
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFungal community analysis in seawater of the Mariana Trench as estimated by Illumina HiSeq†
Zhi-Peng Wang‡
b,
Zeng-Zhi Liu‡c,
Yi-Lin Wangd,
Wang-Hua Bic,
Lu Liuc,
Hai-Ying Wangb,
Yuan Zhengb,
Lin-Lin Zhange,
Shu-Gang Hue,
Shan-Shan Xu *c and
Peng Zhang*a
*c and
Peng Zhang*a
aTobacco Research Institute, Chinese Academy of Agricultural Sciences, Qingdao, 266101, China. E-mail: zhangpeng@caas.cn
bKey Laboratory of Sustainable Development of Polar Fishery, Ministry of Agriculture and Rural Affairs, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, 266071, China
cSchool of Medicine and Pharmacy, Ocean University of China, Qingdao, 266003, China. E-mail: xushanshan328@163.com
dCollege of Science, China University of Petroleum, Qingdao, Shandong 266580, China
eCollege of Chemistry & Environmental Engineering, Shandong University of Science & Technology, Qingdao, 266510, China
First published on 28th February 2019
Abstract
This study assessed the diversity and distribution of fungal communities in thirteen marine seawater samples from four sites (L1, L3, L4 and L7) of the Mariana Trench, with a depth range of 1000–4000 meters, using Illumine Hiseq sequencing with fungal-specific primers targeting the internal transcribed spacer (ITS) region of the ribosomal rRNA gene. Sedimentary fungal communities showed high diversity with 209![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 880 reads belonging to 91 operational taxonomic units (OTUs). Of these OTUs, 45 belonged to the Ascomycota, 37 to Basidiomycota, 3 to Chytridiomycota, 1 to Glomeromycota, 1 to Cryptomycota, and 4 to unknown fungi. The major fungal orders included Saccharomycetales and Sporidiobolales. The commonly found fungal genera were Candida, Malassezia and Cryptococcus. These results suggest the existence of diverse fungal communities in the Mariana Trench marine seawater, which can be considered as a useful community model for further ecological and evolutionary study of fungi in the trench.
880 reads belonging to 91 operational taxonomic units (OTUs). Of these OTUs, 45 belonged to the Ascomycota, 37 to Basidiomycota, 3 to Chytridiomycota, 1 to Glomeromycota, 1 to Cryptomycota, and 4 to unknown fungi. The major fungal orders included Saccharomycetales and Sporidiobolales. The commonly found fungal genera were Candida, Malassezia and Cryptococcus. These results suggest the existence of diverse fungal communities in the Mariana Trench marine seawater, which can be considered as a useful community model for further ecological and evolutionary study of fungi in the trench.
Introduction
As is well known, fungi produce a vast range of secondary metabolites, some of which possess high value in pharmaceutical applications, such as penicillin.1 More specifically, marine fungi are also believed to be a good resource for novel biological natural products.2 As previously described, some interesting compounds, such as cytoglobosins and halovirs, had been isolated from marine fungi.1 Marine fungi are a taxonomically diverse group and exist in various niches within marine ecosystems including marine sediments, sea water, corals, algae, coastal environments and even some extreme environments.3,4 They not only serve as major decomposers of organic matters in marine ecosystems5 but also have been reported as parasites, pathogens, and mutualists of other marine organisms.3,6–8Deep-sea environments have extreme settings, including extreme low temperatures, low nutrient, absence of sunlight, and high hydrostatic pressure.9 Recent studies showed great microbial diversity in sediments of many deep-sea environments, such as the Peru Trench,10 Pacific Ocean,8,11 India Ocean,12,13 Challenger Deep,14 Arctic Ocean,3 Saint Helena Bay,15 and South China Sea.16,17 In these deep-sea environments, there are abundant and diverse fungal resources.
The average depth of oceans is 4000 meters, and the deepest recorded part of the ocean is 11000 meter depth down the Mariana Trench in the central west Pacific Ocean.18 The Mariana Trench is a non-accretionary convergent plate margin between the Philippine sea plate and subducting Pacific plate.19 Due to the special ecological environments around the trench, it may contain abundant fungal resources. In the last decades, there were several studies on the diversity of microbes including actinomycetes,20 bacteria, fungi and Picoeukaryotic isolated from sediments and seawaters in the Pacific Ocean and Mariana Trench8,11,14,20–22 by culture and molecular approches.
The advent of high-throughput sequencing technology (HiSeq) have promoted the significant advances in microbial community analysis. With the increased computational capabilities, HiSeq can simultaneously acquire the sequences of large numbers of DNA fragments. By adapting the HiSeq technologies, culture-independent targeted PCR amplification and sequencing of the ITS rRNA gene continues to offer a powerful and economic way to gain insight into the fungal community in deep-sea samples. Although the HiSeq have been used to reveal the presence of diverse mocrobiol in sediments and seawater of the Pacific Ocean even the Mariana Trench,11,21,22 the study of fungal diversity using the Hiseq method performed for analyzing fungal diversities in the Mariana Trench seawater is still less. In the present study, we investigated fungal-community structures in deep seawater of the Mariana Trench with different depths (seawater depth ranging from 1000 to 4000 m) from four locations (L1, L3, L4 and L7, Fig. 1) using Illumina Hiseq technique, which is used to study the environmental microbial diversity and community composition difference by detecting the sequence variation and abundance of the target region. This approach contains a large amount of sequencing and a short cycle, which can provide more comprehensive information on the microorganisms in various environmental samples.
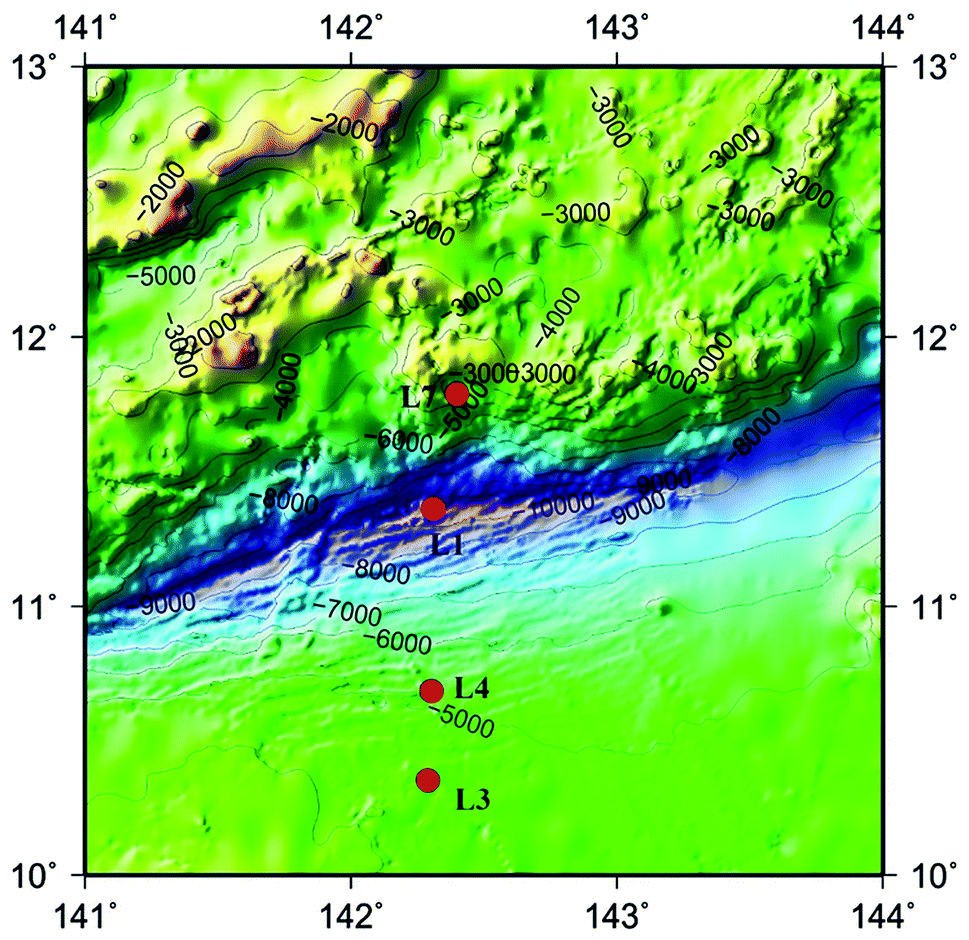 | ||
| Fig. 1 Map of Mariana Trench showing the sites where seawater samples were collected in this study. Four sampling sites are indicated by red dots. | ||
Results
Sequence data
The raw data from 13 seawater samples consisted of 210290 sequences, of which 209880 sequences were retained after removing sequences with different tags at each end for quality filtering and denoising. After removing singletons, chimeric sequences and OTUs of nonfungal organisms or those with unreliable BLAST matches (max score below 200 or aligned query sequence below 200 bp), a total of 91 fungal OTUs from 209880 sequences were included in the final matrix. The length of these sequences ranged from 241 to 315 bp. The number of OTUs in different sediment samples ranged from 34 to 75 (Table 1).
| Sampling site | Sampling code | Sampling date | Coordination | Depth (m) | Temperature (°C) | Salinity (psu) | OTU | Shannon | Simpson | Chao1 | ACE | Goods coverage |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SiteL1 | M1.1 | 2016.9.12 | 11°21.47′N; 142°24.794′E | 1000 | 4.79 | 34.53 | 65 | 2.25 | 0.64 | 59.20 | 55.84 | 0.996 |
| M1.2 | 2016.9.12 | 11°21.776′N; 142°28.921′E | 2000 | 2.30 | 34.62 | 64 | 2.61 | 0.74 | 45.77 | 51.67 | 0.997 | |
| M1.3 | 2016.9.12 | 11°21.47′N; 142°24.794′E | 3000 | 1.68 | 34.66 | 54 | 2.91 | 0.79 | 38.50 | 35.76 | 0.998 | |
| M1.4 | 2016.9.12 | 11°21.136′N; 142°25.823′E | 4000 | 1.49 | 34.68 | 50 | 3.28 | 0.83 | 92.5 | 64.75 | 0.995 | |
| SiteL3 | M3.1 | 2016.9.14 | 10°21.359′N; 142°16.413′E | 1000 | 4.81 | 34.54 | 70 | 2.64 | 0.74 | 58.14 | 59.38 | 0.995 |
| M3.2 | 2016.9.14 | 10°21.442′N; 142°16.545′ | 2000 | 2.26 | 34.63 | 49 | 2.60 | 0.74 | 89.50 | 80.94 | 0.995 | |
| M3.4 | 2016.9.14 | 10°21.384′N; 142°16.809′E | 4000 | 1.49 | 34.67 | 42 | 1.78 | 0.59 | 37.55 | 39.32 | 0.997 | |
| SiteL4 | M4.2 | 2016.9.14 | 10°41.150′N; 142°18.197′E | 2000 | 2.19 | 34.63 | 44 | 2.54 | 0.69 | 45.75 | 49.42 | 0.996 |
| M4.3 | 2016.9.14 | 10°41.150′N; 142°18.197′E | 3000 | 1.68 | 34.66 | 43 | 2.62 | 0.79 | 31.42 | 29.43 | 0.998 | |
| M4.4 | 2016.9.14 | 10°41.150′N; 142°18.197′E | 4000 | 1.47 | 34.69 | 60 | 3.75 | 0.88 | 57.39 | 55.85 | 0.997 | |
| SiteL7 | M7.1 | 2016.9.16 | 11°47.355′N; 142°23.915′E | 1000 | 4.89 | 34.54 | 75 | 2.74 | 0.71 | 67.45 | 66.38 | 0.996 |
| M7.2 | 2016.9.16 | 11°47.355′N; 142°23.915′E | 2000 | 2.31 | 34.63 | 36 | 2.76 | 0.78 | 25.50 | 26.70 | 0.999 | |
| M7.3 | 2016.9.16 | 11°47.355′N; 142°23.915′E | 3000 | 1.66 | 34.67 | 34 | 2.33 | 0.76 | 31.17 | 28.84 | 0.998 |
Fungal diversity and community structure in seawater
Based on BLASTn searches in GenBank, 91 OTUs were identified at different levels of taxonomic precision, and most of these OTUs had their best matches against GenBank accessions. Information of these 91 OTUs is presented in Table S2.† These 91 OTUs spanned 4 phyla, 20 classes, 28 orders, 37 families, 42 genera and 43 species. Out of the 91 OTUs, 45 belonged to the Ascomycota, 37 to Basidiomycota, 3 to Chytridiomycota,1 to Glomeromycota, 1 to Cryptomycota and 4 to unknown fungi (Fig. 2 and Table S2†).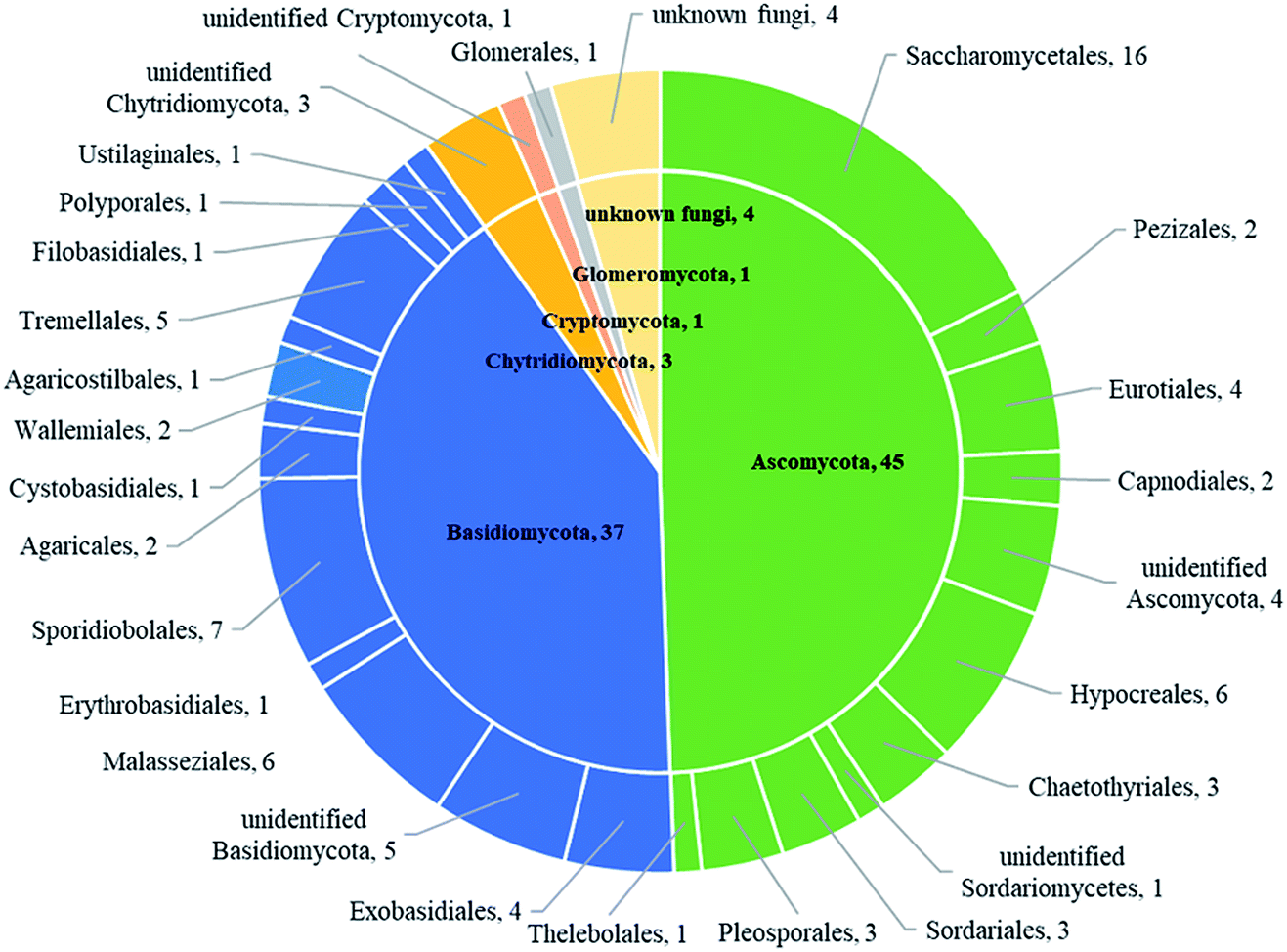 | ||
| Fig. 2 The taxonomic distribution of 91 OTUs at the phylum and order level. | ||
Sequences matching with Ascomycota had high affinities with 6 known classes (Sordariomycetes, Dothideomycetes, Saccharomycetes, Eurotiomycetes, Leotiomycetes and Pezizomycetes), with Sordariomycetes being the most abundant and diverse class (Table 2). Sequences matching with Basidiomycota had high affinities with 9 known classes (Malasseziomycete, Wallemiomycetes, Exobasidiomycetes, Microbotryomycetes, Cystobasidiomycetes, Ustilaginomycetes, Agaricomycetes, Tremellomycetes and Moniliellomycetes) with Malasseziomycete being the most diverse and abundant class (Table 2). Sequences from Ascomycota matched 11 known orders (Hypocreales, Sordariales, Pleosporales, Capnodiales, Saccharomycetales, Chaetothyriales, Eurotiales, Thelebolales, Pezizales, Venturiales, and Hypocreomycetidae), with Hypocreales being the most abundant and the most diverse, followed by Saccharomycetales. Sequences from Basidiomycota matched 14 known orders (Malasseziales, Sporidiobolaceae, Agaricales, Polyporales, Agaricostilbales, Cystobasidiales, Erythrobasidiales, Tremellales, Exobasidiales, Wallemiales, Ustilaginales, Entylomatales, Moniliellales, and Filobasidiales), with Malasseziales being the most abundant and diverse order. Fungal communities in sample M1.4 were most diverse and stretched 23 orders that were detected in the present study, followed by M7.1 which had 22 orders. Besides, fungal diversity in sampling site L1 was most diverse and had 26 orders that were detected, followed by L7 which contained 25 orders, and there were 24 orders in both L3 and L4.
| Site L1 | Site L3 | Site L4 | Site L7 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M1.1 | M1.2 | M1.3 | M1.4 | M3.1 | M3.2 | M3.4 | M4.2 | M4.3 | M4.4 | M7.1 | M7.2 | M7.3 | |
| a The 13 columns provide a taxonomic overview of the fungal communities found in each of the 13 seawater samples, which are represented as the percentage of reads. | |||||||||||||
| Ascomycota | 61.53 | 69.91 | 69.42 | 57.32 | 54.61 | 53.84 | 62.81 | 17.28 | 48.88 | 39.20 | 18.13 | 35.41 | 39.55 |
| Sordariomycetes | 53.82 | 3.73 | 17.40 | 21.78 | 40.05 | 43.44 | 55.32 | 4.18 | 24.64 | 12.55 | 3.06 | 14.08 | 22.95 |
| Hypocreales | 53.81 | 3.72 | 17.40 | 21.44 | 40.02 | 43.43 | 55.31 | 4.18 | 24.64 | 12.46 | 2.86 | 14.08 | 22.95 |
| Sordariales | 0.01 | 0.01 | — | 0.34 | 0.03 | 0.01 | 0.01 | — | — | 0.09 | 0.12 | — | — |
| Hypocreomycetidae | — | — | — | — | — | — | — | — | — | — | 0.08 | — | — |
| Dothideomycetes | 1.54 | 1.01 | 41.64 | 3.28 | 1.10 | 1.53 | 0.35 | 3.68 | 9.79 | 5.32 | 3.06 | 9.50 | 15.46 |
| Pleosporales | 1.02 | 0.41 | 39.63 | 1.03 | 0.26 | 0.13 | 0.18 | 0.17 | 0.04 | 0.83 | 1.91 | 0.05 | 15.13 |
| Capnodiales | 0.52 | 0.55 | 1.99 | 2.22 | 0.81 | 1.40 | 0.17 | 3.51 | 9.74 | 3.72 | 1.03 | 9.44 | 0.33 |
| Venturiales | — | 0.05 | 0.02 | 0.03 | 0.03 | 0.01 | 0.05 | — | 0.01 | 0.77 | 0.12 | 0.01 | — |
| Saccharomycetes | 2.64 | 43.16 | 1.31 | 16.06 | 3.04 | 2.53 | 3.15 | 2.35 | 0.26 | 12.74 | 5.31 | 9.22 | 0.28 |
| Saccharomycetales | 2.64 | 43.16 | 1.31 | 16.06 | 3.04 | 2.53 | 3.15 | 2.35 | 0.26 | 12.74 | 5.31 | 9.22 | 0.28 |
| Eurotiomycetes | 2.56 | 2.28 | 8.58 | 4.73 | 8.42 | 5.68 | 3.41 | 6.54 | 13.52 | 3.76 | 4.59 | 0.47 | 0.80 |
| Chaetothyriales | 0.13 | 0.44 | 3.05 | 0.43 | 2.44 | 0.15 | 2.70 | 0.12 | 7.35 | 0.23 | 0.36 | 0.13 | 0.35 |
| Eurotiales | 2.43 | 1.84 | 5.53 | 4.30 | 5.98 | 5.53 | 0.71 | 6.42 | 6.17 | 3.53 | 4.23 | 0.34 | 0.45 |
| Leotiomycetes | — | 0.02 | — | — | — | — | — | — | — | — | — | 1.23 | — |
| Thelebolales | — | 0.02 | — | — | — | — | — | — | — | — | — | 1.23 | — |
| Pezizomycetes | — | 0.01 | — | — | 0.05 | — | — | — | — | — | — | — | — |
| Pezizales | — | 0.01 | — | — | 0.05 | — | — | — | — | — | — | — | — |
| Unidentified Ascomycota | 0.97 | 19.70 | 0.49 | 11.46 | 1.95 | 0.65 | 0.53 | 0.58 | 0.67 | 4.83 | 2.11 | 0.91 | 0.06 |
| Basidiomycota | 36.85 | 24.15 | 8.30 | 36.84 | 14.98 | 40.02 | 4.93 | 17.11 | 18.68 | 40.99 | 77.61 | 63.65 | 59.12 |
| Malasseziomycete | 33.74 | 23.14 | 7.27 | 35.38 | 14.21 | 37.28 | 4.56 | 16.82 | 18.57 | 38.86 | 76.13 | 47.43 | 56.97 |
| Malasseziales | 33.74 | 23.14 | 7.27 | 35.38 | 14.21 | 37.28 | 4.56 | 16.82 | 18.57 | 38.86 | 76.13 | 47.43 | 56.97 |
| Microbotryomycetes | 2.35 | 0.09 | 0.07 | 0.23 | 0.10 | 0.02 | 0.06 | 0.06 | — | 0.12 | 0.17 | 0.12 | — |
| Sporidiobolaceae | 2.35 | 0.09 | 0.07 | 0.23 | 0.10 | 0.02 | 0.06 | 0.06 | — | 0.12 | 0.17 | 0.01 | — |
| Agaricomycetes | 0.19 | 0.04 | — | 0.06 | 0.16 | 2.48 | 0.01 | — | 0.02 | 0.61 | — | — | |
| Agaricales | — | 0.04 | — | 0.02 | — | 2.48 | 0.01 | — | — | 0.02 | 0.61 | — | — |
| Polyporales | 0.19 | — | — | 0.04 | 0.16 | — | — | — | — | — | — | — | — |
| Agaricostilbomycetes | — | — | — | — | — | — | — | — | — | 0.01 | 0.10 | — | — |
| Agaricostilbales | — | — | — | — | — | — | — | — | — | 0.01 | 0.10 | — | — |
| Cystobasidiomycetes | 0.13 | 0.05 | 0.04 | 0.14 | 0.33 | 0.03 | 0.09 | 0.07 | 0.01 | 1.63 | 0.67 | — | — |
| Cystobasidiales | 0.12 | 0.05 | 0.04 | 0.10 | 0.33 | 0.02 | 0.07 | 0.05 | 0.01 | 0.89 | 0.62 | — | — |
| Erythrobasidiales | 0.01 | — | — | 0.04 | — | 0.01 | 0.02 | 0.02 | — | 0.74 | 0.05 | — | — |
| Tremellomycetes | 0.13 | 0.22 | 0.80 | 0.17 | 0.09 | 0.09 | 0.10 | 0.11 | — | 0.21 | 0.38 | — | 0.31 |
| Tremellales | 0.13 | 0.22 | 0.80 | 0.17 | 0.09 | 0.09 | 0.10 | 0.11 | — | 0.21 | 0.38 | — | 0.31 |
| Filobasidiales | 0.01 | — | — | — | — | — | — | — | — | — | 0.01 | — | — |
| Ustilaginomycetes | 0.07 | 0.05 | 0.01 | 0.01 | — | 0.02 | 0.01 | — | 0.01 | — | — | 0.02 | 1.74 |
| Ustilaginales | 0.07 | 0.05 | 0.01 | 0.01 | — | 0.02 | 0.01 | — | 0.01 | — | — | 0.02 | 1.74 |
| Exobasidiomycetes | 0.10 | 0.05 | 0.01 | 0.66 | 0.04 | 0.05 | — | — | 0.08 | — | 0.03 | 15.69 | 0.08 |
| Exobasidiales | 0.09 | — | 0.01 | 0.57 | — | 0.02 | — | — | 0.01 | — | — | 0.01 | 0.01 |
| Entylomatales | 0.01 | 0.50 | — | 0.09 | 0.04 | 0.03 | — | — | 0.07 | — | 0.03 | 15.68 | 0.07 |
| Wallemiomycetes | 0.13 | 0.06 | 0.10 | 0.19 | 0.05 | 0.05 | 0.10 | 0.05 | 0.01 | 0.04 | 0.11 | — | 0.02 |
| Wallemiales | 0.13 | 0.06 | 0.10 | 0.19 | 0.05 | 0.05 | 0.10 | 0.05 | 0.01 | 0.04 | 0.11 | — | 0.02 |
| Moniliellomycetes | — | — | — | — | — | — | — | — | — | 0.16 | — | — | — |
| Moniliellales | — | — | — | — | — | — | — | — | — | 0.16 | — | — | — |
| Chytridiomycota | — | — | — | 0.04 | 0.02 | — | — | — | — | 0.01 | 0.01 | — | — |
| Glomeromycota | 0.05 | 0.12 | 0.18 | 0.30 | 0.07 | 0.09 | 0.12 | 0.07 | — | 0.05 | 0.09 | — | 0.21 |
| Unknown fungi | — | 0.01 | 0.01 | — | 0.16 | 0.02 | — | — | 0.56 | 0.01 | 0.01 | 0.16 | 0.18 |
| Others | 1.57 | 5.81 | 22.09 | 5.50 | 30.16 | 6.03 | 32.14 | 65.54 | 31.88 | 19.74 | 4.15 | 0.78 | 0.94 |
Of the 42 genera detected in the present study, the dominant genera were Malassezia (62376 reads in 13 samples), Cochliobolus (16008 reads in 13 samples) and Yamadazyma (10337 reads in 11 samples). The most common fungal species (>500 reads in more than 10 samples) were Malassezia globosa (18949 reads in 13 samples), Curvularia lunata (16008 reads in 13 samples), Cladosporium herbarum (3397 reads in 13 samples), Emericellopsis maritima (3181 reads in 13samples), Wickerhamomyces anomalus (2354 reads in 13 samples), Hortaea werneckii (2332 reads in 13 samples), Aspergillus penicillioides (1591 reads in 13 samples), Talaromyces purpureogenus (1503 reads in 13 samples), Sarocladium kiliense (965 reads in 13 samples), Rhodotorula mucilaginosa (795 reads in 11 samples), Sporidiobolus pararoseus (641 reads in 10 samples) and Candida etchellsii (569 reads in 11 samples) (Tables S1 and S2†).
Out of the 91 OTUs, 41 had the matching sequences with high similarity (≥97%). These matching sequences were derived from fungi found in widely-ranged environments, including plant tissues, soil, food, and air which indicated that most of these fungal OTUs were not seawater specific and were distributed widely in both non-marine and marine habitats. In addition, the other 50 fungal OTUs had the matching sequences with similarity below 97%, which suggests that they are species to be unidentified.
Distribution of fungal communities among different seawater samples
The Good's coverage estimator, Chao 1, and Shannon's indices were used to evaluate and compare the diversity of the fungal communities among the 13 seawater samples (Table 1). The Good's coverage estimator ranged from 99.5 to 99.9%. The Chao1 index (31.42–89.50) and Shannon's index (H′ = 1.78–3.75) indicated that the level of diversity varied among the 13 seawater samples.To gain insight into the differences of fungal communities among the 13 seawater samples from these four sampling sites, we applied network analyses of the 91 OTUs to highlight their distributions (Fig. 3). On one hand, the data showed that richness of fungal taxa in site L1, L3, L4 and L7 was 79, 75, 74 and 82 OTUs (Fig. 3), representing 39, 34, 39 and 39 genera, respectively, indicating that highest fungal richness occurred in site L7, L1, L4. On the other hand, the results showed that sampling site M7.1 had the highest number of OTUs. 75 OTUs were detected in this site, while only 36 and 34 OTUs were detected in M7.2 and M7.3, respectively, which was similar with sampling site L1 and L3. These data indicated that the fungal diversity in 1000 m seawater was higher than deeper seawater in site L1, L4 and L7. In contrast, 60 OTUs were detected in M4.4 while only 44 and 43 OTUs detected in M4.2 and M4.3, which indicated that the fungal diversity in deeper seawater was higher than 1000 m seawater in sampling site L4.
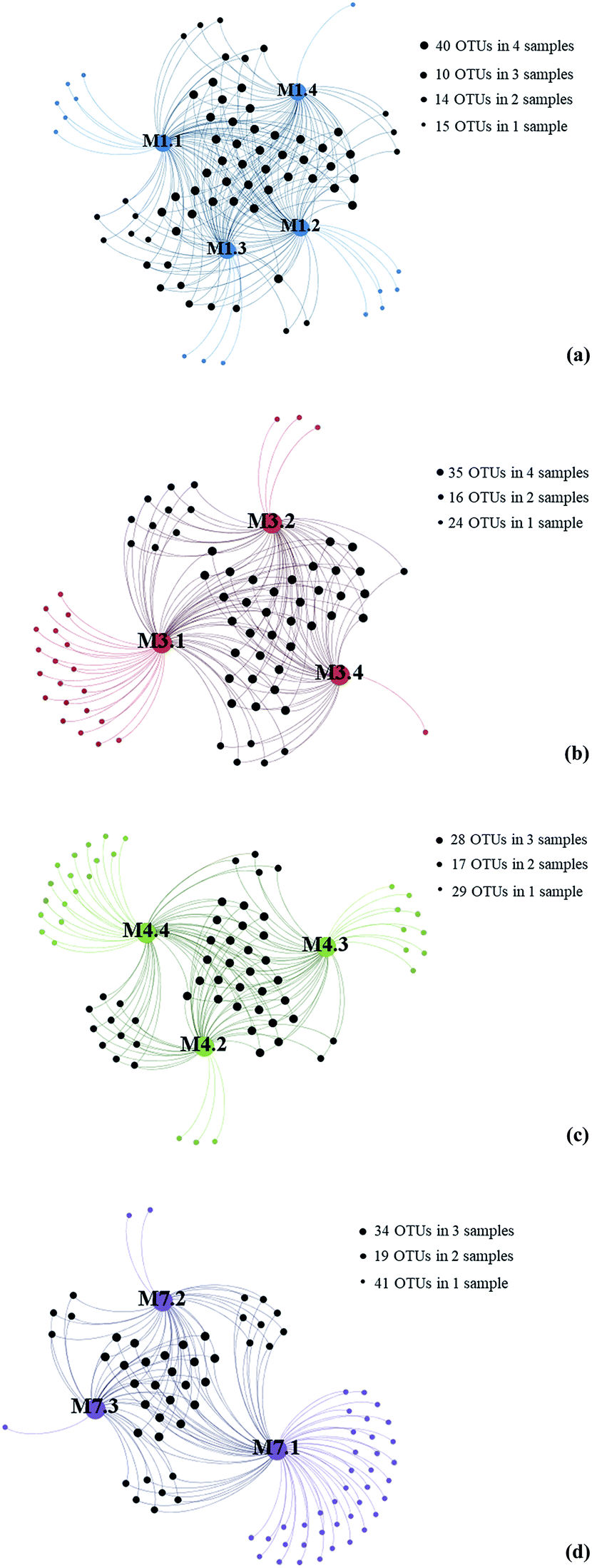 | ||
| Fig. 3 Gephi network diagrams illustrating the fungal OTUs recovered from seawater samples taken from the same sampling sites. (a) 79 OTUs occurring within M1.1, M1.2, M1.3 and M1.4 of the sampling site L1 (blue color); (b) 75 OTUs occurring within M3.1, M3.2 and M3.4 of the sampling site L3 (red color); (c) 74 OTUs occurring within M4.3, M4.3 and M4.4 of the sampling site L4 (green color); (d) 82 OTUs occurring within M7.1, M7.2 and M7.3 of the sampling site L7 (purple color). | ||
To further investigate the relationship between depth and fungal diversity, we also applied network analyses of the 91 OTUs to highlight their distributions based on the depth of the samples (Fig. 4). The results showed that there were 86 OTUs within the samples from 1000 m (Fig. 4a), 73 OTUs within the samples from 2000 m (Fig. 4b), 65 OTUs within the samples from 3000 m (Fig. 4c), and 69 OTUs within the samples from 4000 m (Fig. 4d), which showed higher fungal diversity in 1000 m samples. These data confirmed that the fungal diversity in the Mariana Trench is very richness.
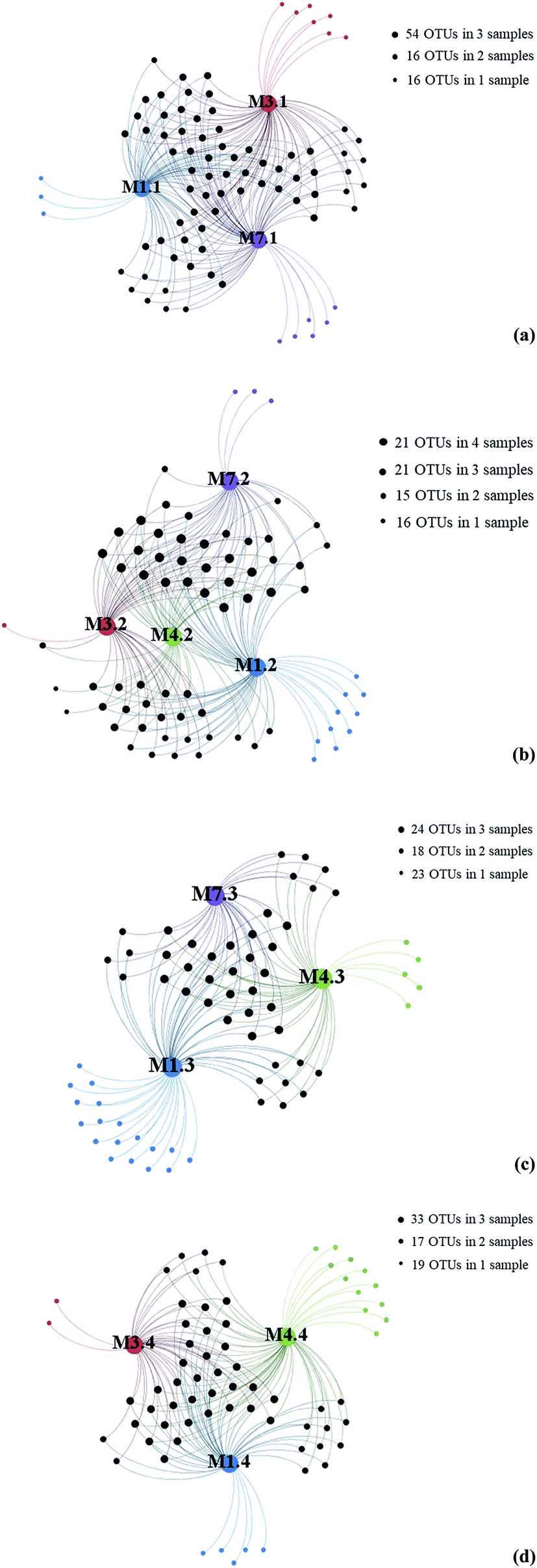 | ||
| Fig. 4 Gephi network diagrams illustrating the fungal OTUs recovered from seawater samples with same water depths. (a) 86 OTUs occurring within M1.1, M3.1 and M7.1 samples; (b) 73 OTUs occurring within M1.2, M3.2, M4.2 and M7.2 samples; (c) 65 OTUs occurring within regions of the M1.3, M4.3 and M7.3 samples; (d) 69 OTUs occurring within regions of the M1.4, M3.4 and M4.4 samples; blue color dot represent samples from L1; red color dot represent samples from L3, green color dot represent samples from L4, purple color dot represent samples from L7. | ||
Discussion
In previous studies, the fungal diversity of the Mariana Trench sediments was detected,11 while the fungal diversity in the Mariana Trench seawater is poorly understood, which impedes our understanding of the ecological importance of fungi in the marine ecosystem of the Mariana Trench. In this study, 91 fungal taxa at the OTU level were revealed in 4 different sites and various depth seawater in the Mariana Trench, greatly advancing our understanding of the fungal diversity in the Mariana Trench. Moreover, several sequences, especially in sample M3.1 and M4.3 (more than one third of the sequences), could not match the database and failed to be assigned to any known taxa, indicating the existence of considerable unknown biodiversity in the Mariana Trench.Members of Ascomycota were more frequently identified in the Mariana Trench seawater than those of Basidiomycota, while members of Chytridiomycota and Glomeromycota represented only a small proportion of the seawater fungal communities, which is similar with the fungal diversity of the Mariana Trench sediments.11 Interestingly, Ascomycota was the dominate phylum in samples from L1 and L3, and Basidiomycota was the dominant phylum in samples from L4 and L7. The phylum Chytridiomycota has been detected in some deep-sea environments, such as methane cold seep23 and Izu-Ogasawara Trench,24 but no Chytridiomycota sequences were detected in the sediment samples collected from the Pacific Ocean as previously described.11 However, in this study, three putative Chytridiomycota OTUs (OTU 59, OTU 79 and OTU 81) were detected in the Mariana Trench seawater (Tables S1 and S2†). Our results suggest that the fungal communities in the deep-sea seawater from the Mariana Trend appears to be dominated by Ascomycota and Basidomycota, while other fungal taxonomic groups are rare.
In Ascomycota, members of Sordariomycetes, Dothideomycetes and Eurotiomycetes have been frequently found in deep-sea sediments in India,25 the Pacific Ocean,8 Arctic fjords3 and in subtropical Chinese seas,26 indicating that these three classes are ubiquitous in marine environments. In our study, Dothideomycetes was dominant in sample M1.3, and Saccharomycetes was dominant in M1.2 and M1.4 (Table 2). And Hypocreales was found dominant in site L3 (including M3.1, M3.2 and M3.4) and M1.1, which was quite different from other researches. And the order Pleosporales, members of Dothideomycetes, was dominant in sample M7.3 (Table 2), which may be essential to survival in marine environments.24,26
In our study, the most abundant class of Basidiomycota was Malasseziomycete, primarily represented by Malassezia sequences. Interestingly, Malassezia was dominant in samples from site L7, with a percentage of more than sixty (Table 2). And Entylomatales was found in sample M7.2, being the second large fungi in this sample, which is rarely found in other researches.
And it is very interesting to find that Glomeromycota sequences are widely distributed in the seawater samples although the sequence abundance was low (Table 2). Glomeromycota, also called arbuscular mycorrhizal fungi, are typical fungal symbionts colonizing the roots of vascular plants and widely-spread on land.27 Recently, the phylum Glomeromycota has been also detected in marine environments, including Arctic, marine sediments,3 and hydrothermal ecosystems.28 And it was not detected in the Mariana Trench in previous reports. These results may indicate the strong adaptability of Glomeromycota and potential significance in marine ecology. But if Glomeromycota act as decomposers needed more research.
Of the 42 fungal genera uncovered in our study, most of them are widespread in terrestrial and marine habitats. The genera Malassezia, Rhodotorula and Trichosporon are extremophilic yeasts that were frequently found in the Arctic, Antarctic and alpine habitats.29 Moreover, the genera Malassezia and Trichosporon are well known pathogens or parasites of marine animals, indicating that they may also be opportunistic pathogens of deep-sea animals.12 Other phylotypes of fungi in this study included Candida, Cryptococcus, Tilletiopsis, Rhodotorula and Erythrobasidium, which were also reappeared in previous studies on fungal diversity in deep-sea sediments.10–12,16,30 Moreover, some of the genera detected in our study are plant pathogens, including Candida, Ceriporia, Exobasidium, Lophiostoma, Moniliella and Mucor. In Eurotiomycetes, Aspergillus are known to be ubiquitous in various marine substrates as well as in marine sponge invertebrates,31 which was detected in the Mariana Trench seawaters. And Aspergillus was also reported that it may produce biologically active natural products,32 indicating that the Mariana Trench may have more fungal resources that can produce active substances. Other genera like Cochliobolus, Emericellopsis, Wickerhamomyces, Yamadazyma and Hortaea were also abundant in the Mariana Trench, while genera Yarrowia, Gymnopus, Kondoa, Neurospora, Mucor, Peziza and Acremonium were rare but indeed exist in the Marina Trench seawater. These data indicated that the fungal resources in the Mariana Trench seawater is rich and diverse.
Conclusion
In summary, our results indicate that there are many fungal taxa resources in seawater of the Mariana Trench, even in the deep-sea (4000 m in this study), most of which possibly originate from terrestrial environments. Although many of the sequences in our study are unidentified, it shows that the Mariana Trench possesses great potential in discovering fungal resources and active substances. It is necessary to explore new methods and technologies to investigate these fungal resources.Experimental
Materials and methods
Library preparation and sequencing. ITS genes of distinct regions (ITS1/ITS2, Arc V4) were amplified used specific primer (18S V4: 528F-706R) with the barcode. All PCR reactions were carried out with Phusion® High-Fidelity PCR Master Mix (New England Biolabs). Mix same volume of 1× loading buffer (contained SYB green) with PCR products and operate electrophoresis on 2% agarose gel for detection. Then, mixture PCR products was purified with GeneJETTM Gel Extraction Kit (Thermo Scientific). Sequencing libraries were generated using Ion Plus Fragment Library Kit 48 rxns (Thermo Scientific) following manufacturer's recommendations. The library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific). At last, the library was sequenced on an Ion S5TM XL platform and 400 bp/600 bp single-end reads were generated.
Sequences analysis were performed by Uparse software.37 Sequences with ≥97% similarity were assigned to the same Operational Taxonomic Units (OTUs). Representative sequence for each OTU was screened for further annotation. Alpha diversity is applied in analyzing complexity of species diversity for a sample through 5 indices, including Observed-species, Chao1, Shannon, Simpson, Good-coverage. All the indices in our samples were calculated with QIIME (version 1.7.0)38 and displayed with R software (version 2.15.3). Beta diversity analysis was used to evaluate differences of samples in species complexity, Beta diversity on both weighted and unweighted unifrac were calculated by QIIME software (version 1.7.0).
Author contributions
Z.-P. W. performed the sample collection, and wrote this manuscript; Z.-Z. L. performed DNA extraction and help to write this manuscript; Y.-L. W. and W.-H. B. analyzed the data; L. L. and H.-Y. W performed the microbial enrichment; Y. Z., L.-L. Z. and S.-G. H. performed checking of the DNA library; P. Z. and S.-S. X. supervised the research work and revised the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (grant no. 31500032), Natural Science Foundation of Shandong Province (grant no. ZR2017BC029), Central Public interest Scientific Institution Basal Research Fund (2017RC-YJ01, 20603022017006, 20603022019011), Qingdao Shinan District Science and Technology Development Fund Project (2016-3-005-ZH) and Qingdao Science and Technology Project (17-3-3-59-nsh).References
- S. K. Pabba, B. Samatha, M. R. Prasad, S. H. Nidadavolu and M. A. S. Charya, Antibacterial Activity of Marine derived Fungi Collected from South East Coast of Tamilnadu, India, J. Microbiol. Biotechnol. Res., 2017, 1, 86–89 Search PubMed.
- K. Liberra and U. Lindequist, Marine fungi--a prolific resource of biologically active natural products?, Die Pharmazie, 1995, 50, 583–588 CAS.
- Z. Tao, N. F. Wang, Q. Z. Yu, Y. L. Hong and Y. Y. Li, Diversity and distribution of fungal communities in the marine sediments of Kongsfjorden, Svalbard (High Arctic), Sci. Rep., 2015, 5, 14524 CrossRef PubMed.
- E. B. G. Jones, J. Sakayaroj, S. Suetrong, S. Somrithipol and K. Pang, Classification of marine Ascomycota, anamorphic taxa and Basidiomycota, Fungal Divers., 2009, 35, 1–187 Search PubMed.
- K. D. Hyde, E. B. G. Jones, E. M. Leano, S. B. Pointing, A. D. Poonyth and L. L. P. Vrijmoed, Role of fungi in marine ecosystems, Biodiversity & Conservation, 1998, 7, 1147–1161 Search PubMed.
- T. L. Campion-Alsumard, S. Golubic and K. Priess, Fungi in corals: symbiosis or disease? Interaction between polyps and fungi causes pearl-like skeleton biomineralization, Mar. Ecol.: Prog. Ser., 1995, 117, 137–147 CrossRef.
- C. Raghukumar, Fungal Parasites of the Marine Green Algae, Cladophora and Rhizoclonium, Bot. Mar., 1986, 29, 289–298 CrossRef.
- W. Xu, K. Pang and Z. Luo, High fungal diversity and abundance recovered in the deep-sea sediments of the Pacific Ocean, Microb. Ecol., 2014, 68, 688–698 CrossRef CAS PubMed.
- Y. Nagano, T. Nagahama, Y. Hatada, T. Nunoura, H. Takami, J. Miyazaki, K. Takai and K. Horikoshi, Fungal diversity in deep-sea sediments–the presence of novel fungal groups, Fungal Ecology, 2010, 3, 316–325 CrossRef.
- V. P. Edgcomb, D. J. Beaudoin, R. J. Gast, J. F. Biddle and A. Teske, Marine subsurface eukaryotes: the fungal majority, Environ. Microbiol., 2011, 13, 172–183 CrossRef CAS PubMed.
- W. Xu, Z. H. Luo, S. Guo and K. L. Pang, Fungal community analysis in the deep-sea sediments of the Pacific Ocean assessed by comparison of ITS, 18S and 28S ribosomal DNA regions, Deep Sea Res., Part I, 2016, 109, 51–60 CrossRef CAS.
- X. Zhang, G. Tang, X. Xu, X. Nong and S. Qi, Insights into Deep-Sea Sediment Fungal Communities from the East Indian Ocean Using Targeted Environmental Sequencing Combined with Traditional Cultivation, PLoS One, 2014, 9, 109–118 Search PubMed.
- C. Raghukumar, S. Raghukumar, G. Sheelu, S. M. Gupta, B. N. Nath and B. R. Rao, Buried in time: culturable fungi in a deep-sea sediment core from the Chagos Trench, Indian Ocean, Deep Sea Res., Part I, 2004, 51, 1759–1768 CrossRef CAS.
- T. Nunoura, M. Nishizawa, M. Hirai, S. Shimamura, P. Harnvoravongchai, O. Koide, Y. Morono, T. Fukui, F. Inagaki and J. Miyazaki, Microbial Diversity in Sediments from the Bottom of the Challenger Deep, the Mariana Trench, Microbes Environ., 2018, 33, 186–194 CrossRef PubMed.
- M. Mouton, F. Postma, J. Wilsenach and A. Botha, Diversity and characterization of culturable fungi from marine sediment collected from St. Helena Bay, South Africa, Microb. Ecol., 2012, 64, 311–319 CrossRef PubMed.
- X. Lai, L. Cao, H. Tan, S. Fang, Y. Huang and S. Zhou, Fungal communities from methane hydrate-bearing deep-sea marine sediments in South China Sea, ISME J., 2007, 1, 756–762 CrossRef CAS PubMed.
- X. Y. Zhang, Y. Zhang, X. Y. Xu and S. H. Qi, Diverse Deep-Sea Fungi from the South China Sea and Their Antimicrobial Activity, Curr. Microbiol., 2013, 67, 525–530 CrossRef CAS PubMed.
- R. N. Glud, F. Wenzhofer, M. Middelboe, K. Oguri, R. Turnewitsch, D. E. Canfield and H. Kitazato, High rates of microbial carbon turnover in sediments in the deepest oceanic trench on Earth, Nat. Geosci., 2013, 6, 284–288 CrossRef CAS.
- Y. Ohara, M. K. Reagan, K. Fujikura, H. Watanabe, K. Michibayashi, T. Ishii, R. J. Stern, I. Pujana, F. Martinez and G. Girard, A serpentinite-hosted ecosystem in the Southern Mariana Forearc, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2831–2835 CrossRef CAS PubMed.
- W. Pathom-Aree, J. E. M. Stach, A. C. Ward, K. Horikoshi, A. T. Bull and M. Goodfellow, Diversity of actinomycetes isolated from Challenger Deep sediment (10,898 m) from the Mariana Trench, Extremophiles, 2006, 10, 181–189 CrossRef CAS PubMed.
- H. Jing, Y. Zhang, Y. Li, W. Zhu and H. Liu, Spatial Variability of Picoeukaryotic Communities in the Mariana Trench, Sci. Rep., 2018, 8, 15357 CrossRef PubMed.
- Z. Xu, M. Wang, W. Wu, Y. Li, Q. Liu, Y. Han, Y. Jiang, H. Shao, A. Mcminn and H. Liu, Vertical Distribution of Microbial Eukaryotes From Surface to the Hadal Zone of the Mariana Trench, Front. Microbiol., 2018, 9, 1–13 CrossRef PubMed.
- T. Nagahama, E. Takahashi, Y. Nagano, M. A. Abdelwahab and M. Miyazaki, Molecular evidence that deep-branching fungi are major fungal components in deep-sea methane cold seep sediments, Environ. Microbiol., 2011, 13, 2359–2370 CrossRef CAS PubMed.
- Y. Naganoa, Fungal diversity in deep-sea extreme environments, Fungal Ecology, 2012, 5, 463–471 CrossRef.
- P. Singh, C. Raghukumar, P. Verma and Y. S. Shouche, Phylogenetic diversity of culturable fungi from the deep-sea sediments of the Central Indian Basin and their growth characteristics, Fungal Divers., 2010, 40, 89–102 CrossRef.
- W. Li, M. M. Wang, X. G. Wang, X. L. Cheng, J. J. Guo, X. M. Bian and L. Cai, Fungal communities in sediments of subtropical Chinese seas as estimated by DNA metabarcoding, Sci. Rep., 2016, 6, 26528 CrossRef CAS PubMed.
- S. J. Lee, M. Kong, P. Harrison and M. Hijri, Conserved Proteins of the RNA Interference System in the Arbuscular Mycorrhizal Fungus Rhizoglomus irregulare Provide New Insight into the Evolutionary History of Glomeromycota, Genome Biol. Evol., 2018, 10, 328–343 CrossRef CAS PubMed.
- T. L. Calvez, G. Burgaud, S. Mahe, G. Barbier and P. Vandenkoornhuyse, Fungal Diversity in Deep-Sea Hydrothermal Ecosystems, Appl. Environ. Microbiol., 2009, 75, 6415–6421 CrossRef PubMed.
- P. Buzzini and R. Margesin, Cold-adapted Yeasts. Biodiversity, Adaptation Strategies and Biotechnological Significance, Springer, Berlin, Heidelberg, 2014 Search PubMed.
- V. P. Edgcomb, D. Beaudoin, R. Gast, J. F. Biddle and A. Teske, Marine subsurface eukaryotes: the fungal majority, Environ. Microbiol., 2011, 13, 172–183 CrossRef CAS PubMed.
- Q. Li and G. Wang, Diversity of fungal isolates from three Hawaiian marine sponges, Microbiol. Res., 2008, 164, 233–241 CrossRef PubMed.
- Z. S. Yu, B. H. Zhang, W. Sun, F. L. Zhang and Z. Y. Li, Phylogenetically diverse endozoic fungi in the South China Sea sponges; and their potential in synthesizing bioactive natural products suggested; by PKS gene and cytotoxic activity analysis, Fungal Divers., 2013, 58, 127–141 CrossRef.
- M. Martin, Cutadapt removes adapter sequences from high-throughput sequencing reads, EMBnet J., 2011, 17, 10–12 CrossRef.
- Q. Christian, P. Elmar, Y. Pelin, G. Jan, S. Timmy, Y. Pablo, P. Jorg and G. C. Frank Oliver, The SILVA ribosomal RNA gene database project: improved data processing and web-based tools, Nucleic Acids Res., 2013, 41, 590–596 Search PubMed.
- R. C. Edgar, B. J. Haas, J. C. Clemente, Q. Christopher and K. Rob, UCHIME improves sensitivity and speed of chimera detection, Bioinformatics, 2011, 27, 2194 CrossRef CAS PubMed.
- B. J. Haas, D. Gevers, A. M. Earl, M. Feldgarden, D. V. Ward, G. Giannoukos, D. Ciulla, D. Tabbaa, S. K. Highlander and E. Sodergren, Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons, Genome Res., 2011, 21, 494 CrossRef CAS PubMed.
- R. C. Edgar, UPARSE: highly accurate OTU sequences from microbial amplicon reads, Nat. Methods, 2013, 10, 996–998 CrossRef CAS PubMed.
- J. G. Caporaso, J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman, E. K. Costello, N. Fierer, A. G. Pena, J. K. Goodrich and J. I. Gordon, QIIME allows analysis of high-throughput community sequencing data, Nat. Methods, 2010, 7, 335–336 CrossRef CAS PubMed.
- M. Bastian, S. Heymann and M. Jacomy, Gephi: An Open Source Software for Exploring and Manipulating Networks, International conference on weblogs and social media, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10142f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |