatalytic activity and long-term stability towards ORR in alkaline electrolytes, and good tolerance to CO poisons. Hu et al.14 further explored the ORR activity of boron and nitrogen co-doped carbon nanotubes (BN-CNTs). As for the two kinds of BN-CNTs, bonded or separated B and N, it was found that only the separated one can turn the inert CNTs into ORR electrocatalysts from both experimental and theoretical results. This progress demonstrates the crucial role of the doping microstructure on ORR performance, which is significant for further exploring the advanced C-based metal-free electrocatalysts. The enhanced ORR activity of carbon-based materials through B and N doping further stimulates research interest in other doping systems. Yang and his collaborators15 reported sulfur/selenium-doped graphene through a simple and economical approach. They confirmed the potential use of S-graphene in direct methanol and alkaline fuel cells. Wei et al.16 successfully prepared phosphorus-doped graphene nanosheets and found that the P atoms were substitutional incorporated into the carbon framework and were partially oxidized, which created new active sites for ORR. Gong et al.17 fabricated nitrogen- and phosphorus-doped biocarbon via a simple and low-cost biosynthesis method using yeast cells as a precursor. The as-prepared biocarbon exhibits an onset potential of −0.076 V (vs. Ag/AgCl) and a negative shift of only about 29 mV in the half-wave potential of the biocarbon compared to commercial Pt/C. In addition, other two-dimensional materials have also been widely used as catalysts in various reactions, such as water splitting,18 CO oxidation19 and ORR.20,21 However, the key ORR activity mechanisms of metal-free materials are still incomparable with those of Pt-based catalysts, which stimulate the on-going debate on active sites and reaction pathway for ORR. In view of the significant influence of raw materials, preparation methods, post-treatment processes on the structure and properties of carbon-based catalysts and the lack of in situ characterization techniques, more promising high activity ORR catalysts could be predicted by screening more heteroatom-doped carbon materials with density functional theory (DFT) method. Although Me–N–C has been a very promising ORR catalyst, it is not the focus of this article.22–24
The research and design of single-atom catalysts (SACs) requires a combination of the geometric and electronic properties of the single atoms themselves, as well as the geometric and electronic properties of the substrate. We have systematically studied the 3d metal atoms-B/C/N modified defective graphene25 and A(III–VI) group atoms-MoS2 with in-plane S defect.26 In this work, we will systematically study the geometric and electronic properties of IIIA, IVA, VA and VIA monoatomic doped single and double vacancies graphene (short as X-SV and X-DV) catalysts. The stability and ORR activities of X-SV/DV have been further evaluated. Density functional theory (DFT) calculations are used to quantify the binding energies and reaction energetics of surface species involved in ORR.
2. Method
2.1 The thermodynamics of the ORR
The ORR activities on active sites of heteroatom-doped defective graphene were studied in detail. In alkaline environment, the oxygen molecular could be reduction to OH−, and the detail reaction scheme is shown as below:| | |
O2 (g) + H2O (l) + e− + * → OOH* + OH−
| (1) |
| | |
O* + H2O (l) + e− → OH* + OH−
| (3) |
where * refers to a possible active site in X-SV/DV model. (l) and (g) refer to the liquid and gas phases, respectively. *O, *OH and *OOH are the adsorbed intermediates.
For each step, the reaction free energy ΔG is defined as the difference between free energies of the initial and final states and is given by the expression,27
| | |
ΔG1 = ΔGOOH* − 4.92 eV + ΔG (pH) + eU
| (5) |
| | |
ΔG2 = ΔGO* − ΔGOOH* + ΔG (pH) + eU
| (6) |
| | |
ΔG3 = ΔGOH* − ΔGO* + ΔG (pH) + eU
| (7) |
| | |
ΔG4 = −ΔGOH* + ΔG (pH) + eU
| (8) |
The Gibbs free energies of eqn (5)–(8) (ΔG) are related to the adsorption energies of the various intermediate species. The adsorption free energy changes of these intermediate species are determined using,28 ΔGads = ΔEDFTads + ΔZPE − TΔS, where ΔEDFTads can be calculated relative to H2O and H2. The absorption energies were calculated as follows, ΔEOH* = E(OH*) − E(*) − (EH2O − 1/2EH2), ΔEOOH* = E(OOH*) − E(*) − (2EH2O − 3/2EH2), ΔEO* = E(O*) − E(*) − (EH2O − EH2), EH2O and EH2 are the calculated DFT energies of H2O and H2 molecules in the gas phase using the approaches outlined by Nørskov et al.28,29 ΔZPE and TΔS are the zero point energy difference and the entropy change between the absorbed state and the free state, i.e., the gas phase, respectively, and T is the temperature (298.15 K in this work). e is the elementary charge and U is the potential difference between the electrode and the normal hydrogen electrode (NHE). The free energy change of H+ is derived according to ΔGpH = kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(10) × pH, (kB is Boltzmann's constant, and pH = 13 for alkaline medium). Because the high-spin ground state of an oxygen molecule is difficult to describe in DFT calculations, the free energy of O2 (g) is derived as GO2 (g) = 2GH2O (l) − 2GH2 (g) + 4.92 eV.
ln(10) × pH, (kB is Boltzmann's constant, and pH = 13 for alkaline medium). Because the high-spin ground state of an oxygen molecule is difficult to describe in DFT calculations, the free energy of O2 (g) is derived as GO2 (g) = 2GH2O (l) − 2GH2 (g) + 4.92 eV.
2.2 Computational details
The spin-polarized DFT calculations were performed by the DMol3 code embedded in Materials Studio30 with long-range dispersion correction via Grimme's scheme.31 The generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE)32 functional is employed to describe exchange and correlation effects. The all electrons core treat method is implemented. The double numerical atomic orbital augmented by a polarization function is chosen as the basis set.33 During geometrical optimization, the basis set cut-off was chosen to be 5.2 Å. The convergence tolerances for the geometry optimization were set to 10−5 Ha for the energy, 0.002 Ha Å−1 for the force, and 0.005 Å for the displacement. The electronic SCF tolerance was set to 10−6 Ha. A Fermi smearing parameter of 0.005 Ha was used in the calculations. The reciprocal space was sampled with a 6 × 6 × 1 k-points grid generated automatically using the Monkhorst–Pack method34 for the relaxation calculations and a (18 × 18 × 1) k-points grid was used for electronic structure computations. The conductor-like screening model (COSMO)35 is used to simulate the aqueous environment, where the dielectric constant is set as 78.54 (water). First-principles molecular dynamics (MD) was performed with NVE method. A time step of 1 fs was used and the temperature was set to 353.15 K. We have conducted a convergence test for important parameters (orbital cut-off quality and k-point set) and slab size, and the results indicate that present parameters and p(4 × 4) slab are fine enough. More detail information is shown in Table S1.† Our DFT calculated lattice parameter for graphite is 2.47 Å which is consistent with the experimental value and other computational results.36 More than 15 Å thick vacuum is added to avoid the artificial interactions between graphene and its images. In all of the structure optimization calculations, all the atoms are fully relaxed.
3. Results and discussions
3.1 The geometric and electronic properties
The geometry-optimized graphene sheet has an in-plane C–C distance of 1.42 Å, which is consistent with the previous results.25,36 The single and double vacancies of graphene were constructed through removing one and two neighbouring atoms, respectively. When a single hetero-atom is introduced into the defect position in graphene, the C atoms with dangling bonds at the defect will capture and anchor the hetero-atom. The geometric and electronic properties of X-SV/DV (X = B, C, N, O; Al, Si, P, S; Ga, Ge, As, Se; In, Sn, Sb, Te; Tl, Pb, Bi) catalysts have been analysed in details.
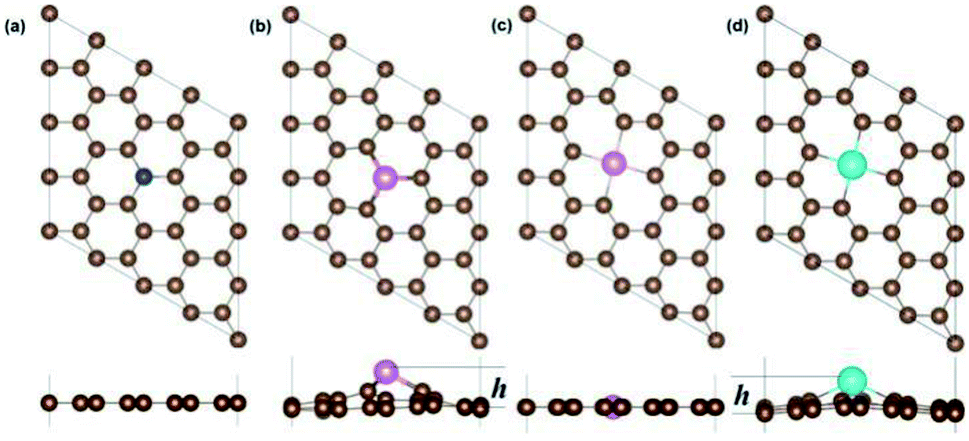
3.1.1 Geometric properties. Depending on the size of the hetero-atoms and defect, the X-SV have two different structures, one being a planar structure, as shown in Fig. 1(a), including B, C, N, O. The atomic radius of B (0.88), N (0.70) and O (0.73) are close to the C (0.77), so the planar structure can be maintained after replacing the C atom position. According to Table S2,† the impurity atoms (B, N, O) are kept in the same plane as the graphene, and the height difference is zero. Second, the convex structure, as shown in Fig. 1(b), includes Al, Si, P, S; Ga, Ge, As, Se; In, Sn, Sb, Te; Tl, Pb and Bi. For a dopant atom with a large atomic radius, such as Al (1.43), Si (1.17), Ga (1.35), and In (1.67) etc., the single atom vacancy defect is not sufficient to accommodate such a large impurity atom, so the heteroatom will protrude from the graphene plane and form a convex structure. The difference in height between the impurity atoms and the plane of the graphene is related to the radius of the impurity atoms. For most systems, the larger the atomic radius, the longer the X–C bond length as well as the greater the height of the X atoms, as shown in Table S2.† For X-DV systems, the situation is similar. It was found that some X-DV systems have a planar structure (X = Al, Si, P, S, Ga, Ge, As) and other X-DVs have a non-planar structure. The reason is similar to the X-SV system. See Table S3† for specific data.
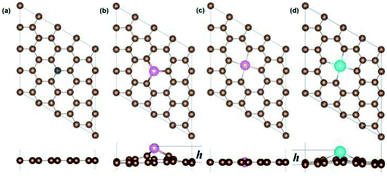 |
| | Fig. 1 The optimized geometry structure of N-SV (a), Al-SV(b), Al-DV (c) and In-DV (d). | |
3.1.2 Electronic properties. In addition, we studied the density of states and Mulliken charge of the X-SV/DV system. It is found that, except for the N&O doping system, the X atoms are positively charged, and the adjacent C atoms coordinating with X are negatively charged, as shown in Tables S2 and S3.† The greater the difference in electronegativity between the X atom and the C atom, the more pronounced the charge transfer between the two. The density of states analysis of the X atom reveals that the average energy (the sp electrons band center) changes regularly. For the X atom of the same period, the center of the sp band satisfies B > C > N > O, as shown in Table S2.† The trend of X-DV is similar to X-SV.
3.2 The stability
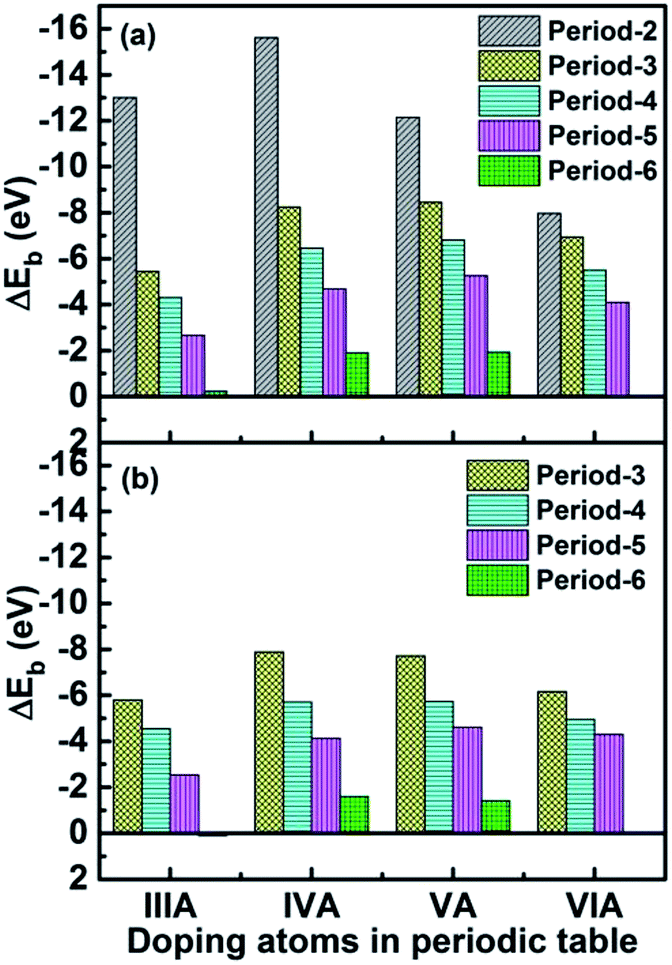
Stability is an important parameter for judging catalyst performance. To evaluate the stability of the catalyst, the bonding energy between the X atom and the defective graphene in the X-SV/DV catalyst was defined as following:| | |
ΔEb = EX-SV/DV − ESV/DV − EX
| (9) |
where EX-SV/DV, ESV/DV and EX are the total energies of the single atom doped graphene systems, defective graphene and an isolated X atoms, respectively. It was found that the X-SV bonding energy for most systems is greater than the corresponding X-DV. The stability of the catalyst is closely related to the size and electronic structure of the dopant atoms. We found that for small atomic radius doping SV systems, the stability decrease in the order: C(p2) > B(p1) > N(p3) > O (p4), with regard to large atomic radius doping SV system, the change trends are somewhat different (as shown in Table S4†): P(p3) > Si(p2) > S(p4) > Al(p1) [As(p3) > Ge(p2) > Se(p4) > Ga(p1)], [Sb(p3) > Sn(p2) > Te(p4) > In(p1)], [Bi(p3) > Pb(p2) > Tl(p1)]. For large-sized doped DV systems: Si(p2) > P(p3) > S(p4) > Al(p1) [As(p3) > Ge(p2) > Se(p4) > Ga(p1)], [Sb(p3) >Te(p4) > Sn(p2) > In(p1)], [Tl(p1) > Bi(p3) > Pb(p2)], the regularity of the system is poor, and the atomic radius is also used. The electronic structure is explained: the density of states (DOS) calculation shows that there is a clear orbital overlap between the X atom and the C atom, and there is a covalent bond between the two. The calculation of differential charge density and Mulliken charge shows that there is a significant charge transfer between X and C, and there is a significant ionic bond between the two. Therefore, the X–C forms a hybrid bond. There is no significant spin change in the material, so it can be considered as a zero spin material. After introducing the aqueous environment effect, the stability of the catalyst is almost unchanged, as shown in Table S4.† Furthermore, first-principles molecular dynamics in a period of 1000 fs (1 ps) calculations at the temperature of 353.15 K were conducted for Tl-SV (ΔEb = −0.23 eV), In-SV (ΔEb = −2.66 eV) and Ga-SV (ΔEb = −4.35 eV). It was found that only Ga and C atoms in Ga-SV can maintain bonding with slight distortion, and the structures of other systems were destroyed, as shown in Fig. S1.† This indicates that only catalysts with a bonding energy greater than 4 eV could remain stable under PEMFC operating conditions.
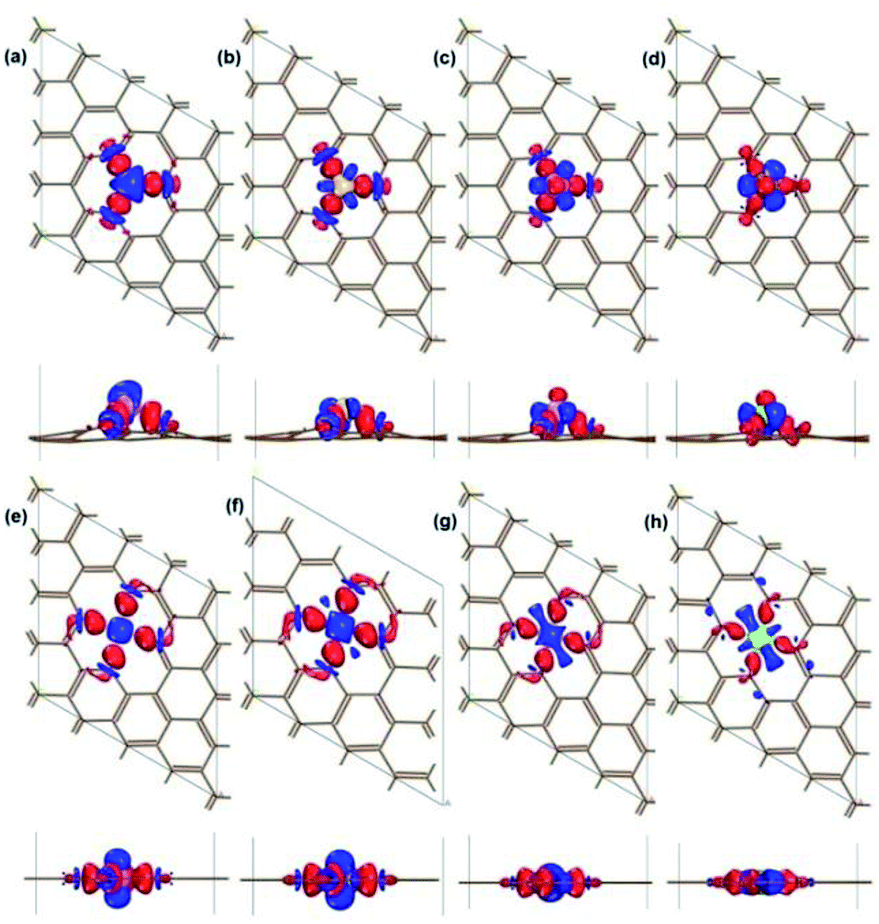
The binding properties of X atom on vacancy are analysed by calculating the charge density difference in real space. The charge density difference of X–V is defined as following equation and shown in Fig. 3:
where the charge density is calculated in the same supercell for all quantities,
ρX–V is the total charge density of the X-vacancy system, and
ρV and
ρX are the charge densities of noninteracting pristine vacancy and X atom, respectively. The charge density difference quantifies the redistribution of electron charge due to the interaction between adatom and vacancy. The accumulated charge density around the X–C bonds indicates that X is strongly bound on the site vacancy of graphene, in accordance with the high exothermic adsorption energy in
Fig. 2 and Table S4,
† implying that the covalent bonding is formed between X and C atoms. In addition,
Fig. 3 shows a charge transfer from X to C atom, which is consistent with the electronegativity difference between C and X atoms. The Mulliken charge of the X-vacancy systems is calculated in Table S3,
† which also indicates that a great amount of electrons are transferred from X to substrate. The amount of charge transformed from X to C decrease in the order Al > Si > P > S, as observed in
Fig. 3 and Table S2
† for both X-SV and X-DV. Also, the X-DV systems are more significant due to more X–C bonds. This phenomenon indicates the obvious ionic bond characteristic.
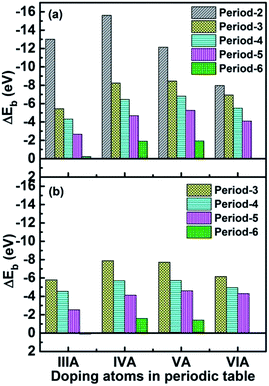 |
| | Fig. 2 The binding energies between X atoms and defective graphene for X-SV (a) and X-DV (b). | |
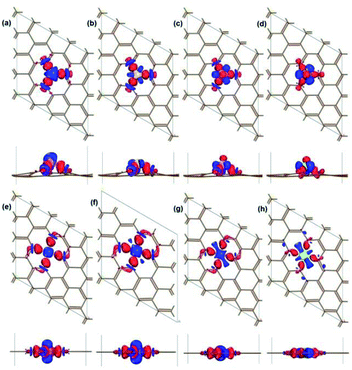 |
| | Fig. 3 Top view and side view of the charge density difference for Al/Si/P/S atom adsorbed on vacancy complexes: Al-SV (a), Si-SV (b), P-SV (c), S-DV (d), Al-DV (e), Si-DV (f), P-DV (g); S-DV (h). The isosurface value is 0.06 e Å−3. The red colour represents electron accumulation, while the blue colour represents electron depletion. The intensity of colour depends on the amount of electron change: the darkest red marks the most accumulation; the darkest blue marks the most depletion. Brown, C atom; purple, Al atom; yellow, Si atom; pink, P atom; green, S atom. | |
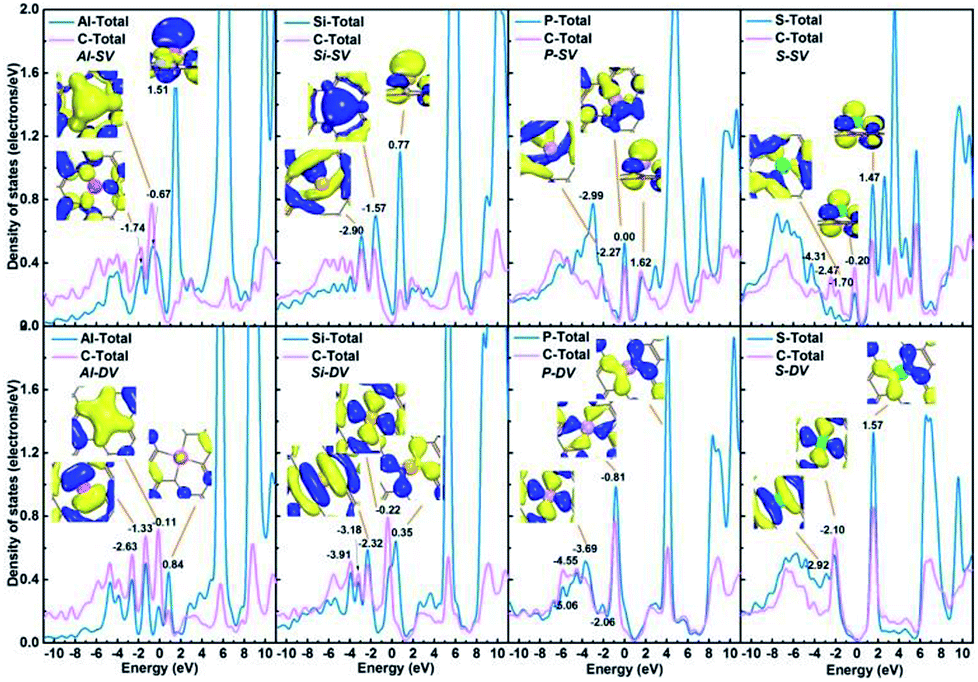
What's more, there exist significant electronic density overlap between X and substrate, as shown in Fig. 4. This phenomenon indicates the obvious covalent bond feature. The corresponding orbits at specific energy levels have also been given in the inset pictures in Fig. 4.
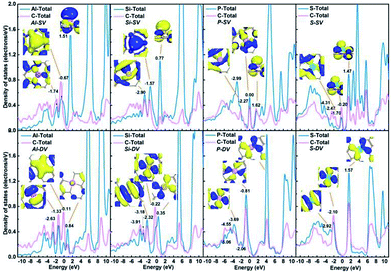 |
| | Fig. 4 PDOS of Al-SV, Si-SV, P-SV, S-SV, Al-DV, Si-DV, P-DV; S-DV. The corresponding orbits at specific energy levels are shown in the inset pictures. | |
3.3 The ORR activities of X-SV/X-DV
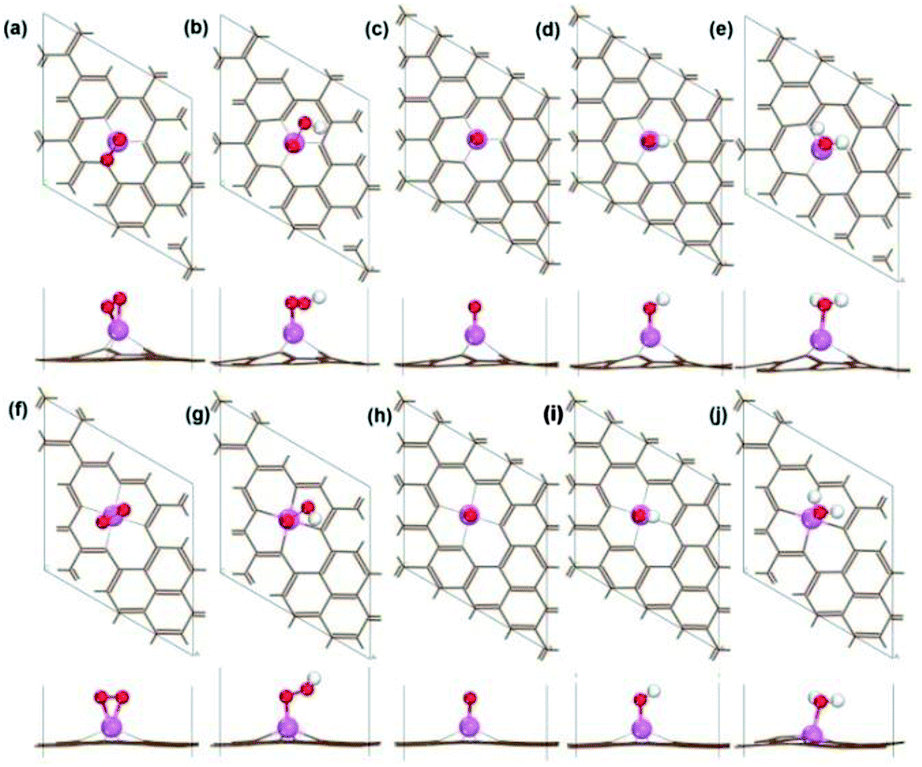
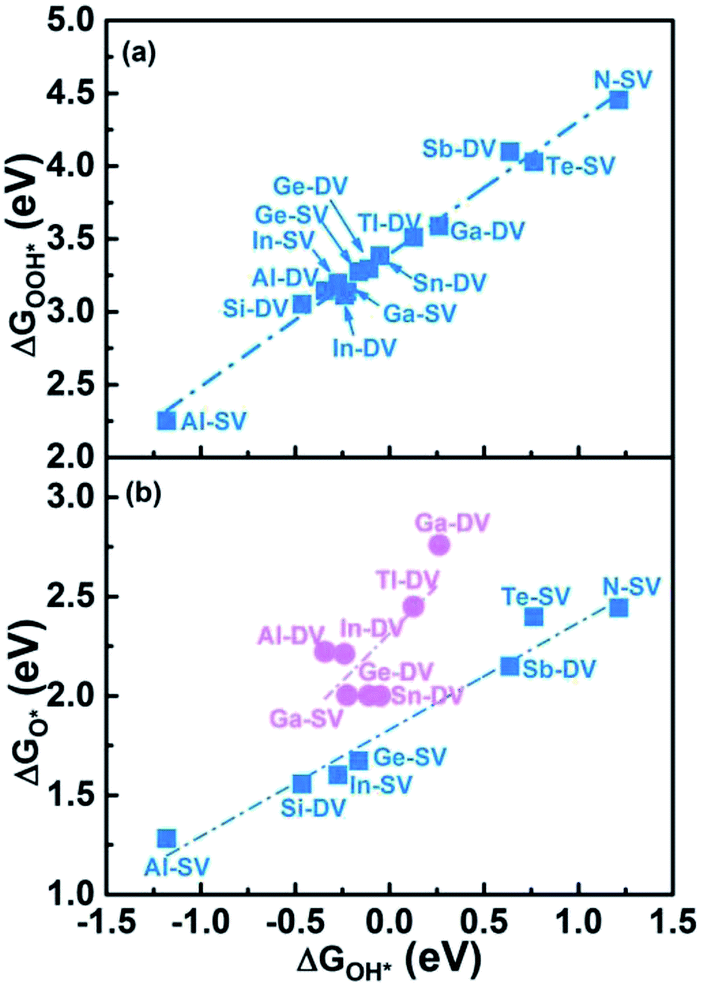
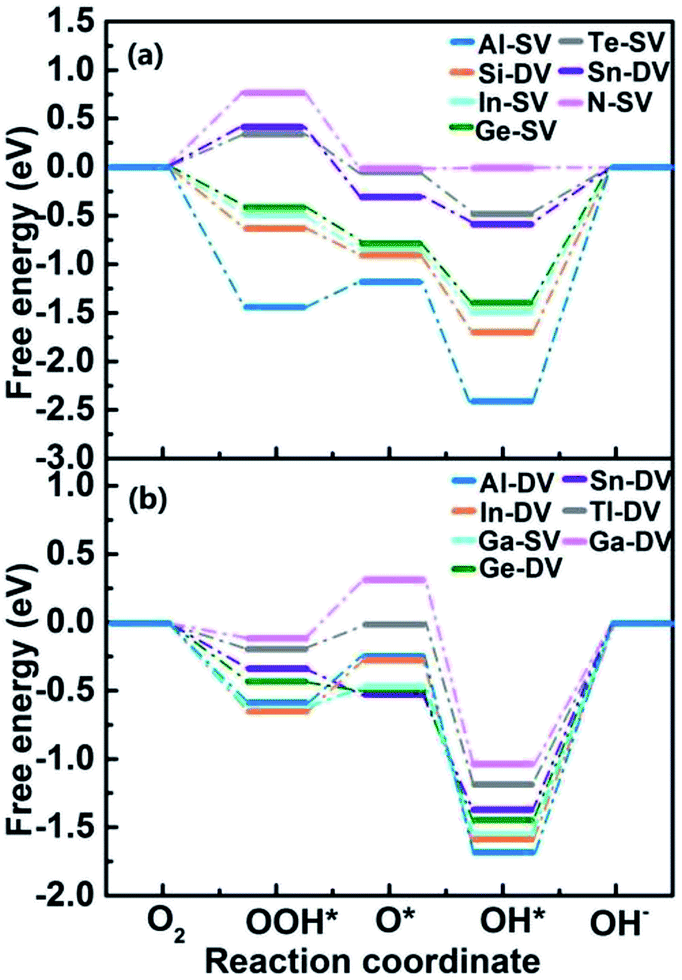
We selected 14 catalysts (according to the adsorption energy of O2 in the range of (0.3 eV, 1.0 eV)) and analyzed the adsorption behavior of reactants (O2), intermediates (O*, OH* and OOH*) and products (H2O) involved in the ORR reaction on the impurity atoms site. The optimized structures of oxygenated species on Al-SV and Al-DV have been shown in Fig. 5. The results show that the adsorption structure of oxygen-containing species is different for different substrates of the same impurity atom or different impurity atoms of the same substrate. Fig. 6(a) shows the adsorption free energies of OOH* and OH* for parts of X-SV and X-DV. The free energies of OOH* are linearly related to that of OH* by y = 0.91x + 3.40. Fig. 6(b) shows the adsorption free energies of OH* and O* for parts of X-SV and X-DV structures. There is also a scaling relation with a slope of 1.04 between OH* and O* except some sites where oxygen atom is adsorbed in Al-DV, Ga-SV and so on. The free energy of reaction (1)–(4) can be calculated using eqn (5)–(8). According to the relationship between ΔGOH* and ΔGO*, the catalysts are divided into two parts, and the detailed reaction free energy for each elemental steps were shown in Fig. 7(a) (linear relationship for Al-SV et al.) and Fig. 7(b) (nonlinear association for Al-DV et al.). For Al-SV et al. catalysts, it is found that the ORR either limited by step 1 (O2 → OOH*) or step 4 (OH* → OH−), due to the weak or strong binding energy between oxygenated species and X-graphene, respectively. As for step 1, the energy barriers increase in the order Al-SV < Si-DV < In-SV < Ge-SV < Te-SV < Sb-DV < N-SV. An opposite trend is observed in step 4, that is Al-SV > Si-DV > In-SV > Ge-SV > Te-SV > Sn-DV > N-SV. For the overall reaction, the rate-determining step (RDS) of Te-SV (0.76 eV), Sb-DV (0.41 eV) and N-SV (0.34 eV) are O2 → OOH*, and the RDS of Al-SV (2.41 eV), Si-DV (1.69 eV), In-SV (1.50 eV) and Ge-SV (1.39 eV) are OH* → OH−. For Al-DV et al. catalysts, the RDS of all seven catalysts are OH* → OH−. The energy barriers increase in the order Ga-DV (0.97 eV) < Tl-DV (1.10 eV) < Sn-DV (1.28 eV) < Ge-DV (1.34 eV) < Ga-SV (1.46 eV) < In-DV (1.47 eV) < Al-DV (1.57 eV).
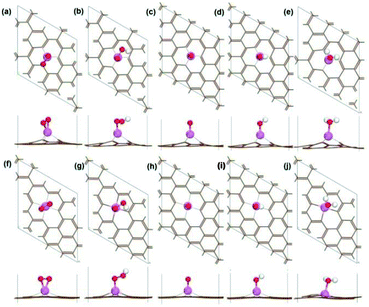 |
| | Fig. 5 The optimized geometry structure of O2 adsorbed Al-SV (a), OOH adsorbed Al-SV (b), O adsorbed Al-SV (c), OH adsorbed Al-SV (d), H2O adsorbed Al-SV (e), O2 adsorbed Al-DV (f), OOH adsorbed Al-DV (g), O adsorbed Al-DV (h), OH adsorbed Al-DV (i), H2O adsorbed Al-DV (j). | |
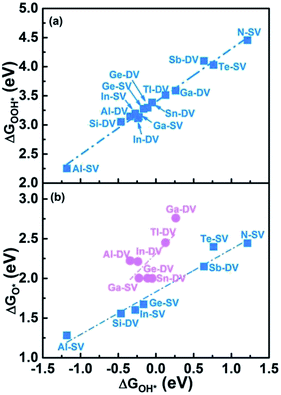 |
| | Fig. 6 The scaling relationship between ΔGOH* and ΔGOOH* (a) as well as ΔGOH* and ΔGO* (b). | |
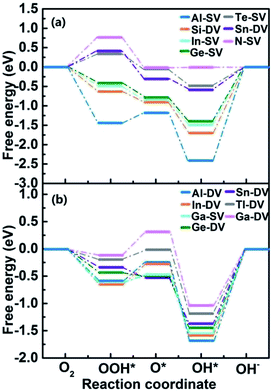 |
| | Fig. 7 The schematic Gibbs free energy diagrams of ORR for X-SV/DV with (a) or without (b) liner relationship. | |
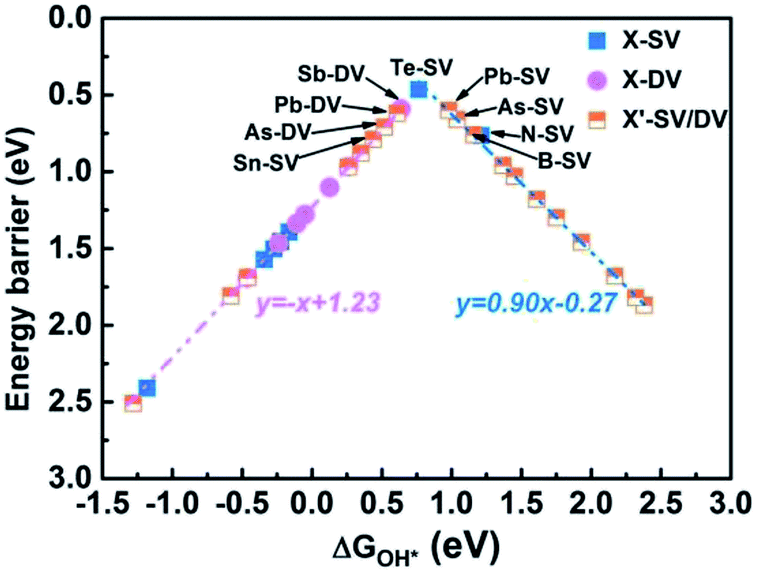
We further analyzed the RDS of the remaining 20 catalysts. First of all, we calculated the ΔGOH* of all 20 catalysts. According to the scaling relationship between ΔGOOH* and ΔGOH* (ΔGOOH* = 0.91ΔGO* + 3.40), the ΔGOOH* were then obtained (as shown in Table S7†). From the above, we can conclude that the RDS is either step 1 (determined by ΔG1 = ΔGOOH* − 4.92 eV + ΔG(pH) + eU) or step 4 (determined by ΔG4 = −ΔGOH* + ΔG(pH) + eU). When pH = 13, UNHE = 0.44 V, we could obtained ΔG1 and ΔG4 based on ΔGOH*. We compare the values of ΔG1 and ΔG4 , and the maximum value was used as the energy barrier of RDS. Then, The relationship between ΔGOH* and max energy barrier was summarized and shown in Fig. 8, Tables S8 and S9.† We found a “volcanic curve” relationship between ΔGOH* and energy barrier. When the binding energy was too strong between OH* and SAC (ΔGOH* < 0.76 eV), the oxygen reduction reaction was limited by OH* → OH−, and the energy barrier decrease with ΔGOH*. As shown in Fig. 8, the identified catalyst energy barriers are lower than 0.8 eV with pH = 13 and UNHE = 0.44. For the left side ones, the energy barrier increase in the order: Te-SV (0.47 eV) < Sb-DV (0.59 eV) < Pb-DV (0.62 eV) < As-DV (0.71 eV) < Sn-SV (0.79 eV), consistent with their increasing ΔGOH* values. Further analysis of the difference between ΔG1 and ΔG1(ΔΔG = ΔG4 − ΔG1), the same trend was found: Te-SV (0.13 eV) < Sb-DV (0.18 eV) < Pb-DV (0.36 eV) < As-DV (0.53 eV) < Sn-SV (0.68 eV). This indicates that, the closer to the vertex of “volcanic curve”, the closer the values between ΔG1 and ΔG4, and the smaller the energy barrier difference between the two elementary reactions. However, the farther the catalysts from the vertex of “volcanic curve”, the bigger the gap between RDS and other steps. When the binding energy was too weak between OH* and SAC (ΔGOH* > 0.76 eV), the oxygen reduction reaction was limited by O2 → OOH*, and the energy barrier decrease with ΔGOH*. The trend in right side is opposite to the left one. An ideal catalyst should be able to facilitate oxygen reduction just above the equilibrium potential, but requires all the four electrons transfer steps to have reaction free energies of the same magnitude at zero potential (i.e., 4.92 eV/4 = 1.23 eV). This is equivalent to all the reaction free energies being zero at the equilibrium potential, 1.23 V for acid medium and 0.44 V for alkaline medium with pH = 13. The reaction free energy ΔG1, ΔG2, ΔG3 and ΔG4 have the following relationship regardless of pH and potential correction:
| | |
ΔG1 + ΔG2 = ΔGO* − 4.92 eV
| (11) |
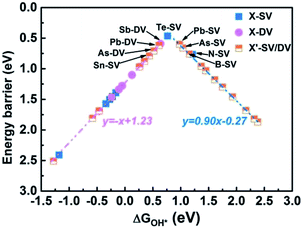 |
| | Fig. 8 The “volcanic curve” relationship between ΔGOH* and energy barrier for all 34 catalysts. | |
Eqn (11) and (12) are competing with each other due to the opposite sign. Therefore, when ΔG1, ΔG2, ΔG3 and ΔG4 are equal, the RDS obtains the minimum value. As we have mentioned, when ΔΔG is the smallest, the lowest energy barrier obtained.
We also calculated the adsorption free energy and reaction free energy for all catalyst with aqueous environment. Although there are some differences in the absolute value of free energy, the general trend is almost same. Therefore, we put all related results in ESI (Fig. S2–S4†) for comparison and will not discuss here.
We also compared our results with other experimental and DFT results. Zhang et al.37 conducted research on four different S-doped graphene quantum dot structures using the DFT method. Their calculations indicates that carbon atoms located at the zigzag edges or close to the SO2 doping structure possess high positive charge density or spin density, and the reaction can be carried out simultaneously through the four-electron path and the two-electron path on these carbon atoms. According to our calculations, however, both S-SV (0.97 eV) and S-DV (1.49 eV) have higher energy barriers at 0.9 V, and the rate-determining step is the same, both of which are O2(g) hydrogenation to form OOH*. We also compared the ORR activity of N-SV catalyst with the results of Li et al.38 It was found that when consider the same reaction path, the rate-limiting step was same (O2 (g) + (H+ + e−) + * → OOH*). However, our obtained overpotential values (0.78 V) was much higher than the ref. 38. The differences in doping structure lead to differences in activity. The comparison of phosphorus doped graphene between our work and the others have also been done.39,40 The results obtained by Kaukonen et al.39 indicate that single P atoms embedded into divacancies in graphene can be good candidates for ORR catalysts, which is consistent with our calculation results (0.55 eV at 0.9 V for P-DV). The DFT results of Zhang et al.40 (P-SV) show that the reduction step from the second OH to H2O is the rate-determining step, which has a larger barrier of 0.88 eV, which is also consistent with our results, the same rate-limiting step and high energy barrier (1.36 eV) at 0.9 V. The difference in specific values is duo to the different method of evaluating barriers. We even compared with platinum-based ORR catalysts and found that the most active Te-SV has a reaction barrier comparable to Pt(211).41,42 In this article, we did not consider all possible active sites in detail, such as not considering carbon atoms adjacent to heteroatom. Therefore, the conclusions we give in this paper are a preliminary judgment. Next, we will specifically study the catalyst with high activity in a more detailed manner. By further analysing the various reaction pathways and all possible active sites, our ultimate goal is to finally confirm the high activity characteristics of the catalysts and achieve laboratory preparation of highly active catalysts.
4. Conclusions
In summary, we have systematically investigated the geometric property, electronic property, stability and ORR activity of 34 SACs based on defective graphene (short as X-SV or X-DV, X= B, N, O; Al, Si, P, S; Ga, Ge, As, Se; In, Sn, Sb, Te; Tl, Pb, Bi) using dispersion-corrected density functional theory method. The oxygen reduction activity of 14 catalysts was analysed in details, and the Gibbs free energy of each element reaction was calculated. According to the scaling relationship between ΔGOOH* and ΔGOH*, the rate-determining steps of the remaining 20 catalysts were further analyzed. The results show that when the ORR reaction proceeds in the path O2 → OOH* → O* → OH* → OH−, the reaction energy barrier of Te-SV, Sb-DV, Pb-SV (unstable), Pb-DV (unstable), As-SV, As-DV, B-SV, N-SV and Sn-SV is lower than 0.8 eV. Our result provides a theoretical basis for further exploration of carbon-based SACs or co-doped catalysts for ORR.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Supported by the “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA21090300 and National Natural Science Foundation of China, Grant No. 91834301. The DFT calculations utilized resources at the High Performance Computing Center, Jilin University.
References
- Z. W. Chen, D. Higgins, A. P. Yu, L. Zhang and J. J. Zhang, A review on non-precious metal electrocatalysts for PEM fuel cells, Energy Environ. Sci., 2011, 4(9), 3167–3192 RSC.
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Recent Advances in Electrocatalysts for Oxygen Reduction Reaction, Chem. Rev., 2016, 116(6), 3594–3657 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. D. Wei, Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction, Chem. Soc. Rev., 2015, 44(8), 2168–2201 RSC.
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. J. Dai, An oxygen reduction electrocatalyst based on carbon nanotube-graphene complexes, Nat. Nanotechnol., 2012, 7(6), 394–400 CrossRef CAS PubMed.
- H. B. Wang, T. Maiyalagan and X. Wang, Review on Recent Progress in Nitrogen-Doped Graphene: Synthesis, Characterization, and Its Potential Applications, ACS Catal., 2012, 2(5), 781–794 CrossRef CAS.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, Nitrogen-Doped Graphene as Efficient Metal-Free Electrocatalyst for Oxygen Reduction in Fuel Cells, ACS Nano, 2010, 4(3), 1321–1326 CrossRef CAS PubMed.
- S. Y. Wang, L. P. Zhang, Z. H. Xia, A. Roy, D. W. Chang, J. B. Baek and L. M. Dai, BCN Graphene as Efficient Metal-Free Electrocatalyst for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2012, 51(17), 4209–4212 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Two-Step Boron and Nitrogen Doping in Graphene for Enhanced Synergistic Catalysis, Angew. Chem., Int. Ed., 2013, 52(11), 3110–3116 CrossRef CAS PubMed.
- C. H. Choi, S. H. Park and S. I. Woo, Binary and Ternary Doping of Nitrogen, Boron, and Phosphorus into Carbon for Enhancing Electrochemical Oxygen Reduction Activity, ACS Nano, 2012, 6(8), 7084–7091 CrossRef CAS PubMed.
- S. B. Yang, L. J. Zhi, K. Tang, X. L. Feng, J. Maier and K. Mullen, Efficient Synthesis of Heteroatom (N or S)-Doped Graphene Based on Ultrathin Graphene Oxide-Porous Silica Sheets for Oxygen Reduction Reactions, Adv. Funct. Mater., 2012, 22(17), 3634–3640 CrossRef CAS.
- J. T. Jin, F. P. Pan, L. H. Jiang, X. G. Fu, A. M. Liang, Z. Y. Wei, J. Y. Zhang and G. Q. Sun, Catalyst-Free Synthesis of Crumpled Boron and Nitrogen Co-Doped Graphite Layers with Tunable Bond Structure for Oxygen Reduction Reaction, ACS Nano, 2014, 8(4), 3313–3321 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Nitrogen-Doped Carbon Nanotube Arrays with High Electrocatalytic Activity for Oxygen Reduction, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- Z. H. Sheng, H. L. Gao, W. J. Bao, F. B. Wang and X. H. Xia, Synthesis of boron doped graphene for oxygen reduction reaction in fuel cells, J. Mater. Chem, 2012, 22, 390–395 RSC.
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, Can Boron and Nitrogen Co-doping Improve Oxygen Reduction Reaction Activity of Carbon Nanotubes?, J. Am. Chem. Soc., 2013, 135(4), 1201–1204 CrossRef CAS PubMed.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. a. Chen and S. Huang, Sulfur-Doped Graphene as an Efficient Metal-free Cathode Catalyst for Oxygen Reduction, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed.
- R. Li, Z. D. Wei, X. L. Gou and W. Xu, Phosphorus-doped graphene nanosheets as efficient metal-free oxygen reduction electrocatalysts, RSC Adv., 2013, 3(25), 9978–9984 RSC.
- X. Gong, S. S. Liu, C. Y. Ouyang, P. Strasser and R. Z. Yang, Nitrogen- and Phosphorus-Doped Biocarbon with Enhanced Electrocatalytic Activity for Oxygen Reduction, ACS Catal., 2015, 5(2), 920–927 CrossRef CAS.
- C. Chowdhury, S. Karmakar and A. Datta, Monolayer Group IV-VI Monochalcogenides: Low-Dimensional Materials for Photocatalytic Water Splitting, J. Phys. Chem. C, 2017, 121(14), 7615–7624 CrossRef.
- S. Karmakar, C. Chowdhury and A. Datta, Noble-Metal-Supported GeS Monolayer as Promising Single-Atom Catalyst for CO Oxidation, J. Phys. Chem. C, 2018, 122(26), 14488–14498 CrossRef CAS.
- C. Chowdhury and A. Datta, Silicon-Doped Nitrogen-Coordinated Graphene as Electrocatalyst for Oxygen Reduction Reaction, J. Phys. Chem. C, 2018, 122(48), 27233–27240 CrossRef CAS.
- C. Chowdhury and A. Datta, Doped boron nitride surfaces: potential metal free bifunctional catalysts for non-aqueous Li-O-2 batteries, Phys. Chem. Chem. Phys., 2018, 20(24), 16485–16492 RSC.
- N. Larouche, R. Chenitz, M. Lefevre, E. Proietti and J. P. Dodelet, Activity and stability in proton exchange membrane fuel cells of iron-based cathode catalysts synthesized with addition of carbon fibers, Electrochim. Acta, 2014, 115, 170–182 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, High-Performance Electrocatalysts for Oxygen Reduction Derived from Polyaniline, Iron, and Cobalt, Science, 2011, 332(6028), 443–447 CrossRef CAS PubMed.
- L. Yu, X. L. Pan, X. M. Cao, P. Hu and X. H. Bao, Oxygen reduction reaction mechanism on nitrogen-doped graphene: a density functional theory study, J. Catal., 2011, 282(1), 183–190 CrossRef CAS.
- X. M. Zhang, S. S. Yu, H. Chen and W. T. Zheng, TM atoms on B/N doped defective graphene as a catalyst for oxygen reduction reaction: a theoretical study, RSC Adv., 2015, 5(101), 82804–82812 RSC.
- X. Zhang, S. Shi, T. Gu, L. Li and S. Yu, The catalytic activity and mechanism of oxygen reduction reaction on P-doped MoS2, Phys. Chem. Chem. Phys., 2018, 20(27), 18184–18191 RSC.
- N. Yang, L. Li, J. Li, W. Ding and Z. D. Wei, Modulating the oxygen reduction activity of heteroatom-doped carbon catalysts via the triple effect: charge, spin density and ligand effect, Chem. Sci., 2018, 9(26), 5795–5804 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin and T. Bligaard, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886 CrossRef.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martinez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Norskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, Chemcatchem, 2011, 3(7), 1159–1165 CrossRef CAS.
- B. Delley, From molecules to solids with the DMol(3) approach, J. Chem. Phys., 2000, 113(18), 7756–7764 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Delley, An all-electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B, 1976, 13(12), 5188–5192 CrossRef.
- A. Klamt and G. Schuurmann, Cosmo - a New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient, J. Chem. Soc., Perkin Trans. 2, 1993,(5), 799–805 RSC.
- D. E. Jiang, B. G. Sumpter and S. Dai, Unique chemical reactivity of a graphene nanoribbon's zigzag edge, J. Chem. Phys., 2007, 126(13), 134701 CrossRef PubMed.
- L. Zhang, J. Niu, M. Li and Z. Xia, Catalytic Mechanisms of Sulfur-Doped Graphene as Efficient Oxygen Reduction Reaction Catalysts for Fuel Cells, J. Phys. Chem. C, 2014, 118(7), 3545–3553 CrossRef CAS.
- M. T. Li, L. P. Zhang, Q. Xu, J. B. Niu and Z. H. Xia, N-doped graphene as catalysts for oxygen reduction and oxygen evolution reactions: Theoretical considerations, J. Catal., 2014, 314, 66–72 CrossRef CAS.
- M. Kaukonen, A. V. Krasheninnikov, E. Kauppinen and R. M. Nieminen, Doped Graphene as a Material for Oxygen Reduction Reaction in Hydrogen Fuel Cells: A Computational Study, ACS Catal., 2013, 3(2), 159–165 CrossRef CAS.
- X. Zhang, Z. Lu, Z. Fu, Y. Tang, D. Ma and Z. Yang, The mechanisms of oxygen reduction reaction on phosphorus doped graphene: a first-principles study, J. Power Sources, 2015, 276, 222–229 CrossRef CAS.
- H. Wang and W. An, Promoting the oxygen reduction reaction with gold at step/edge sites of Ni@AuPt core–shell nanoparticles, Catal. Sci. Technol., 2017, 7(3), 596–606 RSC.
- H. Wang, W. An, X. Y. Liu and C. H. Turner, Oxygen reduction reaction on Pt(111), Pt(221), and Ni/Au1Pt3(221) surfaces: probing scaling relationships of reaction energetics and interfacial composition, Chem. Eng. Sci., 2018, 184, 239–250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00167k |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Suli Wang*a and
Gongquan Sun
b,
Suli Wang*a and
Gongquan Sun