
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect production of olefins via syngas conversion over Co2C-based catalyst in slurry bed reactor
Xinxing Wangab,
Tiejun Lina,
Jie Lia,
Fei Yuab,
Dong Lva,
Xingzhen Qia,
Hui Wanga,
Liangshu Zhong *ac and
Yuhan Sunac
*ac and
Yuhan Sunac
aCAS Key Laboratory of Low-Carbon Conversion Science and Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201203, P. R. China. E-mail: zhongls@sari.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100049, P. R. China
cSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201203, P. R. China
First published on 30th January 2019
Abstract
Direct production of olefins via syngas conversion over a Co2C-based catalyst was investigated in a slurry bed reactor (SBR). It was found that the total selectivities to olefins and oxygenates reached 88.8C% at a CO conversion of 29.5% at 250 °C, 5 bar and H2/CO = 0.5. The hydrocarbon distribution greatly deviated from the classical Anderson–Schulz–Flory (ASF) distribution, with only 2.6C% methane selectivity was obtained. XRD and TEM characterization verified that the Co2C nanoprisms with special exposed facts of (101) and (020) constitutes the Fischer–Tropsch to olefins (FTO) active site. The catalytic activity increased gradually with rising the reaction temperature, while the product distribution almost kept unchanged under various reaction condition in SBR. Compared to the reaction in FBR, the Co2C-based catalyst exhibited relative better catalytic performance during FTO process in SBR. Specifically, a higher CO conversion, a lower methane selectivity and a higher total selectivities to olefins and oxygenates were achieved in SBR. In addition, the catalyst can be in situ reduced in slurry bed reactor at mild temperature (300 °C) and no obvious deactivation was found within nearly 100 h time-on-stream, which suggested a promising route for the direct production of olefins via syngas in industrial application.
1. Introduction
Olefins including lower olefins (C2–4=) and longer-chain olefins (C5+=), are extensively used to synthesize a wide range of products such as polymers, solvents, drugs, cosmetics and detergents.1–5 Traditionally, they are produced by thermal or catalytic cracking of a broad range of petroleum products, such as naphtha, gas oil, condensates and light alkanes. Due to the depletion of the limited petroleum reserves, it is necessary to develop new processes for the production of olefins from alternative feedstocks.6–8 Syngas, a mixture of CO and H2, can be obtained from various carbon-containing sources, such as coal, nature gas and biomass.9,10 Syngas conversion provides an alternative nonpetroleum route for olefins production. The methods for synthesis of olefins from syngas can be divided into indirect process and direct process. The indirect process mainly refers to the methanol to olefins (MTO or DMTO) technology, where an intermediate such as methanol or dimethyl ether is synthesized from syngas at first, then dehydrated to form lower olefins using zeolite catalysts.11,12 Due to the simplified operation and low energy consumption, direct production of olefins from syngas has attracted increasing attention. The typical routes of direct production of olefins from syngas include bifunctional catalytic reactions and the Fischer–Tropsch to olefin (FTO) process. Jiao et al. reported a ZnCrOx/MSAPO bifunctional catalyst to produce olefins from syngas. Under the condition of 400 °C, 25 bar and a H2/CO ratio 1.5, C2–4= selectivity reached as high as 80% with the CO conversion of 17%.6 Cheng et al. reported a Zn–Zr/SAPO-34 catalyst with a CO conversion of 10% and C2–4= selectivity of 74C% at 400 °C and 1 MPa.7 In addition, the integration manner and nanoscale proximity of the two components are crucial for the catalytic performance. Fischer–Tropsch synthesis (FTS) is well known for producing fuels and various chemicals from syngas.13–18 Paraffins, olefins, oxygenates (mainly alcohols and aldehydes) and other products are always co-produced during FTS process, depending on the catalyst systems and reaction conditions. As olefins are more value-added than paraffins, Fischer–Tropsch to olefins (FTO) reaction attracts more and more interests from both academia and industry. Promoted Fe-based Fischer–Tropsch catalysts have been widely studied for the FTO reaction. Torres Galvis et al. prepared a serial of supported iron catalysts and found it exhibited high selectivity toward lower olefins (∼61% C).19 By investigating the effects of Na and sulfur, they found Na increased the chain growth probability, while sulfur reduced the coverage of hydrogen on the catalyst surface, resulting in the lower methane selectivity and higher olefin selectivity.19–21 Zhai et al. developed a Zn and Na promoted Fe5C2 catalysts, which exhibited an outstanding activity and high selectivity for olefins (up to 79%) at 340 °C.22 The Zn severed as a structure promoter, while the Na suppressed the hydrogenation of double bonds and promoted the olefins selectivity. However, Fe-based FTO reactions usually require very high temperature (higher than 300 °C), which may lead to fast catalyst deactivation due to particles sintering and carbon deposition on the catalyst surface.Recently, we discovered Co2C nanostructures possess strong facet effects during syngas conversion process. Co2C nanoprisms with special exposed facets of (101) and (020) exhibit excellent catalytic performance for FTO.23,24 At 250 °C and 1 bar, the C2–4= selectivity was as high as 60.8C%, while methane selectivity was less than 5C%, and O/P value among the C2–C4 products was as high as 30. Catalyst characterization and theoretical calculation indicated that preferentially exposed (101) and (020) facets favor olefins production and inhibit methane formation. The effects of alkali and manganese on the catalytic performance of Co2C catalysts for FTO reactions were specially studied.24,25 It was found that Na, acting as an electronic donor to cobalt, promoted the formation of Co2C. Mn not only acted as a typical electronic or structural promoter but also had a strong influence on the morphology control of Co2C nanoprisms. In addition, the reduction process also had a great impact on the structure–performance of Co2C-based FTO catalyst.26 Weak interaction of cobalt-support facilitates the formation of the CoMn composite oxide in supported cobalt catalysts, which contributes to the generation of Co2C nanoprisms.27
Two types of reactors including fixed bed reactor (FBR) and slurry bed reactor (SBR) are widely employed in FTS in industry.28–31 In the case of FBR, it is hard to avoid temperature runaway due to its limited heat-transfer capacity. In addition, the catalyst size in FBR should be chosen carefully. In order to decrease pressure drop, large size for pellet catalyst should be used. However, it would lead to mass-transfer limitation and weaken the catalytic performance. Compared with fixed bed reactor, slurry bed reactor possesses the following advantages:32–39 (1) better heat-transfer capacity by the usage of liquid solvent; (2) easier operation for catalyst replacement during industrial production; (3) higher yield per reactor volume; and (4) relatively lower investment capital and operating cost. Similar with the FTS, the FTO process is also a highly exothermic reaction. In order to overcome the drawbacks of FBR, it is necessary to study the FTO catalytic performance in SBR. Due to these significant differences between FBR and SBR, the catalytic performance at different reactors should be different. The study of FTO process in SBR was meaningful for its industrialized application.
Up to now, most of reported FTO studies are tested in FBR, and very few works concern the utilization of SBR. Gao et al. observed the improvement in olefins selectivity by adding polar liquid into a SBR over Fe-based catalyst, and the olefins fraction in hydrocarbon reached up to 70 wt% at 240 °C and 3 MPa.40,41 Rausch et al. used the ammonia containing syngas as feedstock for FTO reaction over iron- and cobalt-based catalyst in SBR, and the selectivity to α-olefins was found to significantly increase at the expense of catalyst activity.42 Herein, we aim to explore the possibility of Co2C-based FTO catalyst used in SBR. The influence of different reactors on the catalytic performance of Co2C-based catalyst was studied. In addition, the effect of reaction conditions in SBR for FTO process and stability test were also investigated in detail.
2. Experiment
2.1 Catalyst preparation
The catalyst precursors were prepared by the co-precipitation of an aqueous solution of Co(NO3)2·6H2O and Mn(NO3)2 (Sinopharm Chemical Reagent Co., Ltd) as described previously. Generally, a solution with a total [Co + Mn] cation concentration of 2 M (Co/Mn = 2, atom ratio) was contacted with a basic solution of sodium carbonate (Na2CO3, Sinopharm Chemical Reagent Co., Ltd) by dropwise addition of both solutions into a beaker with 200 ml deionized water under vigorously magnetic stirring. The pH value was maintained at 8.0 ± 0.1 and the temperature was kept at 30 ± 1 °C during the precipitation process. After aging for 2 hours at 60 °C, the obtained suspension was centrifuged and washed with deionized water for 8 times, then dried at 80 °C for 12 hours. The prepared sample was denoted as CoMn-precursor. Then the samples were calcined in a furnace at 350 °C for 5 hours under static air. The calcined sample was ground and sieved to 40–60 mesh for test in FBR and SBR.2.2 Catalyst characterization
X-ray diffraction (XRD) patterns of catalysts were recorded on a Rigaku Ultima IV X-ray powder diffractometer using Cu Kα radiation at 40 kV and 40 mA. The samples were scanned from 10° to 90° with a rate of 4° min−1 (1.2 s per 0.02°) in the continuous scanning mode. Phase identification was carried out according to the PDF4-2015 database. The standard PDF cards used to identify the phase constitution of catalyst included CoCO3 (PDF#01-78-0209), MnCO3 (PDF#01-086-0173), MnCo2O4.5 (PDF#00-032-0297), MnO (PDF#01-075-0625), CoO (PDF#01-071-1178) and Co2C (PDF#03-065-1457). The crystallite sizes were estimated using the Scherrer equation.Transmission electron microscopy (TEM) measurements were performed on a JEOL JEM 2011 transmission electron microscope, and the accelerating voltage was 200 kV. Samples for TEM test were prepared by dispersing the powder in ethanol followed by ultra-sonication. One droplet of the suspension was dropped on a carbon-coated copper grids for measurement. The particle size distribution was determined by random statistics of more than 250 particles in TEM images.
2.3 Catalytic evaluation
Catalytic evaluation was carried out in both FBR and SBR. The fixed bed reactor consisted of a 316 stainless steel tubular reactor with inner diameter of 9 mm, in which 1.5 g of catalyst (40–60 mesh) mixed with 3 g silica of the same size was placed. FTO experiments in SBR were conducted in a 1 L stainless steel vessel (diameter = 80 mm), and 20 g of the same catalyst was loaded into the reactor vessel and suspended in 500 g Ploywax-500 (Baker Hughes, Inc.). Prior to the catalytic test, the catalyst was reduced in situ at 300 °C for 10 h in a flow of 10 vol% CO diluted with N2 and the WHSV (weight hourly space velocity) was kept at 6000 ml h−1 gcat−1. The reducing pressure was atmospheric in FBR and 5 bar in SBR. The ramping rate during the reduction process for FBR and SBR was 1 °C min−1. A stirring motor was used in SBR with the stirring speed of 800 rpm. After reduction, the temperature was dropped to the setting reaction temperature. A mixture of 97 vol% syngas with different H2/CO ratio (0.5, 0.75 and 1) and 3 vol% N2 (as an internal standard) was introduced into the reactor as feed gas. The gas flow was adjusted by calibrated mass flow controllers (Brooks 5850 series mass flow controllers). The pressure of the system was kept at desired value with a pressure controller (TESCOM).The tail gas from both FBR and SBR, after passing through a hot trap (120 °C) and a cold trap (0 °C), was analysed on-line by gas chromatography (Agilent 7890B) equipped with a flame ionization (FID) and thermal conductivity detector (TCD). The liquid products and solid wax products, collected from cold trap and hot trap, were analysed off-line by GC (Shimadzu GC2010 plus). The detailed analysis process had been described in our previous work.43–45 The catalytic activity and selectivity at the stable stage was used for discussion (after 50 h of time on stream). The mass balance, carbon balance and oxygen balance were calculated according to the formula as following:
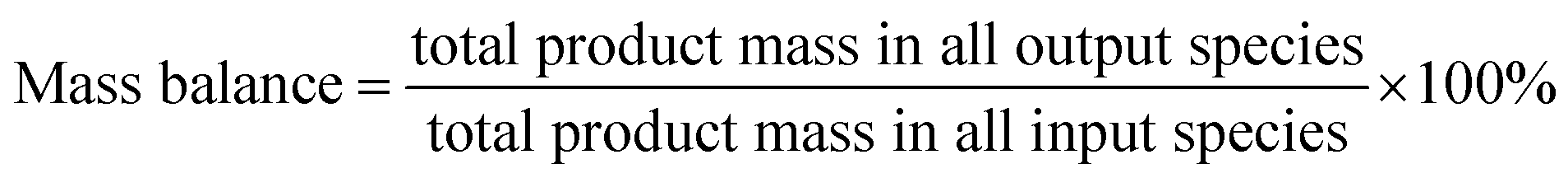
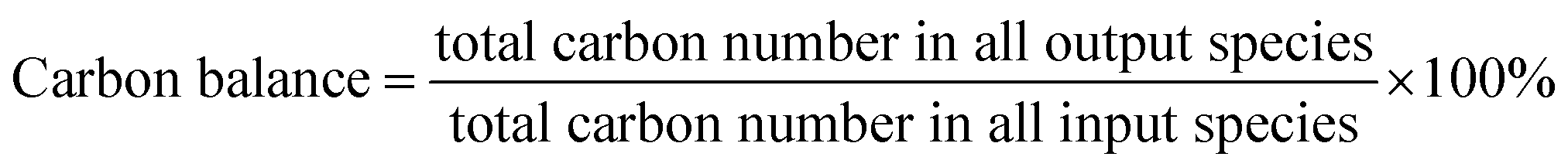
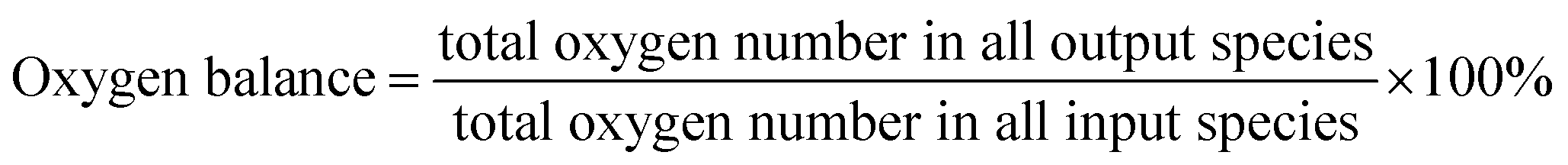
The mass balance, carbon balance and oxygen balance calculated in each test were between 95% and 105%.
For a typical FTO reaction, CO conversion (XCO) and products selectivity (SCO2, Si) were calculated according to the formula as following:
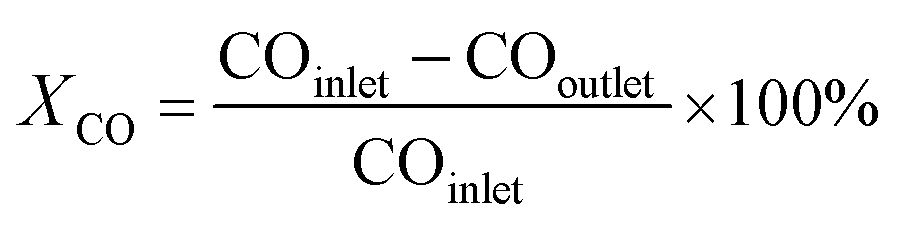
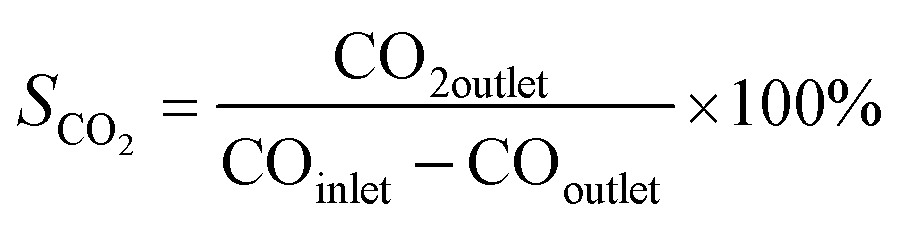
where COinlet and COoutlet represent moles of CO at the inlet and outlet, CO2 outlet represents moles C of CO2 at the outlet, Ni represents the molar fraction of product i (hydrocarbon or oxygenate), and ni is the carbon number of product i.
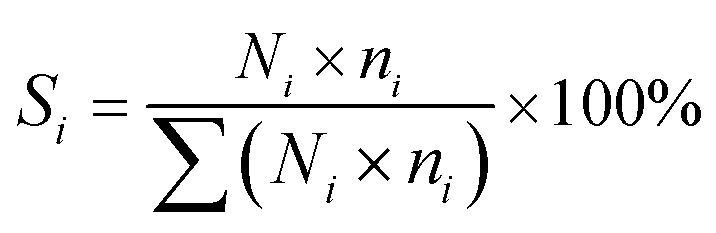
The catalytic performance changed dramatically during the initial stage, it was hard to perform a full analysis of all products in the short time period. In this case, the product distribution was obtained by the tail gas analysis (N2, CO, CO2 and C1 to C7 hydrocarbons). (C5+ + Oxy.) selectivity was calculated according to the following formula:(C5+ + Oxy.) selectivity = 100% − CO2 selectivity − C1–4 selectivity.where C5+ represents hydrocarbons with 5 or more than 5 carbons, Oxy. represents oxygenate products (mainly aldehydes and alcohols).
3. Results and discussion
3.1 Characterization
Fig. 1 shows the XRD patterns of various samples at different stages. As shown in Fig. 1a, the phase of CoMn-precursor was CoMn composite carbonate.46 After calcination at 350 °C for 5 h, the sample showed the diffraction peaks ascribed to Co2MnO4.5 (Fig. 1b). For the samples reduced both in FBR and SBR (Fig. 1c and d), diffraction peaks ascribing to CoxMn1−xO phase with lower metal valence were found.23,27,46 The average crystallite size of CoxMn1−xO was 11.2 and 12.6 nm in FBR and SBR, respectively, according to the calculation from Scherrer's equation using the (111) peak located at 2θ = 36.1°. For the spent catalysts (Fig. 1e and f), Co2C and Mn2CO3 were observed. The average crystallite size of Co2C was 18.8 and 20.1 nm in FBR and SBR, respectively. Due to the existence of residual SiO2 in FBR and solvent wax in SBR, the diffraction peaks of SiO2 and polywax were also detected in the reduced and spent catalysts.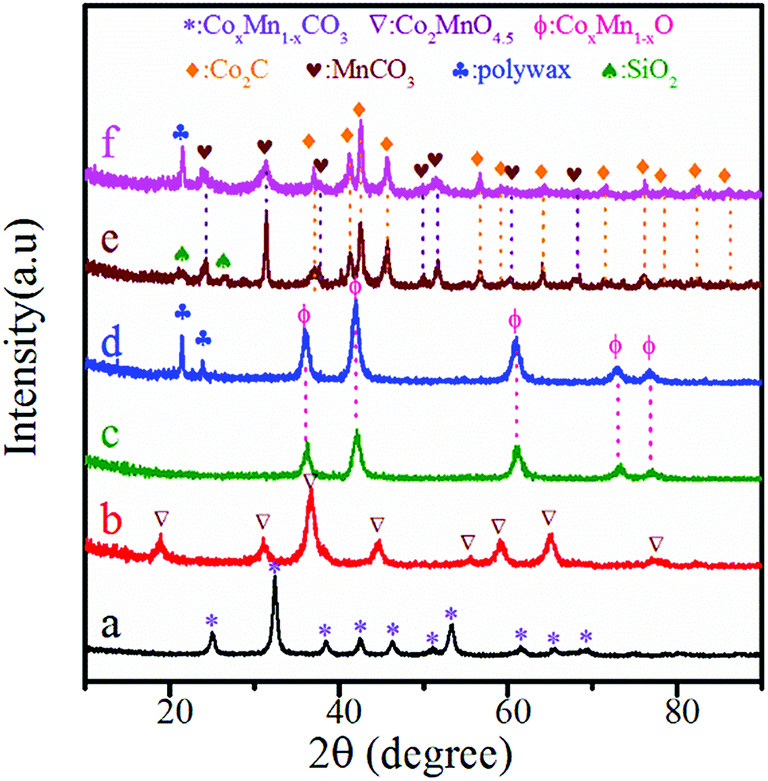 | ||
| Fig. 1 XRD patterns of CoMn catalysts after drying (a), calcination (b), reduction in FBR (c) and SBR (d) as well as reaction in FBR (e) and SBR (f). | ||
TEM images and particle size distribution for the dried and calcined samples are displayed in Fig. 2a–f. The dried samples exhibited an aggregation of CoMn composite carbonate nanoparticles with average size of 6.7 nm. The corresponding HRTEM image presented a lattice distance with d spacing of 2.78 Å, representing the (104) facet of the CoMn composite carbonate. After calcination at 350 °C for 5 h (Fig. 2d–f), sphere-like nanoparticles were observed with size distribution of 5–20 nm. Lattice distance with d spacing of 2.45, 2.90 and 4.61 Å was observed, corresponding to the (311), (220) and (111) facets of Co2MnO4.5, respectively. The TEM images and particle size distribution of reduced CoMn catalysts in FBR and SBR were compiled in Fig. 2g–l. All the reduced samples showed a homogeneous distribution of sphere-like CoxMn1−xO nanoparticle, and the d spacing of 2.13 and 2.42 Å represented the (200) and (111) facets, respectively. The average particle size of CoxMn1−xO in FBR (11.6 nm) was close to that in SBR (12.9 nm), which was in accordance with the XRD characterization.
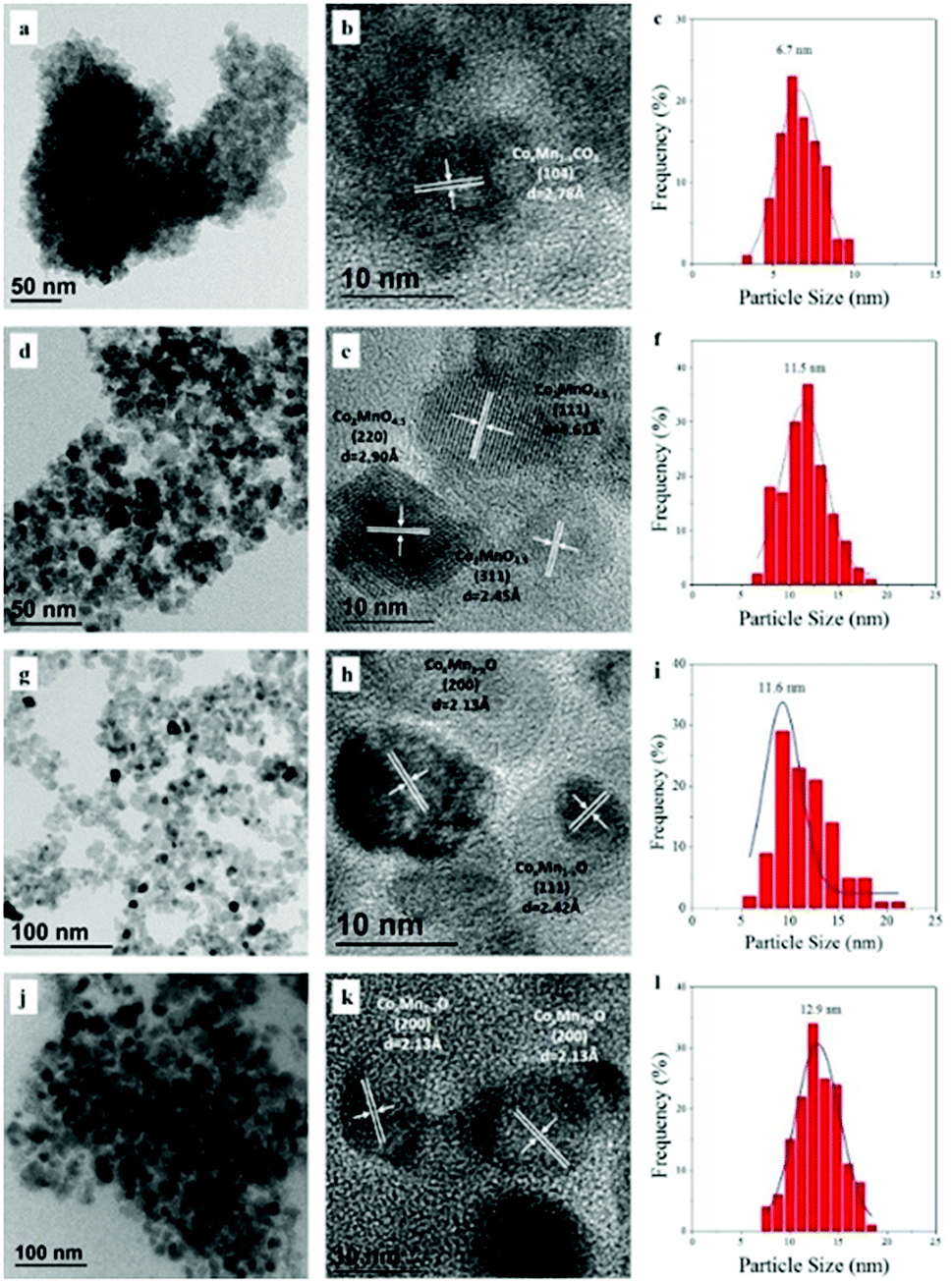 | ||
| Fig. 2 TEM images and particle size distribution for CoMn catalyst after dried (a–c), calcination (d–f) as well as reduction in FBR (g–i) and SBR (j–l). | ||
For the spent catalysts, as shown in Fig. 3, plenty of nanoprisms were observed for the samples withdrawn from both FBR and SBR. The magnified images of randomly selected area are shown in Fig. 3b, c, e and f, respectively. The nanoprisms with a lattice spacing of 2.18 and 2.43 Å corresponding to the (020) and (101) facets of Co2C were detected. Our previous studies demonstrated that Co2C nanostructures possessed strong facet effects during syngas conversion, and the Co2C (101) surface benefited olefins formation, while the formation of methane was unfavorable on both Co2C (101) and Co2C (020).23–25
 | ||
| Fig. 3 TEM images of the spent CoMn catalysts withdrawn from FBR (a–c) and SBR (d–f). | ||
3.2 Comparison of catalytic performance in SBR and FBR
The CoMn catalysts were firstly evaluated in both SBR and FBR to determine the influence of reactors on the catalytic activity and product selectivity at different reaction temperatures. As shown in Table 1, the catalytic activity was higher in SBR than that in FBR under the same reaction conditions. CO conversion of CoMn catalyst in SBR reached 29.5% at 250 °C, while only 22.1% was obtained in FBR. With the increase of the reaction temperature, CO conversion increased in both SBR and FBR. It was interesting to find that the activity of CoMn catalyst in SBR was more sensitive to reaction temperature, which increased quickly to 42.8% as temperature raised to 270 °C. While for FBR, CO conversion only slightly increased to 27.6% at 270 °C. CO2 selectivity varied between 37.7–44.6C% in FBR, while that in SBR kept nearly at 50C%. The high water-gas-shift (WGS) activity of Co2C-based catalysts was also reported in literatures, which suggested that syngas with low H2/CO ratio derived from coal or biomass is more appropriate to be the feedstock.28–31 In addition, the methane selectivity in FBR increased quickly from 2.6 to 6.1C% as rising the reaction temperature from 250 to 270 °C, while that in SBR only slight increased from 2.6 to 3.2C%. Moreover, the O/P ratio obtained in SBR was always slightly higher than that in FBR, suggesting that the SBR can alleviate the olefins re-adsorption and further hydrogenation. The CoMn catalyst in SBR was more beneficial for the formation of olefins and oxygenates as value-added products. The total selectivities to the desired olefins and oxygenates reached 88.8C% at 250 °C, and only slightly dropped to 86.2C% as rising the reaction temperature to 270 °C in SBR. For the FBR, the total selectivities to the desired olefins and oxygenates was lower than 84.0C% at different reaction temperatures.| Reactors | T (°C) | P (bar) | H2/CO | WHSV (ml h−1 gcat−1) | XCO (C%) | SCO2 (C%) | Product selectivity (C%, CO2-free) | Olefin/paraffin ratio | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Olefin | Oxy.a | Olefin + Oxy. | Paraffinb (CH4) | C2 | C3 | C4 | O/P | |||||||
| a Oxy., oxygenates (including alcohol and aldehyde).b The value in brackets represent the methane selectivity in total product (C%, CO2-free). | ||||||||||||||
| FBR | 250 | 5 | 0.5 | 2000 | 22.1 | 37.7 | 68.2 | 15.1 | 83.3 | 16.7 (2.6) | 4.4 | 11.3 | 7.7 | 4.0 |
| 260 | 5 | 0.5 | 2000 | 25.7 | 43.2 | 67.2 | 16.7 | 83.9 | 16.1 (4.6) | 2.7 | 10.8 | 7.4 | 4.2 | |
| 270 | 5 | 0.5 | 2000 | 27.6 | 44.6 | 66.3 | 17.4 | 83.7 | 16.3 (6.1) | 2.7 | 11.0 | 7.3 | 4.1 | |
| SBR | 250 | 5 | 0.5 | 2000 | 29.5 | 49.4 | 68.1 | 20.7 | 88.8 | 11.2 (2.6) | 7.7 | 11.5 | 8.0 | 6.1 |
| 260 | 5 | 0.5 | 2000 | 35.9 | 47.4 | 67.1 | 18.9 | 86.0 | 14.0 (2.7) | 5.4 | 11.3 | 7.7 | 4.8 | |
| 270 | 5 | 0.5 | 2000 | 42.8 | 49.2 | 65.7 | 20.5 | 86.2 | 13.8 (3.2) | 4.9 | 11.6 | 7.8 | 4.8 | |
| 250 | 5 | 0.5 | 1000 | 39.4 | 49.2 | 70.1 | 16.8 | 86.9 | 13.1 (3.2) | 5.7 | 11.1 | 7.5 | 5.4 | |
| 250 | 5 | 0.5 | 4000 | 15.5 | 46.6 | 65.1 | 24.0 | 89.1 | 10.9 (2.4) | 9.9 | 11.5 | 8.0 | 6.0 | |
| 250 | 5 | 0.75 | 4000 | 19.0 | 47.0 | 63.1 | 23.9 | 87.0 | 13.0 (3.2) | 7.5 | 9.5 | 6.6 | 4.9 | |
| 250 | 5 | 1.0 | 4000 | 23.3 | 47.2 | 55.4 | 29.5 | 84.9 | 15.1 (3.4) | 6.1 | 7.2 | 4.9 | 3.7 | |
| 250 | 7.5 | 0.5 | 4000 | 17.7 | 49.5 | 58.0 | 28.0 | 86.0 | 14.0 (2.8) | 7.2 | 7.8 | 5.3 | 4.1 | |
| 250 | 10 | 0.5 | 4000 | 18.8 | 49.7 | 58.6 | 25.6 | 84.2 | 15.8 (3.6) | 6.0 | 6.9 | 4.7 | 3.7 | |
As mentioned above, the SBR provides a better catalytic performance than FBR at different reaction temperatures. The influence of other operating conditions, such as WHSV, H2/CO ratio and pressure, on the catalytic performance in SBR was also investigated in detail. To study the effect of WHSV, the FTO catalytic performance was tested at 250 °C, 5 bar and H2/CO = 0.5 with WHSV ranged from 1000 to 4000 ml h−1 gcat−1. As shown in Table 1, CO conversion dramatically decreased from 39.4 to 15.5% with increasing WHSV. The selectivities for olefins and methane also dropped from 70.1 to 65.1C%, and 3.2 to 2.4C%, respectively. The oxygenate selectivity, however, increased from 16.8 to 24.0C%. The total selectivities to olefins and oxygenates slightly increased from 86.9 to 89.1C%.
The H2/CO ratio of feed gas and reaction pressure always possess a great effect on the catalytic performance for both cobalt- and iron-based catalyst.43–48 As showed in Table 1, a higher H2/CO ratio and reaction pressure contributed to a higher CO conversion, while CO2 selectivity stayed at around 50C%. Due to the enhancement of hydrogenation of CoMn catalyst at high H2/CO molar ratio or high reaction pressure, the olefin selectivity and O/P value decreased, while the paraffin and methane selectivity increased with the increase of H2/CO ratio and reaction pressure. However, the total product selectivities to olefins and oxygenates only slightly decreased.
Fig. 4 shows the chain growth probability of CoMn catalyst in SBR and FBR at different reaction conditions. A remarkable deviation from the typical ASF distribution was observed for CoMn catalyst performed in both SBR and FBR. The dash linear line was the ideal ASF line according to the chain growth probability obtained by the component with the carbon number of 3–7. The value of C1 and C2 negatively deviated from the value by ideal ASF prediction. A modified distribution model has been proposed based on the independent chain growth probability of α1 for C1 to C2, α2 for C2 to C3 and α3 for the growth of longer chains (n ≥ 3).46 The independent chain growth probability of α1, α2 and α3 of CoMn catalyst under various reaction conditions were listed in Table 2.
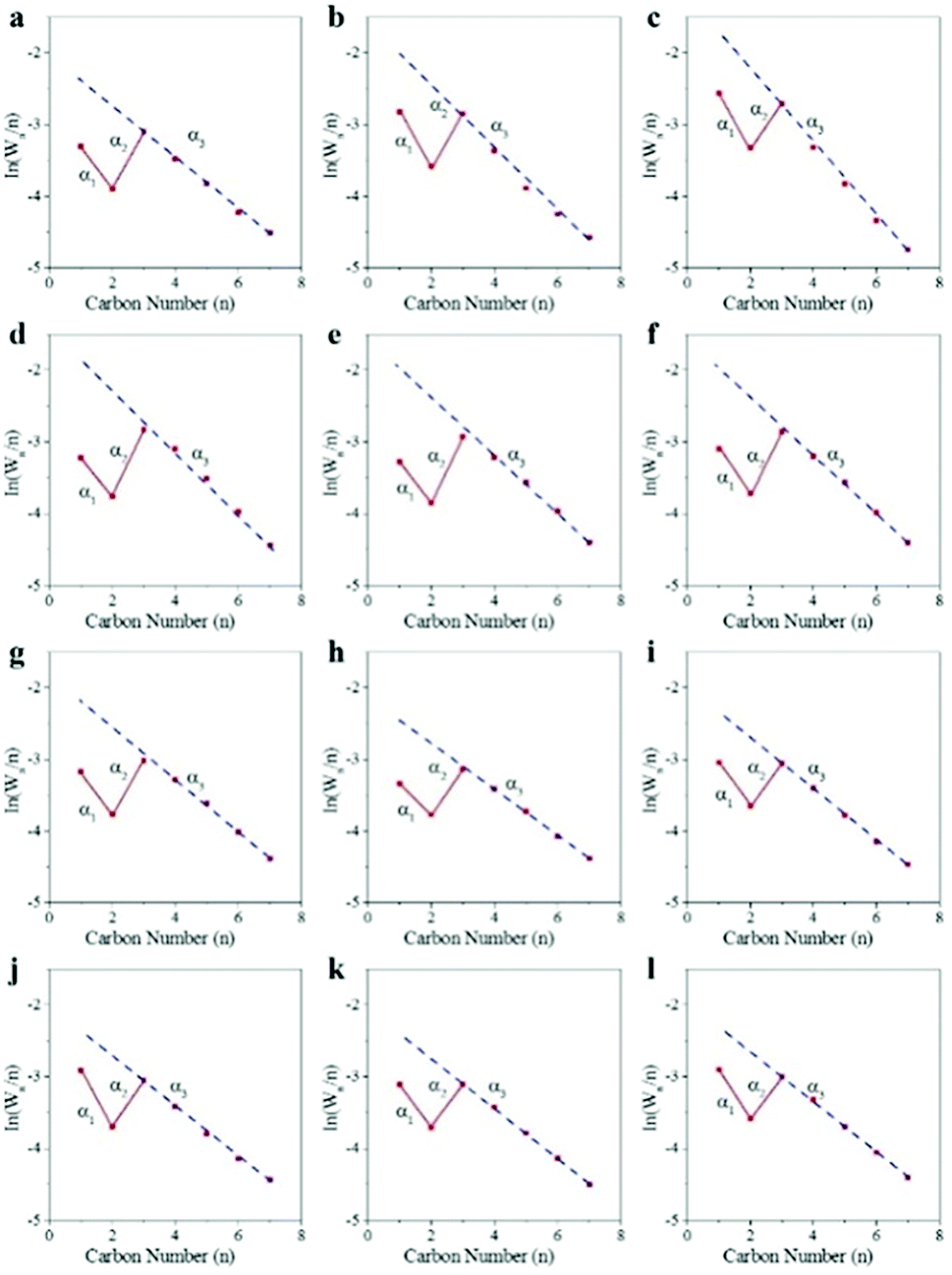 | ||
| Fig. 4 The chain growth probability of hydrocarbons over CoMn catalyst under various reaction conditions. (a) 250 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in FBR; (b) 260 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in FBR; (c) 270 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in FBR; (d) 250 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (e) 260 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (f) 270 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (g) 250 °C, 5 bar, 1000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (h) 250 °C, 5 bar, 4000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (i) 250 °C, 5 bar, 4000 ml h−1 gcat−1 and H2/CO = 0.75 in SBR; (j) 250 °C, 5 bar, 4000 ml h−1 gcat−1 and H2/CO = 1 in SBR; (k) 250 °C, 7.5 bar, 4000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (l) 250 °C, 10 bar, 4000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR. | ||
| Reactors | P (bar) | H2/CO | WHSV (ml h−1 gcat−1) | Temp. (°C) | Chain growth probability | ||
|---|---|---|---|---|---|---|---|
| α1 | α2 | α3 | |||||
| FBR | 5 | 0.5 | 2000 | 250 | 0.84 | 0.90 | 0.70 |
| 5 | 0.5 | 2000 | 260 | 0.78 | 0.87 | 0.65 | |
| 5 | 0.5 | 2000 | 270 | 0.75 | 0.84 | 0.60 | |
| SBR | 5 | 0.5 | 2000 | 250 | 0.84 | 0.89 | 0.70 |
| 5 | 0.5 | 2000 | 260 | 0.85 | 0.90 | 0.65 | |
| 5 | 0.5 | 2000 | 270 | 0.83 | 0.89 | 0.64 | |
| 5 | 0.5 | 1000 | 250 | 0.82 | 0.88 | 0.71 | |
| 5 | 0.5 | 4000 | 250 | 0.84 | 0.88 | 0.72 | |
| 5 | 0.75 | 4000 | 250 | 0.84 | 0.88 | 0.71 | |
| 5 | 1.0 | 4000 | 250 | 0.80 | 0.87 | 0.70 | |
| 7.5 | 0.5 | 4000 | 250 | 0.81 | 0.87 | 0.71 | |
| 10 | 0.5 | 4000 | 250 | 0.78 | 0.86 | 0.70 | |
Our previous study demonstrated Co2C nanoprisms effectively suppressed methane formation, because the barrier for methane formation from CH2 and H on Co2C (020) surface was the highest for all the selected facets.23 As for the C2 component, re-absorption of ethylene would be the main reason for the value of the C2 component to be much lower than the prediction.49–51 Due to the deviation for the C1 and C2 components, both α1 and α2 were higher than α3. With the increase of reaction temperature from 250 °C to 270 °C, the three chain growth probability α1, α2 and α3 in FBR dropped quickly from 0.84 to 0.75, 0.90 to 0.84 and 0.70 to 0.60, respectively. However, the effect of reaction conditions on chain growth probability in SBR is insignificant. Under the selected reaction conditions in SBR, the chain growth probability of α1 and α2 almost remain unchanged, and kept around 0.84 and 0.89 respectively. And the chain growth probability of α3 decreased from 0.7 to 0.64 as rising the reaction temperature from 250 °C to 270 °C. While the chain growth probability almost kept unchangeable at various WHSV for SBR. Due to the enhancement of hydrogenation of CoMn catalyst at high H2/CO molar ratio or high reaction pressure, the chain growth probability of α3 slightly decreased with the increase of H2/CO molar ratio or reaction pressure.
Fig. 5 shows the detailed distributions of the produced hydrocarbons under various reaction conditions. The distribution of hydrocarbons was in a narrow range with high selectivity to olefins. The formation of methane was greatly suppressed while the hydrocarbons with carbon number less than 20 accounted for more than 98 wt% in SBR. In addition, the olefins fraction reached 85 wt% at 250 °C. As rising the reaction temperature from 250 to 270 °C, the methane content only slightly increased from 3.7 to 4.5 wt%, and the olefin fraction almost kept steady around 84 wt% in SBR. However, the methane content increased greatly from 3.7 to 7.7 wt%, and the olefin fraction dropped from 80.0 to 72.9 wt% in FBR. The total olefin/paraffin (O/P) ratio kept at around 4.1 in FBR, and 4.8 in SBR. For the distribution of olefins, due to the decrease of chain growth probability under higher reaction temperature, C2–C10 olefin fraction in FBR increased from 78.6 to 90.4 wt% as rising the reaction temperature from 250 to 270 °C. However, a stable olefin distribution was observed in SBR, and the C2–C10 olefin fraction only slightly fluctuated between 87.4 and 91.5 wt%.
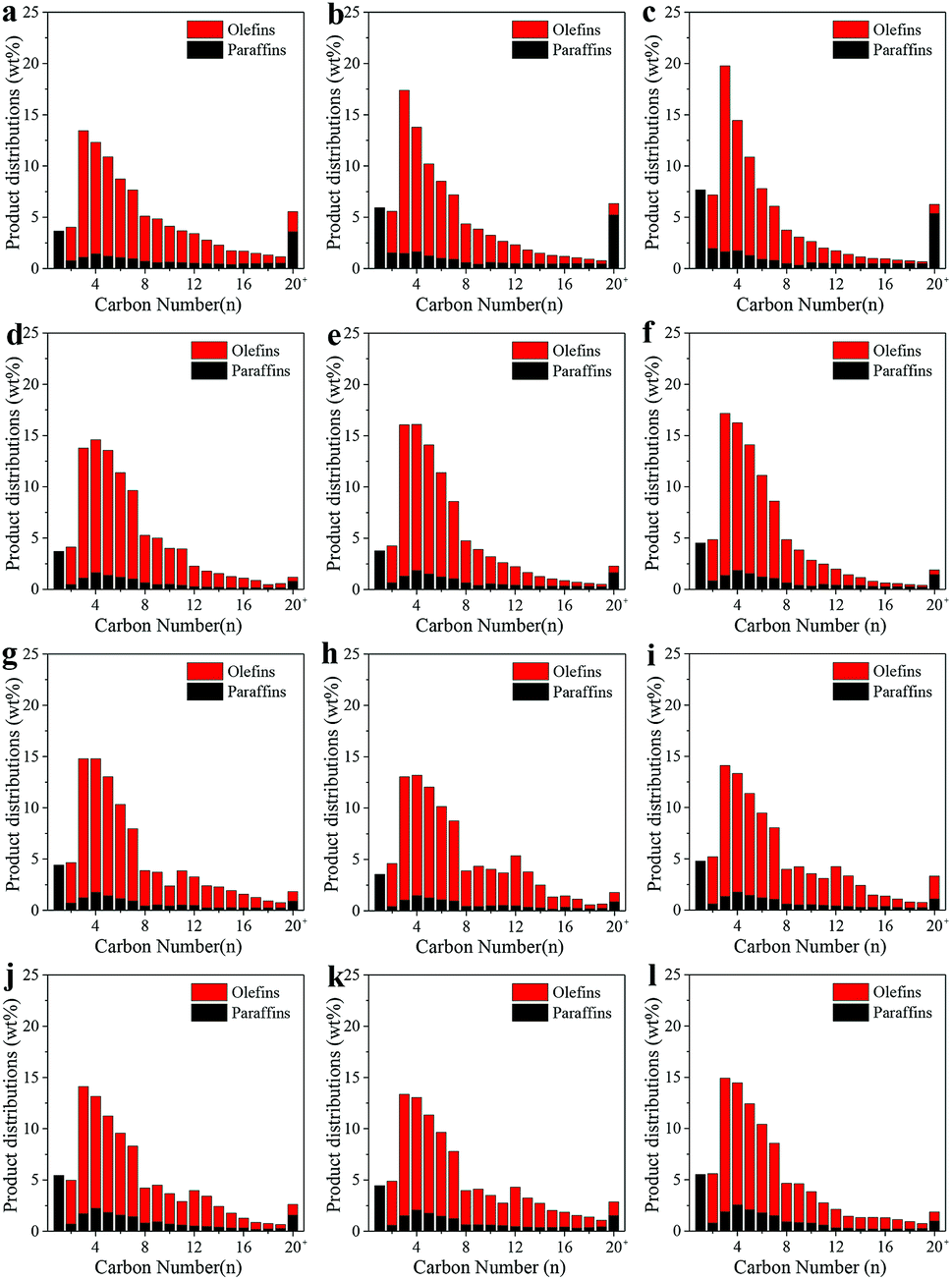 | ||
| Fig. 5 The distributions of hydrocarbons on CoMn catalyst under various reaction conditions. (a) 250 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in FBR; (b) 260 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in FBR; (c) 270 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in FBR; (d) 250 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (e) 260 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (f) 270 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (g) 250 °C, 5 bar, 1000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (h) 250 °C, 5 bar, 4000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (i) 250 °C, 5 bar, 4000 ml h−1 gcat−1 and H2/CO = 0.75 in SBR; (j) 250 °C, 5 bar, 4000 ml h−1 gcat−1 and H2/CO = 1 in SBR; (k) 250 °C, 7.5 bar, 4000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR; (l) 250 °C, 10 bar, 4000 ml h−1 gcat−1 and H2/CO = 0.5 in SBR. | ||
The influence of other operating conditions, such as WHSV, H2/CO ratio and pressure, on the detailed distributions of hydrocarbons in SBR was also investigated. The methane fraction in hydrocarbons decreased from 4.4 to 3.6 wt% as rising the WHSV from 1000 to 4000 ml h−1 gcat−1. The olefin fraction almost remained unchanged (∼84 wt%) at different WHSV. With the rising the H2/CO ratio from 0.5 to 1, the methane fraction in hydrocarbons increased from 3.55 to 5.45 wt% while the olefin fraction decreased from 85.7 to 78.6 wt%. When the reaction pressure increased from 5 to 10 bar, the methane fraction in hydrocarbons increased from 3.55 to 5.51 wt% while the olefin fraction decreased from 85.7 to 78.8 wt%.
3.3 Study of catalytic stability
Catalytic stability is an important parameter for industrial application. Herein, the stability of CoMn catalyst in SBR was investigated at 250 °C, 5 bar, H2/CO = 0.5 and a WHSV of 2000 ml h−1 gcat−1. Similar to that in FBR, CO conversion and product selectivity evolved dramatically during the initial stage (Fig. 6).23 After introducing the syngas, high selectivity to both C5+ and oxygenate was achieved, while the selectivities for C2–4= and CO2 were rather low in the first 20 h. In the following stage (20–40 h), CO conversion and CO2 selectivity increased dramatically. As for the distribution of the hydrocarbons, methane production first increased and then remained stable at around 2.6C%. In addition, the production of C2–4= increased at the expense of C5+ production. After 40 h, the catalytic performance seemed to be stable. CO conversion stabilized at about 30% and CO2 selectivity remained at about 45C%. Methane fraction stayed at around 2.6C%, C2–4= selectivity reached 30C% while selectivity to both C5+ and oxygenate remained at about 65C%. The ratio of C2=/C2o, C3=/C3o and C4=/C4o increased greatly in the first 20 h, and reached the highest point of 28, 47 and 29 at 20 h, then it decreased and kept at around 9, 13 and 8, respectively. The hydrocarbons distribution as a function of time-on-stream was plotted in Fig. 6c, which followed the typical ASF distribution in the first 20 h. Then the ln(Wn/n) value of C1 and C2 dropped, and the ln(Wn/n) value of C1 showed a deviation from the typical ASF distribution and was much lower than the predicted value. Fig. 6d shows the variation of chain growth probability (α3) for hydrocarbons as a function of time-on-stream. It seemed that the chain growth probability (α3) dropped at the first 20 h, and then increased and kept stable at 0.62 during the reaction process.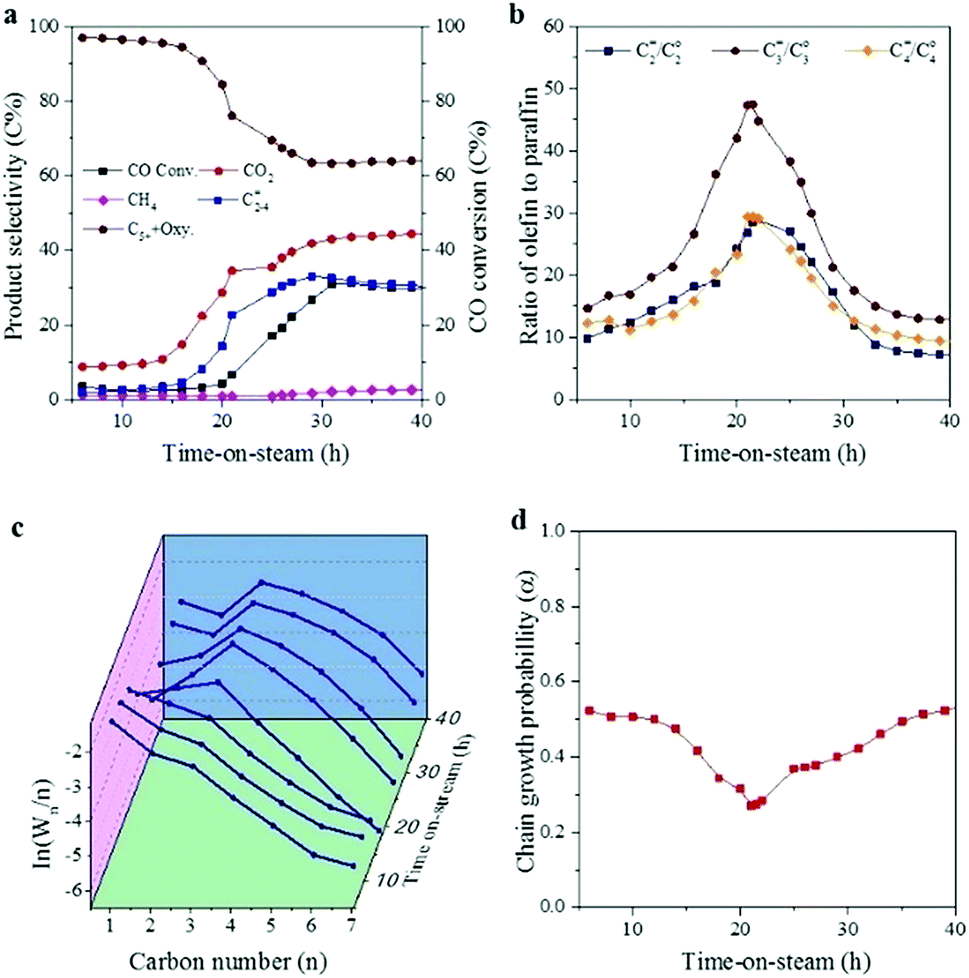 | ||
| Fig. 6 Catalytic performance of CoMn catalyst in the initial stage for the FTO process in SBR. (a) CO conversion and product selectivity as a function of time-on-steam. The selectivities to CH4, CO2, C2–4= and C5+ + Oxy. were calculated based on tail gas analysis. (b) O/P ratio as a function of time-on-stream. (c) Product plots (ln(Wn/n) and n) with time-on-stream. Wn is the weight fraction of a product with n number of carbon atoms. (d) Chain growth probability (α3) as a function of time-on-stream, obtained by fitting the results generated for chains of three to seven carbons using ASF model (reaction conditions: 250 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5). | ||
Fig. 7 shows the variation of CO conversion versus time-on-stream in the following stabilization stage for SBR and FBR. After the first 40 h, CO conversion gradually stabilized at around 24.6% in FBR and 29.6% in SBR. In the following test, CO conversion showed a downtrend in FBR, and decreased to 22.1% at 100 h, while it still kept rather stable around 29.8% in SBR. In addition, the methane selectivity also showed an increasing trend from 2.0 to 3.5C% in FBR, while it slightly dropped from 2.5 to 2.4C% in SBR. The fast deactivation rate in FBR is mainly due to the difficulty to avoid hot spots in FBR. It is noteworthy that the traditional slurry cobalt-based catalysts are always reduced ex situ at about 400 °C to form metallic cobalt as the active phase. Afterwards, the reduced catalyst was transferred into the SBR under inlet gas. The complicated reduced technology will increase the investment of FTS plant. However, the CoMn catalyst can be reduced in situ at mild condition (300 °C and 5 bar) in our study, which is more convenient for the industry application.
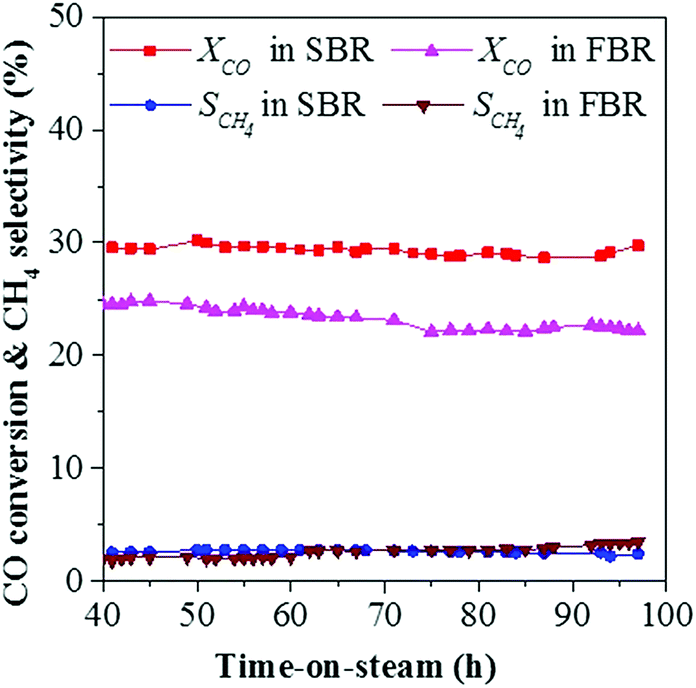 | ||
| Fig. 7 Catalytic stability of CoMn catalysts in FBR and SBR for the FTO process (reaction conditions: 250 °C, 5 bar, 2000 ml h−1 gcat−1 and H2/CO = 0.5). | ||
3.4 Co2C-based FTO process in SBR
As mentioned above, the SBR reactor represents a better catalytic performance than that in FBR. Specifically, a higher CO conversion, a lower methane selectivity and a higher total selectivity to both olefins and oxygenates were obtained in SBR. Some researchers reported that a relative lower catalytic activity was found in SBR than FBR for both cobalt-based and iron-based FTS.34–36 They ascribed this to the less reduction degree of the catalysts used in SBR, and the coverage of catalyst particle by liquid wax which would inhibit the mass-transfer efficiency. In order to enhance the catalytic activity in SBR, rising the reduction temperature or using reduction gas with stronger reduction capability were always adopt.52,53 However, in our study, an obvious higher catalytic activity was observed at the same reaction condition for Co2C-based FTO in SBR than that in FBR. This was due to the different generation route for active phase in FTS and Co2C-based FTO process. The active phase for the traditional FTS catalysts is recognized to be metallic cobalt or iron carbide,13,22 which is always generated during the reduction process. However, the reduction process for Co2C-based FTO catalyst was to form CoMn composited oxide with lower valence, which was subsequently carburized to Co2C nanoprisms with exposed special facets of (101) and (020) when exposing to syngas atmosphere.23–25 The product selectivity was also remarkable influenced by the reactors for the studied Co2C-based FTO catalyst. The SBR showed a selectivity of ∼87C% for the desired olefins and oxygenates, while it dropped to ∼83C% in FBR. The O/P ratio was also always higher in SBR (∼5) than that in FBR (∼4). The distribution of hydrocarbons in SBR was more stable under various reaction temperatures. Methane selectivity only slightly increased as rising the reaction temperature in SBR, while it increased dramatically in FBR. Similar phenomena was also identified in FTS process, indicating that a lower methane selectivity and a higher C5+ selectivity were obtained in SBR than that in FBR.34,35Olefins may re-adsorb upon catalyst surface and undergo secondary reactions such as further hydrogenation to paraffins as final products, or chain growth for longer carbon chain. In our study, the Co2C-based catalyst showed higher olefin selectivity and higher O/P ratio in SBR than that in FBR. Since the FTO process is strong exothermic, the main reason can be ascribed to the better heat transfer efficiency in SBR comparing with FBR. Some researchers found the olefin content in supercritical media was higher than that in traditional reaction phases.54–57 In addition, the olefin products can be effectively extracted and transported out of the catalyst particles by the supercritical fluid before they were re-adsorbed and hydrogenated to paraffin. Gao et al. also found improved olefin selectivity on Fe-based FTS catalyst was obtained by using polar solvent (PEG) as reaction medium, as olefin was more favored to transfer into PEG and remove from the catalyst surface.40,58 In the further study, the chemical process intensification in the SBR for Co2C-based FTO process will be a major consideration.
4. Conclusions
In summary, a Co2C-based catalyst derived from CoMn composite oxides was investigated in slurry bed reactor (SBR) for FTO reaction. At 250 °C, 5 bar and H2/CO = 0.5, the CO conversion reached 29.5%, and the total desire selectivity towards olefins and oxygenates was as high as 88.8C%. The formation of methane was greatly suppressed, resulting in the distribution of hydrocarbons deviated from the typical ASF distribution. After a mild in situ reaction in SBR at 300 °C, the CoMn composite oxide with low-valence were generated, and the Co2C nanoprisms with special exposed facts of (101) and (020) were further formed by subsequent exposing syngas. The effect of reaction temperature, pressure, H2/CO ratio and WHSV were detailed studied. It was found that the activity of CoMn catalyst in SBR was more sensitive to reaction temperature, and increased quickly to 42.8% as rising the reaction temperature to 270 °C. Comparing with that in FBR, a relative higher CO conversion and lower selectivity to methane and paraffin were obtained in SBR, which was mainly due to the better temperature control in SBR. Furthermore, the Co2C-based catalyst in SBR also exhibited higher stability than that in FBR and no obvious deactivation was found during 100 h time-on-stream. As the SBR was more suitable for the strong exothermic reaction, it suggests a promising route for the direct production of olefins via syngas for industrial application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key Research Program of Frontier Sciences, CAS (Grant No. QYZDB-SSW-SLH035), the “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA21020600), the National Key R&D Program of China (2017YFB0602202, 2017YFB0602203, 2018YFB0604700), Natural Science Foundation of China (21573271, 91545112, 21703278, 21776296).References
- H. M. T. Galvis and K. P. D. Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef
.
- W. Chen, T. Lin, Y. Dai, Y. An, F. Yu, L. Zhong, S. Li and Y. Sun, Catal. Today, 2018, 311, 8–22 CrossRef CAS
- Y. An, T. Lin, F. Yu, Y. Yang, L. Zhong, M. Wu and Y. Sun, Sci. China: Chem., 2017, 60, 887–903 CrossRef CAS
- K. Xiao, Z. Bao, X. Qi, X. Wang, L. Zhong, K. Fang, M. Lin and Y. Sun, Chin. J. Catal., 2013, 34, 116–129 CrossRef CAS
- Y. Dai, F. Yu, Z. Li, Y. An, T. Lin, Y. Yang, L. Zhong, H. Wang and Y. Sun, Chin. J. Chem., 2017, 35, 918–926 CrossRef CAS
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou and M. Li, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725–4728 CrossRef CAS PubMed
- A. Corma, E. Corresa, Y. Mathieu, L. Sauvanaud, S. Al-Bogami, M. S. Al-Ghrami and A. Bourane, Catal. Sci. Technol., 2017, 7, 12–46 RSC
- V. S. Sikarwar, M. Zhao, P. Clough, J. Yao, X. Zhong, M. Z. Memon, N. Shah, E. J. Anthony and P. S. Fennell, Energy Environ. Sci., 2016, 9, 2939–2977 RSC
- L. Liu, Y. Cao, D. Ma, Q. Liu and J. Yang, RSC Adv., 2017, 7, 55450–55458 RSC
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS
- J. Li, Y. Wei, J. Chen, S. Xu, P. Tian, X. Yang, B. Li, J. Wang and Z. Liu, ACS Catal., 2015, 5, 661–665 CrossRef
- E. Iglesia, Appl. Catal., A, 1997, 161, 59–78 CrossRef CAS
- Q. Zhang, W. Deng and Y. Wang, J. Energy Chem., 2013, 22, 27–38 CrossRef CAS
- O. Borg, S. Eri, E. Blekkan, S. Storsater, H. Wigum, E. Rytter and A. Holmen, J. Catal., 2007, 248, 89–100 CrossRef CAS
- D. B. Bukur and C. Sivaraj, Appl. Catal., A, 2002, 231, 201–214 CrossRef CAS
- C. K. Roferdepoorter, Chem. Rev., 1981, 81, 447–474 CrossRef CAS
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, Catal. Sci. Technol., 2017, 7, 4580–4598 RSC
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, ChemCatChem, 2012, 4, 835–838 Search PubMed
- H. M. Torres Galvis, A. C. J. Koeken, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Catal., 2013, 303, 22–30 CrossRef CAS
- A. C. Koeken, H. M. Torres Galvis, T. Davidian, M. Ruitenbeek and K. P. de Jong, Angew. Chem., Int. Ed., 2012, 124, 7302–7305 CrossRef
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y. Li, Q. Zhang, X. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–86 CrossRef CAS PubMed
- Z. Li, L. Zhong, F. Yu, Y. An, Y. Dai, Y. Yang, T. Lin, S. Li, H. Wang and P. Gao, ACS Catal., 2017, 7, 3622–3631 CrossRef CAS
- Z. Li, T. Lin, F. Yu, Y. An, Y. Dai, S. Li, L. Zhong, H. Wang, P. Gao and Y. Sun, ACS Catal., 2017, 7, 8023–8032 CrossRef CAS
- Y. An, Y. Zhao, F. Yu, T. Lin, Y. Lu, S. Li, Z. Li, Y. Dai, X. Wang, H. Wang, L. Zhong and Y. Sun, J. Catal., 2018, 366, 289–299 CrossRef CAS
- X. Wang, W. Chen, T. Lin, J. Li, F. Yu, Y. An, Y. Dai, H. Wang, L. Zhong and Y. Sun, Chin. J. Catal., 2018, 39, 1869–1880 CrossRef CAS
- B. H. Davis, Catal. Today, 2002, 71, 249–300 CrossRef CAS
- A. P. Steynberg, M. E. Dry, B. H. Davis and B. B. Breman, in Stud. Surf. Sci. Catal., ed. A. Steynberg and M. Dry, Elsevier, 2004, vol. 152, pp. 64–195 Search PubMed
- F. G. Botes, J. W. Niemantsverdriet and J. van de Loosdrecht, Catal. Today, 2013, 215, 112–120 CrossRef CAS
- E. van Steen, M. Claeys, K. P. Möller and D. Nabaho, Appl. Catal., A, 2018, 549, 51–59 CrossRef CAS
- V. A. D. L. P. O'Shea, M. C. Álvarez-Galván, J. M. Campos-Martín and J. L. G. Fierro, Appl. Catal., A, 2007, 326, 65–73 CrossRef
- S. T. Sie and R. Krishna, Appl. Catal., A, 1999, 186, 55–70 CrossRef CAS
- D. B. Bukur, A. Xiaosu Lang and L. Nowicki, Ind. Eng. Chem. Res., 2005, 44, 6038–6044 CrossRef CAS
- Y. Sun, G. Yang, G. Sun, Z. Sun and L. Zhang, Envrion. Prog. Sustain., 2018, 37, 553–561 CrossRef CAS
- C. N. Satterfield, G. A. H. Jr, H. G. Stenger, J. L. Carter and R. J. Madon, Ind. Eng. Chem. Fundam., 1985, 24, 450–454 CrossRef CAS
- D. B. Bukur, S. A. Patel and X. Lang, Appl. Catal., 1990, 61, 329–349 CrossRef CAS
- S. Chambrey, P. Fongarland, H. Karaca, S. Piché, A. Griboval-Constant, D. Schweich, F. Luck, S. Savin and A. Y. Khodakov, Catal. Today, 2011, 171, 201–206 CrossRef CAS
- J. K. Neathery, R. L. Spicer, D. E. Sparks and B. H. Davis, Stud. Surf. Sci. Catal., 2001, 139, 407–414 CrossRef CAS
- J. Gao, B. Wu, L. Zhou, Y. Yang, X. Hao, J. Xu, Y. Xu, L. Cao and Y. Li, Catal. Commun., 2011, 12, 1466–1470 CrossRef CAS
- J. Gao, W. U. Baoshan, L. Zhou, Y. Yong, H. Xu and Y. Li, Chin. J. Catal., 2011, 32, 1790–1802 CrossRef CAS
- A. K. Rausch, L. Schubert, R. Henkel, E. van Steen, M. Claeys and F. Roessner, Catal. Today, 2016, 275, 94–99 CrossRef CAS
- K. Xiao, X. Qi, Z. Bao, X. Wang, L. Zhong, K. Fang, M. Lin and Y. Sun, Catal. Sci. Technol., 2013, 3, 1591–1602 RSC
- K. Xiao, Z. Bao, X. Qi, X. Wang, L. Zhong, K. Fang, M. Lin and Y. Sun, J. Mol. Catal. A: Chem., 2013, 378, 319–325 CrossRef CAS
- Y. Yang, L. Wang, K. Xiao, T. Zhao, H. Wang, L. Zhong and Y. Sun, Catal. Sci. Technol., 2015, 5, 4224–4232 RSC
- F. Yu, T. Lin, X. Wang, S. Li, Y. Lu, H. Wang, L. Zhong and Y. Sun, Appl. Catal., A, 2018, 563, 146–153 CrossRef CAS
- M. Ding, Y. Yang, Y. Li, T. Wang, L. Ma and C. Wu, Appl. Energy, 2013, 112, 1241–1246 CrossRef CAS
- K. Keyvanloo, S. J. Lanham and W. C. Hecker, Catal. Today, 2016, 270, 9–18 CrossRef CAS
- E. Iglesia, S. C. Reyes and R. J. Madon, J. Catal., 1991, 129, 238–256 CrossRef CAS
- J. Patzlaff, Y. Liu, C. Graffmann and J. Gaube, Catal. Today, 2002, 71, 381–394 CrossRef CAS
- G. V. D. Laan and A. A. C. M. Beenackers, Catal. Rev., 1999, 41, 255–318 CrossRef
- D. B. Bukur, L. Nowicki and S. A. Patel, Can. J. Chem. Eng., 2010, 74, 399–404 CrossRef
- D. B. Bukur, L. Nowicki and X. Lang, Catal. Today, 1995, 24, 111–119 CrossRef CAS
- R. M. M. Abbaslou, J. S. S. Mohammadzadeh and A. K. Dalai, Fuel Process. Technol., 2009, 90, 849–856 CrossRef
- W. Linghu, X. Li, K. Asami and K. Fujimoto, Fuel Process. Technol., 2004, 85, 1121–1138 CrossRef CAS
- K. Yokota and K. Fujimoto, Fuel, 1989, 68, 255–256 CrossRef CAS
- W. Yuan, G. C. Vaughan, C. B. Roberts and M. R. Eden, in Computer Aided Chemical Engineering, ed. E. N. Pistikopoulos, M. C. Georgiadis and A. C. Kokossis, Elsevier, 2011, vol. 29, pp. 1929–1933 Search PubMed
- J. Gao, B. Wu, L. Zhou, Y. Yang, X. Hao, J. Xu, Y. Y. Xu and Y. Li, Ind. Eng. Chem. Res., 2012, 51, 11618–11628 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2019 |