
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecondary metabolites from the endolichenic fungus Ophiosphaerella korrae†
Yue-Lan Lia,
Rong-Xiu Zhub,
Gang Li c,
Ning-Ning Wanga,
Chun-Yu Liua,
Zun-Tian Zhaod and
Hong-Xiang Lou*a
c,
Ning-Ning Wanga,
Chun-Yu Liua,
Zun-Tian Zhaod and
Hong-Xiang Lou*a
aDepartment of Natural Product Chemistry, Key Lab of Chemical Biology (MOE), School of Pharmaceutical Sciences, Shandong University, Jinan 250012, People's Republic of China. E-mail: louhongxiang@sdu.edu.cn; Fax: +86-531-88382019; Tel: +86-531-88382012
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, People's Republic of China
cDepartment of Natural Medicinal Chemistry and Pharmacognosy, School of Pharmacy, Qingdao University, Qingdao 266021, People's Republic of China
dCollege of Life Sciences, Shandong Normal University, Jinan 250014, People's Republic of China
First published on 31st January 2019
Abstract
The isolation of the cytotoxic fractions from the endolichenic fungus Ophiosphaerella korrae yielded six new metabolites, including five polyketides (ophiofuranones A (1) and B (2), with unusual furopyran-3,4-dione-fused heterocyclic skeletons, ophiochromanone (3), ophiolactone (4), and ophioisocoumarin (5)), one sesquiterpenoid ophiokorrin (10), and nine known compounds. Their structures were established on the basis of the analysis of HRESIMS and NMR spectroscopic data. ECD calculations, GIAO NMR shift calculations and single-crystal X-ray diffraction were employed for the stereo-structure determination. A plausible biogenetic pathway for the ophiofuranones A (1) and B (2) was proposed. The cytotoxic assay suggested that the five known perylenequinones mainly contributed to the cytoxicity of the extract. Further phytotoxic studies indicated that ophiokorrin inhibited root elongation in the germination of Arabidopsis thaliana with an IC50 value of 18.06 μg mL−1.
Introduction
Endolichenic fungi are diverse groups of predominantly filamentous fungi that reside asymptomatically in the interior of lichen thalli.1 Since the first report decades ago,2 more and more metabolites biosynthesized by endolichenic fungi have been found with promising pharmacological properties and structural diversity.1,3 During our continuing investigations of novel and bioactive metabolites from endolichenic fungi,4–7 Ophiosphaerella korrae was discovered from the lichen Physciaceae physcia collected from Xinjiang Province, China. O. korrae is well-known as one of the three phythogens of spring dead spot.8Bioscreening established that the EtOAc crude extracts of O. korrae not only exhibited acetylcholinesterase (AChE) inhibitory effects but also had significant inhibition of cell viability against the A549 cell line. Further fractionation of the extract by silica gel column chromatography performed to find relevant biologically active components obtained twenty fractions (A–T). A previous chemical investigation on Fr. D and Fr. E with strong acetylcholinesterase inhibitory effects identified several rare secondary metabolites.9 Herein, we continued our chemical investigation on the three cytotoxic fractions G, I and L (which were active against the A549 cells with IC50 values of 13.36, 23.63 and 25.28 μg mL−1, respectively). Six new metabolites (1–5 and 10), together with nine known metabolites including four aromatic polyketides (6–9) and five perylenequinones (11–15), were obtained (Fig. 1). We found that the five known perylenequinones (11–15), as the major components, are responsible for the cytotoxic activity, as we have reported previously.10 The details of the isolation, structure elucidation, plausible biogenetic pathway and phytotoxic activity of these compounds are reported here.
 | ||
| Fig. 1 Structures of the isolated compounds 1–15. | ||
Results and discussion
Compound 1 was acquired as a white solid and has a molecular formula of C15H18O5 (requiring seven double bond equivalents, DBEs) as deduced from the pseudo-molecular-ion peak at m/z 279.1229 [M + H]+ (calcd. 279.1227) in HRESIMS. The IR spectrum showed the characteristic absorption bands for the hydroxyl (3384 cm−1), carbonyl (1747 cm−1), olefinic (1607 cm−1) and terminal double bond (911 cm−1) functionalities. The 1H NMR spectrum of 1 (Table 1) displayed the characteristic signals of a vinyl group (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) at δH 6.41 (dd, J = 17.2, 10.8 Hz, H-3′), 5.16 (d, J = 10.8 Hz, H-4′a), and 5.35 (d, J = 17.2 Hz, H-4′b), and suggested the presence of four methyl groups (δH 1.32, 1.36, 1.56 and 1.82), one oxygenated methine (δH 4.61), one olefinic proton (δH 5.58), and a hydroxyl group (δH 6.40). The 13C NMR spectrum (Table 1) resolved fifteen carbon signals, which were assigned by the HSQC spectrum as four methyls, one methylene, three methines and seven quaternary carbons. With the aid of the 1H–1H COSY spectrum, two coupling systems were identified as CH(3′)–CH2(4′) and CH(6)–CH3(9) (Fig. 2). The 2-methylbutadiene chain was established according to the key HMBC correlations from H3-8 to C-1′, C-2, and C-3 (δC 196.5) and from H3-5′ to C-1′, C-2′, and C-3' (Fig. 2). The key HMBC correlations of H-6/C-4 and of H3-10/C-6, C-7, and C-7a (δC 195.2), together with three residual quaternary carbons (δC 102.1, 159.0, and 195.2) with the characteristic chemical shifts for an α,β-unsaturated ester/acid,11 indicated a α-pyrone moiety (Fig. 2). Given the downfield chemical shifts and molecular formula, the olefinic carbon C-7a forms an ether bond with C-2 (δC 93.2) to fulfill the DBEs. Finally, the planar structure of 1 was elucidated as a furopyran-3,4-dione-fused heterocyclic core linked with a 2-methylbutadiene side chain. The furopyran-3,4-dione-fused heterocyclic core is unusual in natural products and was further confirmed by comparative analysis of its chemical shift values with those in the known cyclogregatin, which was first isolated from Aspergillus panamensis12 in 1988 by Anke et al. and revised as a type-c furancarboxylic acid derivative by Burghar-Stoll and Brückner in 2012.13 The configuration of the double bond at C1′C2′ was assigned to be E on the basis of the NOESY correlation of H-1′ and H-3' (Fig. 3).
CH2) at δH 6.41 (dd, J = 17.2, 10.8 Hz, H-3′), 5.16 (d, J = 10.8 Hz, H-4′a), and 5.35 (d, J = 17.2 Hz, H-4′b), and suggested the presence of four methyl groups (δH 1.32, 1.36, 1.56 and 1.82), one oxygenated methine (δH 4.61), one olefinic proton (δH 5.58), and a hydroxyl group (δH 6.40). The 13C NMR spectrum (Table 1) resolved fifteen carbon signals, which were assigned by the HSQC spectrum as four methyls, one methylene, three methines and seven quaternary carbons. With the aid of the 1H–1H COSY spectrum, two coupling systems were identified as CH(3′)–CH2(4′) and CH(6)–CH3(9) (Fig. 2). The 2-methylbutadiene chain was established according to the key HMBC correlations from H3-8 to C-1′, C-2, and C-3 (δC 196.5) and from H3-5′ to C-1′, C-2′, and C-3' (Fig. 2). The key HMBC correlations of H-6/C-4 and of H3-10/C-6, C-7, and C-7a (δC 195.2), together with three residual quaternary carbons (δC 102.1, 159.0, and 195.2) with the characteristic chemical shifts for an α,β-unsaturated ester/acid,11 indicated a α-pyrone moiety (Fig. 2). Given the downfield chemical shifts and molecular formula, the olefinic carbon C-7a forms an ether bond with C-2 (δC 93.2) to fulfill the DBEs. Finally, the planar structure of 1 was elucidated as a furopyran-3,4-dione-fused heterocyclic core linked with a 2-methylbutadiene side chain. The furopyran-3,4-dione-fused heterocyclic core is unusual in natural products and was further confirmed by comparative analysis of its chemical shift values with those in the known cyclogregatin, which was first isolated from Aspergillus panamensis12 in 1988 by Anke et al. and revised as a type-c furancarboxylic acid derivative by Burghar-Stoll and Brückner in 2012.13 The configuration of the double bond at C1′C2′ was assigned to be E on the basis of the NOESY correlation of H-1′ and H-3' (Fig. 3).
| a 1H and 13C NMR data recorded at 400 and 100 MHz.b 1H and 13C NMR data recorded at 600 and 150 MHz, respectively. | ||||
|---|---|---|---|---|
| Position | 1a | 2b | ||
| δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | δC, type | |
| 2 | 93.2, C | 93.2, C | ||
| 3 | 196.5, C | 196.4, C | ||
| 3a | 102.1, C | 102.0, C | ||
| 4 | 159.0, C | 159.1, C | ||
| 6 | 4.61 q (6.4) | 80.0, CH | 4.62 q (6.6) | 80.0, CH |
| 7 | 66.8, C | 66.8, C | ||
| 7a | 195.2, C | 195.0, C | ||
| 8 | 1.56 s | 23.4, CH3 | 1.55 s | 24.1, CH3 |
| 9 | 1.32 d (6.4) | 12.8, CH3 | 1.32 d (6.6) | 12.8, CH3 |
| 10 | 1.36 s | 18.4, CH3 | 1.32 s | 18.5, CH3 |
| 1′ | 5.58 s | 126.4, CH | 5.46 s | 124.1, CH |
| 2′ | 140.1, C | 133.3, C | ||
| 3′ | 6.41 dd (17.2, 10.8) | 140.2, CH | 6.77 dd (17.4, 10.8) | 138.7, CH |
| 4′a | 5.16 d (10.8) | 115.5, CH2 | 5.33 dt (10.8, 1.2) | 118.5, CH2 |
| 4′b | 5.35 d (17.2) | 5.40 d (17.4) | ||
| 5′ | 1.82 s | 12.9, CH3 | 1.85 d (1.2) | 19.5, CH3 |
| OH-7 | 6.40 s | 6.39 s | ||
 | ||
| Fig. 2 Key HMBC (red arrows) and 1H–1H COSY (bold black lines) of 1, 3–5 and 10. | ||
 | ||
| Fig. 3 Key NOESY (dash arrows) correlations of 1–4 and 10. | ||
In the NOESY spectrum, the cross-peak of H-6/H3-10 suggested that they were cofacial (Fig. 3). However, the NOESY correlation failed to assign the location of H3-8 relative to H3-10 and H-6. Therefore, ECD calculations for four configurations (2S,6R,7S; 2R,6R,7S; 2S,6S,7R; and 2R,6S,7R) of compound 1 were directly applied to confirm the absolute configuration of 1 (Fig. 4A and S60†). ECD calculations were performed on the B3PW91/TZVP//mPW1PW91/6-311G(d) level of theory. The result showed that the experimental ECD absorption band of 1 had better accuracy with the calculated ECD absorption band of (2S,6R,7S)-1 (Fig. 4A), which was also supported by the GIAO NMR shift calculation at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31G(d) level of theory14,15 (Table S1 and Fig. S64†). As a result, 1 was finally determined as (2S,6R,7S)-7-hydroxy-2,6,7-trimethyl-2-((E)-2-methylbuta-1,3-dien-1-yl)-6,7-dihydro-4H-furo[3,2-c]pyran-3,4(2H)-dione and named ophiofuranone A.
 | ||
| Fig. 4 Experimental and calculated ECD spectra of 1 (A), 2 (B) and 4 (C). | ||
Compound 2 was simultaneously obtained with 1 using RP-HPLC (47% MeOH in H2O) and had a very similar NMR profile to 1. The main difference was observed in the geometric configuration of the C-1'C-2′ double bond. Compound 2 was determined as Δ1′(2′) Z due to the NOESY correlation between H-1′ (δH 5.46) and H3-5′ (δH 1.85) (Fig. 3). The identical NMR data with those of 1 suggested the same relative configuration as 1 in the furopyran-3,4-dione-fused heterocyclic core (Table 1). Moreover, the experimental ECD curve highly matched that of 1 (Fig. 4B), implying the same absolute configurations of the three asymmetric carbons. Thus, 2 was determined as (2S,6R,7S)-7-hydroxy-2,6,7-trimethyl-2-((Z)-2-methylbuta-1,3-dien-1-yl)-6,7-dihydro-4H-furo[3,2-c]pyran-3,4(2H)-dione and named ophiofuranone B.
Compound 3 was in the form of white needles and displayed a molecular formula of C16H20O4, as determined by HRESIMS (m/z 299.1261 [M + H]+, calcd. 299.1259), indicating seven DBEs. The IR spectrum showed absorption bands for ester carbonyl (1727 cm−1) and benzoyl (1684, 1603, and 1582 cm−1) functionalities. Analysis of the 1D NMR data of 3 (Table 2) revealed five methyls, one methylene, two methines (one oxygenated), six aromatic carbons (proved to be a pentasubstituted phenyl), one ketone carbonyl and one ester carbonyl. The presence of a pentasubstituted aromatic ring was supported by the HMBC correlations from H-8 (δH 7.30) to C-6, C-10, C-14, and C-15 and from H2-13 (δH 5.37, 5.33) to C-5, C-6, and C-7 (Fig. 2). The 1H–1H COSY data of 3 revealed the presence of the spin-coupling system CH3(11)–CH(2)–CH(3)–CH3(12) shown by bold black lines in Fig. 2. The aforementioned fragments were assembled into a chromanone core using the HMBC correlations from H3-12 (δH 1.06) to C-4 and from H-2 (δH 4.25) to C-4 and C-10 (Fig. 2). The large coupling constant JH2–H3 (11.9 Hz) observed in the 1H NMR spectrum of 3 reflected the trans-orientation for H-2/H-3, which was also confirmed by the NOESY correlation of H-2/H3-12 and H-3/H3-11 (Fig. 3). A positive n → π* CE at 351 nm (Δε = +0.83) indicated a 2R configuration by analysis of the ECD spectrum (Fig. S29†).16,17 Moreover, the stereochemistry of 3 was further confirmed as 2R,3R by single-crystal X-ray diffraction analysis (CCDC 1855706) (Fig. 5), and 3 was named ophiochromanone.
| Position | 3 | 4 | ||
|---|---|---|---|---|
| δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | δC, type | |
| a 1H and 13C NMR data recorded at 600 and 150 MHz. | ||||
| 2 | 4.25 dq (11.9, 6.6) | 77.8, CH | 4.31 dq (10.2, 6.6) | 78.4, CH |
| 3 | 2.63 dq (11.9, 6.6) | 47.0, CH | 1.56 tq (10.2, 6.6) | 38.7, CH |
| 4 | 196.1, C | 5.14 d (10.2) | 77.5, CH | |
| 5 | 118.6, C | 134.5, C | ||
| 6 | 130.9, C | 120.5, C | ||
| 7 | 131.2, C | 128.6, C | ||
| 8 | 7.30 s | 137.8, CH | 7.08 s | 133.8, CH |
| 9 | 127.0, C | 128.7, C | ||
| 10 | 158.0, C | 147.5, C | ||
| 11 | 1.43 d (6.6) | 19.5, CH3 | 1.40 d (6.6) | 18.7, CH3 |
| 12 | 1.06 d (6.6) | 10.4, CH3 | 1.15 d (6.6) | 13.8, CH3 |
| 13 | 5.37 d (11.2) | 60.2, CH2 | 169.6, C | |
| 5.33 d (11.2) | ||||
| 14 | 2.23 s | 18.1, CH3 | 2.40 s | 15.5, CH3 |
| 15 | 2.16 s | 15.5, CH3 | 2.18 s | 14.7, CH3 |
| OAc-13 | 170.3, C | |||
| 1.97 s | 20.6, CH3 | |||
 | ||
| Fig. 5 The X-ray crystallographic structures of 3, 5, 9 and 10. | ||
Compound 4 was deduced to have the molecular formula C14H16O3 from HRESIMS, indicating seven DBEs. The main difference of the 13C NMR spectra between 3 and 4 was the presence of an ester signal (δC 169.6, C-13) and an oxygenated methine (δC 77.5, C-4) instead of the methylene and keto carbonyl groups in 3 with the absence of an acetoxy signal. On the basis of the 1H–1H COSY and HMBC correlations (Fig. 2), the planar structure of 4 was established to fulfil the DBEs. The presence of the H-2/H3-12/H-4 NOESY correlations (Fig. 3) determined the same side orientation of H-2, H3-12 and H-4, which was also supported by the large coupling constants of JH2–H3 and JH3–H4 (10.2 and 10.2 Hz, respectively). The experimental ECD spectrum of 4 was consistent with the calculated ECD curve of (2R,3R,4S)-4 (Fig. 4C). Ultimately, the structure of 4 was established as shown in Fig. 1 and 4 was named ophiolactone.
Compound 5 was obtained as white needles from MeOH, with the molecular formula of C12H12O4 by HRESIMS. The 1D and 2D NMR spectra (Table 3 and Fig. 2) of 5 closely resembled those of (S)-8-hydroxy-6-methoxy-4,5-dimethyl-3-methylene-isochromen-1-one,18,19 except that a hydroxyl is in the place of a methoxyl at C-6 (δC 164.4). The absolute configuration of 5 was determined as 9S by the uniform ECD CEs [240 nm (Δε = +10.15), 271 (+11.8), 315 (−1.72)] with (S)-8-hydroxy-6-methoxy-4,5-dimethyl-3-methylene-isochromen-1-one. The single-crystal X-ray diffraction using Cu Kα radiation confirmed the assignment of the 9S absolute configuration for 5 (Fig. 5, CCDC 1849055), and this compound was named ophioisocoumarin.
| Position | 5a | 10b | ||
|---|---|---|---|---|
| δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | δC, type | |
| a 1H and 13C NMR data recorded at 600 and 150 MHz in acetone-d6.b 1H and 13C NMR data recorded at 400 and 100 MHz in DMSO-d6, respectively. | ||||
| 1 | 168.0, C | |||
| 2 | 167.6, C | 5.78 s | 127.7, CH | |
| 3 | 98.8, C | 197.4, C | ||
| 4 | 163.3, C | 2.22 m | 41.6, CH2 | |
| 5 | 6.38 s | 101.6, CH | 2.18 m | 33.1, CH |
| 6 | 164.4, C | 41.7, C | ||
| 7 | 113.8, C | 1.65 m | 33.7, CH2 | |
| 8 | 144.3, C | 2.13 m,1.76 m | 34.8, CH2 | |
| 9 | 4.06 q (7.2) | 35.3, CH | 158.7, C | |
| 10 | 158.6, C | 5.63 s | 116.0, CH | |
| 11 | 4.71, 4.72 (each br s) | 95.8, CH2 | 11.89 s | 167.4, COOH |
| 12 | 2.11 s | 10.0, CH3 | 2.09 s | 18.3, CH3 |
| 13 | 1.36 d (7.2) | 22.4, CH3 | 1.91 s | 19.7, CH3 |
| 14 | 0.90 d (6.4) | 15.1 | ||
| 15 | 1.00 s | 19.1 | ||
| OH-4 | 10.89 s | |||
Compound 10 had a molecular formula of C15H22O3 by HRESIMS. The 13C NMR results (Table 3) along with the HSQC data of 10 confirmed the presence of four methyls, three methylenes, one sp3 methine, one quaternary sp3 carbon, one keto carbonyl, one carboxyl and four olefinic carbons. An α,β-unsaturated cyclohexenone ring was elucidated by the HMBC correlations from H3-15 to C-1, C-5, and C-6, from H3-13 to C-1, from H-2 to C-4 and C-6 and from H-5 to C-3 together with the proton spin system CH2(4)–CH(5)–CH3(14) indicated by the 1H–1H COSY correlations (Fig. 2). Another spin system of CH2(7)–CH2(8) together with the HMBC correlations of H3-12/C-8, C-9, C-10 and H-10/C-11 confirmed a branched chain of carboxylic acid. The branched chain was attached to the α,β-unsaturated cyclohexenone ring, which was supported by the key HMBC correlation from H3-15 to C-7. The NOESY correlations of H2-8/H-10 determined the Δ9 double bond as E (Fig. 3). Moreover, H3-14 and H3-15 were assigned to be the same side orientation by the NOESY correlation of H3-14/H3-15 (Fig. 3). According to the X-ray diffraction data (Fig. 5, CCDC 1855023), the absolute configuration of 10 was assigned as 5R,6S, and compound 10 was named ophiokorrin.
Compound 9 was isolated as massive colourless crystals. The NMR profiles of 9 were exactly consistent with those of the known compound clearanol E,20 which was first isolated as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with clearanol D from two of the fungal isolates [a glomeromycete (possibly Entrophospora sp.) and a dothideomycete (possibly Phaeosphaeria sp.)]. The relative configuration of the C3–C8 segment was established as anti by single-crystal X-ray diffraction (Fig. 5 CCDC 1873977). According to the coincident ECD data with those of the calculated (3R,8S)-9, the absolute structure was assigned. Here, the stereochemistry of clearanol E is determined for the first time [mp 80–85 °C; [α]D20 = −26.6 (c 0.1, MeOH)]. The other known compounds were identified as (R)-3,4-dihydro-4,8-dihydroxy-6-methoxy-4,5-dimethyl-3-methyleneisochromen-1-one (6),21 (3R,8S)-dihydroxy-3-hydroxymethyl-6-methoxy-4,5-dimethylisochroman-1-one (7),21 (R)-7-hydroxy-3-((S)-1-hydroxyethyl)-5-methoxy-3,4-dimethylisobenzofuran-1(3H)-one (8),22 hypocrellins A (11), elsinochromes A–C (12–14) and phaeosphaerin C (15)10 by comparison of their spectroscopic data with those reported.
:1 mixture with clearanol D from two of the fungal isolates [a glomeromycete (possibly Entrophospora sp.) and a dothideomycete (possibly Phaeosphaeria sp.)]. The relative configuration of the C3–C8 segment was established as anti by single-crystal X-ray diffraction (Fig. 5 CCDC 1873977). According to the coincident ECD data with those of the calculated (3R,8S)-9, the absolute structure was assigned. Here, the stereochemistry of clearanol E is determined for the first time [mp 80–85 °C; [α]D20 = −26.6 (c 0.1, MeOH)]. The other known compounds were identified as (R)-3,4-dihydro-4,8-dihydroxy-6-methoxy-4,5-dimethyl-3-methyleneisochromen-1-one (6),21 (3R,8S)-dihydroxy-3-hydroxymethyl-6-methoxy-4,5-dimethylisochroman-1-one (7),21 (R)-7-hydroxy-3-((S)-1-hydroxyethyl)-5-methoxy-3,4-dimethylisobenzofuran-1(3H)-one (8),22 hypocrellins A (11), elsinochromes A–C (12–14) and phaeosphaerin C (15)10 by comparison of their spectroscopic data with those reported.
Ophiofuranones A (1) and B (2) are rare furopyran-3,4-dione-fused heterocyclic polyketides. Taking the unique structure features into consideration, a polyketide biosynthetic pathway was proposed (Scheme 1). Originating from propionate and acetate units, the polyketide chain was connected ultimately through a Claisen reaction.23,24 After a sequence of post-PKS modifications, compounds 1 and 2 were biosynthesized separately.
 | ||
| Scheme 1 A proposed biogenetic pathway for compounds 1 and 2. | ||
Given the amounts and structure features of 1–15, five compounds (6–8, 10 and 13) were tested with models of A. thaliana (thale cress, Brassicaceae) root elongation. Only compound 10, as an analogue of the well-known phytohormone abscisic acid,25,26 showed an inhibitory activity on the root elongation in the germination of A. thaliana (Fig. 6) with an IC50 value of 18.06 μg mL−1 (Fig. 6 and S65†).
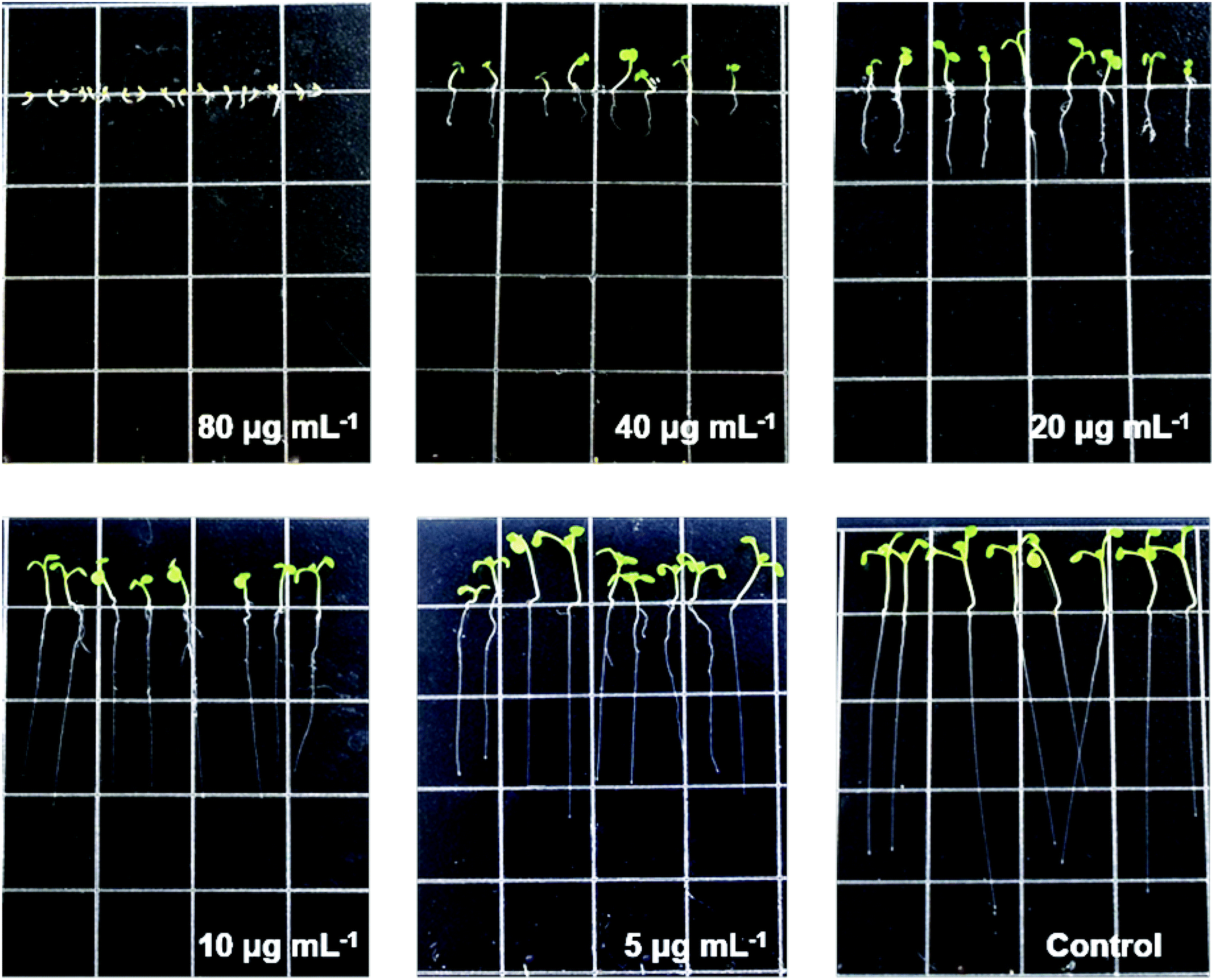 | ||
| Fig. 6 The root elongation of A. thaliana on Petri dishes with different doses of compound 10. | ||
Conclusions
In summary, six new metabolites (1–5 and 10) and nine known compounds (6–9 and 11–15) were isolated and identified from the EtOAc extract of O. korrae. Ophiofuranones A (1) and B (2), possessing an unusual furopyran-3,4-dione-fused heterocyclic skeleton, were assumed to be derived from the PKS pathway. Ophiokorrin (10), as an analogue of the well-known phytohormone abscisic acid, could inhibit root elongation in the germination of A. thaliana according to the results of the phytotoxic assay.Experimental section
General experimental procedures
All NMR spectra were acquired on a Bruker AVIII 400 spectrometer (1H: 400 MHz, 13C: 100 MHz) or a Bruker Avance DRX-600 spectrometer (1H: 600 MHz, 13C: 150 MHz) using TMS as the internal standard. HRESIMS data were obtained using a Thermo Scientific LTQ-Orbitrap XL. UV and CD spectra were obtained from a Chirascan spectropolarimeter. IR spectra were recorded on a Nicolet NEXUS 470 FT-IR (Thermo Fisher Scientific, Inc., Hudson, NH, USA) using KBr discs. Optical rotations were provided on an MCP 200 polarimeter from Anton Paar. Melting points (uncorrected) were observed on an X-6 melting-point apparatus (Beijing TECH Instrument Co. Ltd.). Medium Pressure Liquid Chromatography (MPLC) was carried out on a Lisure EZ Purifier apparatus equipped with a dual-wavelength UV-Vis detector (Lisure Science (Suzhou) Co., ltd.) and an ODS column (column size: 30 × 130 mm). Semipreparative HPLC was accomplished on an Agilent 1100 series instrument with Eclipse XDB-C18 5 μm columns (column size: 9.4 × 250 mm). Silica gel (200–300 mesh; Marine Chemical Ltd., Qingdao, China), Sephadex LH-20 (Pharmacia Biotek, Denmark), and reversed phase C18 silica gel (150–200 mesh, Merck) were used for column chromatography. Pre-coated silica gel GF254 plates (Marine Chemical Ltd., Qingdao, China) were used for TLC. Spots of TLC were visualized under UV light (254 nm) or by spraying with H2SO4/EtOH (1:9, v/v), followed by heating. 1/2 MS medium was purchased from Qingdao Hope Bio-Technology Co., Ltd.
Strain and fermentation
The fungus O. korrae was isolated from the lichen Physciaceae physcia which was collected in Ditch Muzhate, Zhaosu, Xinjiang, China (coordinates of 80°08′–81°30′ E, 43°09′–43°15′ N). The strain, assigned no. 0207b, was identified by the nuclear 18S rDNA sequences (GenBank: KC841077). The voucher strain was deposited in the Key Lab of Chemical Biology of Ministry of Education, Shandong University. O. korrae was cultured on PDA plates at 25 °C for 7 days. Mycelium was inoculated into three 300 mL Erlenmeyer flasks containing 100 mL of PDB medium and cultured at 25 °C for 10 days on a rotary shaker (110 rpm). The fungus O. korrae was grown on a solid sterile rice medium (20 × 500 mL Fernbach culture flasks, each containing 80 g of rice and 120 mL of distilled water) for 40 days at room temperature.Extraction and isolation
The fermented rice substrate was extracted repeatedly with EtOAc (3 × 10 L) and 44 g of crude extract was obtained by evaporating the solvent under vacuum. The crude extract (44 g) was partitioned between water and EtOAc (1:2) three times. The EtOAc extract was separated into 20 fractions (A–T) by silica gel CC, eluted with a step gradient of CH2Cl2–MeOH from 100:0 to 0:100 (v/v).
Fr. G (1.16 g) was separated by Sephadex LH-20 CC twice, eluted with CH2Cl2–MeOH (1:1) and MeOH successively. Fr. G2 (0.49 g) was divided into 7 parts (a–g) by MPLC. Fr. G2c (105.8 mg) was purified by semipreparative reversed-phase (RP) HPLC using 70% MeOH–H2O (flow rate: 1.5 mL min−1) as the solvent system, to afford 5 (5.1 mg, tR = 26.4 min) and 4 (1.4 mg, tR = 38.5 min). Under the same purifying conditions, 6 (12.4 mg, tR = 15.8 min) and 3 (1.0 mg, tR = 45.6 min) were obtained from Fr. G2d (211.2 mg). In addition, Fr. G2g (10.0 mg) afforded 15 (1.5 mg, tR = 16.7 min) and 14 (1.8 mg, tR = 18.0 min) using HPLC (MeOH–CH3CN–H2O, 9:5:86, 1.5 mL min−1).
Fr. I (2.50 g) was subjected to MPLC to give 22 parts (a–v) eluted with MeOH–H2O (10%–100%). Fr. Ig (84.1 mg, from 30%) was chromatographed over Sephadex LH-20 eluted with MeOH to obtain part 2 (63.3 mg), which was further purified to afford 8 (2.6 mg, tR = 31.7 min) by HPLC using 45% MeOH–H2O (1.5 mL min−1). Fr. Ik (81.2 mg, from 40%) was purified to yield 1 (4.6 mg, tR = 40.0 min), 2 (1.2 mg, tR = 42.3 min) and 7 (20.4 mg, tR = 48.6 min) by HPLC using 47% MeOH–H2O (1.5 mL min−1). Fr. Im (100.0 mg, from 50%) was subjected to Sephadex LH-20 eluted with MeOH and then purified to yield 10 (33.3 mg, tR = 30.7 min) by HPLC with 58% MeOH–H2O containing 0.1% acetic acid (1.5 mL min−1). Fr. It (77.7 mg, from 70%) afforded 11 (8.8 mg) and 12 (8.4 mg) over Sephadex LH-20 eluted with MeOH.
Fr. L (1.90 g) was subjected to MPLC eluted with MeOH–H2O (10–100%), and Fr. La–Fr. Lm was obtained. Fr. Lc (59.2 mg, from 45%) was purified by HPLC (34% MeOH–H2O, 1.5 mL min−1) to yield 9 (3.2 mg, tR = 41.5 min). Fr. Lf (39.7 mg, from 70%) yielded 13 (9.2 mg, tR = 11.2 min) using HPLC (90% MeOH–H2O, 1.5 mL min−1).
ε) 211 (3.84), 257 (3.60) nm; ECD (MeOH): 261 (Δε −2.24), 306 (Δε +0.55) nm; IR νmax 3384, 2986, 2939, 1747, 1607, 911 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS: m/z 279.1229 for [M + H]+, calcd. 279.1227 and 296.1495 for [M + NH4]+, calcd. 296.1492, for C15H22NO5+.ε) 221 (3.99) nm; ECD (MeOH): 263 (Δε −3.46), 313 (Δε +0.57) nm; IR νmax 3429, 2984, 2945, 1751, 1593, 1430, 739, 728 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS: m/z 301.1050 for [M + Na]+, calcd. 301.1052, for C15H18O5Na+.ε) 223 (4.18), 260 (3.60), 335 (3.31) nm; ECD (MeOH): 222 (Δε +2.33), 317 (Δε −3.46), 351 (Δε +0.83) nm; IR νmax 2974, 1727, 1684, 1603, 1582 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS: m/z 299.1261 for [M + Na]+, calcd. 299.1259, for C16H20O4Na+.ε) 224 (4.07), 251 (3.57), 310 (3.18) nm; ECD (MeOH): 233 (Δε −0.94), 261 (Δε −2.15), 306 (Δε +0.68) nm; IR νmax 2930, 1737 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS: m/z 233.1170 for [M + H]+, calcd. 233.1172 for C14H17O3+.ε) 216 (4.23), 272 (3.99), 314 (3.74) nm; ECD (MeOH): 240 (Δε +10.15), 271 (Δε +11.8), 315 (Δε −1.72) nm; IR νmax 3314, 1679, 1642, 1612 cm−1; 1H and 13C NMR data, see Table 3; HRESIMS: m/z 221.0805 for [M + H]+, calcd. 221.0814 for C12H13O4+.ε) 250 (3.90); ECD (MeOH): 215 (Δε +5.29), 240 (Δε −3.38), 295 (Δε −0.17) nm; IR νmax 2957, 1709, 1644, 1582 cm−1; 1H and 13C NMR data, see Table 3; HRESI MS: m/z 251.1644 for [M + H]+, calcd. 251.1642 and 273.1642 for [M + Na]+, calcd. 273.1641, C15H22O3Na+.X-ray single crystal diffraction
Ophiochromanone (3) was obtained as white crystals from MeOH using the vapour diffusion method.Ophioisochromanone (5) was obtained as white crystals from MeOH using the vapour diffusion method.
Ophiokorrin (9) was obtained as white crystals from MeOH using the vapour diffusion method.
Ophiokorrin (10) was obtained as white crystals from MeOH using the vapour diffusion method.
Phytotoxic effects on the root elongation of seeds of A. thaliana
Seeds of A. thaliana were cleaned using EtOH–H2O (75:25, v/v) for 3 min, followed by 100% EtOH for 2 min, and finally washed with sterilized H2O (five times) for surface sterilization. Compounds 6–8, 10 and 13 were dissolved in DMSO to a concentration of 40 mg mL−1. Then, different volumes (1.25, 2.5, 5, 10 and 20 μL) of each solution were added to 10 mL of 1/2 MS medium to obtain plates with different concentrations of compounds (5, 10, 20, 40 and 80 μg mL−1). To eliminate the effect of DMSO on the growth of A. thaliana, plates with the same volumes of DMSO were used as blank controls. Ten seeds were distributed on each square Petri dish (10.0 × 10.0 cm) as described before. Three replicates were performed for each concentration. The seeds were placed in a growth chamber at 28 ± 1 °C under 12 h of light and 12 h of darkness. After ten days, the lengths of the seedling roots were measured for statistical analysis.27
Computational details
ECD calculations were carried out as described previously.28 Conformational searches were performed by means of Frog2 online version with a 100 kcal mol−1 van der Waals (VDW) energy window from the global minimum.29 Further geometrical optimization and vibrational evaluation with the Gaussian 09 program were performed using DFT calculations [using the b3pw91 functional and the TZVP basis set]. The singlet electronic excitation energies and rotational strengths in MeOH were calculated by TDDFT at the mPW1PW91/6-311G(d) level of theory. The ECD curve for each conformer was simulated according to the eqn (8d) with a value of σ = 0.3 eV.30 The final ECD spectrum was generated by summing the individual conformer values with respect to their Boltzmann statistics.Gauge-independent atomic orbital (GIAO) calculations of 1H and 13C NMR chemical shifts for the optimized conformers of (2S,6S,7R)-1 and (2S,6R,7S)-1 were accomplished at the mPW1PW91-SCRF(DMSO)/6-311G(2d,p) level in the polarizable continuum solvation model. The calculated NMR data of the lowest energy conformers for (2S,6S,7R)-1 and (2S,6R,7S)-1 were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy. The 1H and 13C NMR chemical shifts for TMS were calculated by the same protocol and used as a reference. The experimental and calculated data were analysed by the improved probability DP4+ method for isomeric compounds. A significantly higher DP4+ probability score for (2S,6R,7S)-1 suggested the correctness of its configuration.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 81630093, 81874293). We thank Mr Hongbo Zheng and Ms Ke Xu for the NMR measurements. Ms Yanan Qiao and Dr Jiaozhen Zhang are also acknowledged for the X-ray diffraction analysis.References
- J. J. Kellogg and H. A. Raja, Phytochem. Rev., 2017, 16, 271–293 CrossRef CAS.
- P. A. Paranagama, E. M. Wijeratne, A. M. Burns, M. T. Marron, M. K. Gunatilaka, A. E. Arnold and A. A. Gunatilaka, J. Nat. Prod., 2007, 70, 1700–1705 CrossRef CAS PubMed.
- B. N. Singh, D. K. Upreti, V. K. Gupta, X. F. Dai and Y. M. Jiang, Trends Biotechnol., 2017, 35, 808–813 CrossRef CAS PubMed.
- X. B. Li, L. Li, R. X. Zhu, W. Li, W. Q. Chang, L. L. Zhang, X. N. Wang, Z. T. Zhao and H. X. Lou, J. Nat. Prod., 2015, 78, 2155–2160 CrossRef CAS PubMed.
- W. Li, W. Gao, M. Zhang, Y. L. Li, L. Li, X. B. Li, W. Q. Chang, Z. T. Zhao and H. X. Lou, J. Nat. Prod., 2016, 79, 2188–2194 CrossRef CAS PubMed.
- F. Xie, W. Q. Chang, M. Zhang, Y. Li, W. Li, H. Z. Shi, S. Zheng and H. X. Lou, Sci. Rep., 2016, 6, 33687 CrossRef CAS PubMed.
- Y. H. Zhou, M. Zhang, R. X. Zhu, J. Z. Zhang, F. Xie, X. B. Li, W. Q. Chang, X. N. Wang, Z. T. Zhao and H. X. Lou, J. Nat. Prod., 2016, 79, 2149–2157 CrossRef CAS PubMed.
- F. J. Flores, S. M. Marek, G. Orquera and N. R. Walker, Crop Sci., 2017, 57, S249–S261 CrossRef CAS.
- Y. L. Li, R. X. Zhu, J. Z. Zhang, F. Xie, X. N. Wang, K. Xu, Y. N. Qiao, Z. T. Zhao and H. X. Lou, ACS Omega, 2018, 3, 176–180 CrossRef CAS PubMed.
- G. Li, H. Y. Wang, R. X. Zhu, L. M. Sun, L. N. Wang, M. Li, Y. Y. Li, Y. Q. Liu, Z. T. Zhao and H. X. Lou, J. Nat. Prod., 2012, 75, 142–147 CrossRef CAS PubMed.
- G. P. Yin, Y. R. Wu, M. H. Yang, T. X. Li, X. B. Wang, M. M. Zhou, J. L. Lei and L. Y. Kong, Org. Lett., 2017, 19, 4058–4061 CrossRef CAS PubMed.
- H. Anke, I. Casser, M. Schrage and W. Steglich, J. Antibiot., 1988, 41, 1681–1684 CrossRef CAS PubMed.
- H. Burghart-Stoll and R. Brückner, Eur. J. Org. Chem., 2012, 3978–4017 CrossRef CAS.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526–12534 CrossRef CAS PubMed.
- A. M. Sarotti, Org. Biomol. Chem., 2018, 16, 944–950 RSC.
- D. Slade, D. Ferreira and J. P. Marais, Phytochemistry, 2005, 66, 2177–2215 CrossRef CAS PubMed.
- H. Cui, M. Ding, D. Huang, Z. R. Zhang, H. T. Liu, H. B. Huang and Z. G. She, RSC Adv., 2017, 7, 20128–20134 RSC.
- W. C. Tayone, S. Kanamaru, M. Honma, K. Tanaka, T. Nehira and M. Hashimoto, Biosci., Biotechnol., Biochem., 2011, 75, 2390–2393 CrossRef CAS PubMed.
- Y. X. Song, J. Wang, S. W. Li, B. Cheng, L. Li, B. Chen, L. Liu, Y. C. Lin and Y. C. Gu, Planta Med., 2012, 78, 172–176 CrossRef CAS PubMed.
- A. L. Gerea, K. M. Branscum, J. B. King, J. You, D. R. Powell, A. N. Miller, J. R. Spear and R. H. Cichewicz, Tetrahedron Lett., 2012, 53, 4202–4205 CrossRef CAS PubMed.
- T. El-Elimat, H. A. Raja, M. Figueroa, J. O. Falkinham and N. H. Oberlies, Phytochemistry, 2014, 104, 114–120 CrossRef CAS PubMed.
- W. C. Tayone, M. Honma, S. Kanamaru, S. Noguchi, K. Tanaka, T. Nehira and M. Hashimoto, J. Nat. Prod., 2011, 74, 425–429 CrossRef CAS PubMed.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, John Wiley & Sons, Ltd, 3rd edn, 2009, pp. 64–131 Search PubMed.
- T. Asai, K. Tsukada, S. Ise, N. Shirata, M. Hashimoto, I. Fujii, K. Gomi, K. Nakagawara, E. N. Kodama and Y. Oshima, Nat. Chem., 2015, 7, 737–743 CrossRef CAS PubMed.
- T. Oritani and H. Kiyota, Nat. Prod. Rep., 2003, 20, 414–425 RSC.
- F. Aquea, F. Federici, C. Moscoso, A. Vega, P. Jullian, J. Haseloff and P. Arce-Johnson, Plant, Cell Environ., 2012, 35, 719–734 CrossRef CAS PubMed.
- P. H. Fan, K. Hostettmann and H. X. Lou, Chemoecology, 2010, 20, 223–227 CrossRef CAS.
- G. Li, S. Kusari, C. Golz, H. Laatsch, C. Strohmann and M. Spiteller, J. Nat. Prod., 2017, 80, 983–988 CrossRef CAS PubMed.
- M. A. Miteva, F. Guyon and P. Tuffery, Nucleic Acids Res., 2010, 38, W622–W627 CrossRef CAS PubMed.
- P. J. Stephens and N. Harada, Chirality, 2010, 22, 229–233 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: spectra of all new compounds (1H NMR, 13C NMR, 2D NMR, HRESIMS, UV, CD, and IR) and computational details. CCDC 1855706, 1849055, 1855023 and 1873977. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra10329a |
| This journal is © The Royal Society of Chemistry 2019 |