
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTarget DNA mutagenesis-based fluorescence assessment of off-target activity of the CRISPR-Cas9 system†
Dan Wanga,
Cuili Niua,
Jingxin Hana,
Dejun Ma a and
Zhen Xi*ab
a and
Zhen Xi*ab
aDepartment of Chemical Biology, State Key Laboratory of Elemento-Organic Chemistry, National Engineering Research Center of Pesticide (Tianjin), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: zhenxi@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China
First published on 19th March 2019
Abstract
The RNA-guided CRISPR/Cas9 system could cleave double-stranded DNA at the on-target sites but also induce off-target mutations in unexpected genomic regions. The base-pairing interaction of sgRNA with off-target DNA was still not well understood and also lacked a direct cell-based assay. Herein we developed a fast target DNA mutagenesis-based fluorescence assay to directly detect the Cas9 activity at different off-target sites in living cells. The results showed that Cas9 nuclease had low tolerance to the nucleotide mismatches in the binding region adjacent to PAM sites, and a tradeoff between Cas9 activity and specificity was also observed compared with the high-fidelity Cas9 variant. The combination of computer-based predictions and this target DNA mutagenesis-based fluorescence assay could further provide accurate off-target prediction guidance to minimize off-target effects to enable safer genome engineering.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems in bacteria and archaea provided an adaptive immunity against foreign gene invasion.1–3 An original version of the CRISPR/Cas9 system needs Streptococcus pyogenes Cas9 protein (SpCas9), CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA) to cleave the complementary DNA sequences followed by a 5′-NGG protospacer adjacent motif (PAM).4–7 A chimeric single-guide RNA (sgRNA) was then engineered as a versatile tool to induce double strand breaks at specific genomic sites, which had been successfully applied in many organisms for crop improvement, pest control, industrial production and therapeutics.8–12As target DNA binding was controlled by the base pairing of the 20 nt guide RNA with the target DNA followed by PAM, the guide RNA and PAM sequence determined the Cas9 specificity.13,14 However, there were numerous sequences similar to target DNA in the genome, which could be also cleaved by Cas9 to induce unexpected off-target lesions.15–20 For example, Cas9 could also cleave the same length of off-target sites containing one to five nucleotide mismatches with the guide RNA sequence.21 Through lowering the base-pairing interaction of sgRNA with non-target DNA, Cas9 nuclease and sgRNA could be modified to decrease off-target effects.22–26 Although several high-fidelity nucleases or truncated sgRNAs were screened to reduce off-targets, a comprehensive study of the base-pairing interaction of sgRNA with off-target sites was underdeveloped and new methods for fast, accurate off-target evaluation were also needed.27
The on-target sites in genomic DNA could not be intentionally mutated to different types of off-target sites in living cells was the main barrier of determining the base-pairing interaction of a given sgRNA with their off-target DNA in living cells. The existing quantitative off-target evaluating approaches like T7EI assay,28 in vitro DNA cleavage assay and cell-based sequencing could not completely evaluate all off-target mutations for the existence of numerous off-target sequences in the genomic DNA or the limitation of any complex chromatin structures.29–33 Meanwhile, several computational methods of predicting potential off-target sites like “CCTop”, “CRISPR-OFF webserver (v1.1)” and “CRISTA” and so on were also needing the following experiments for validation.34–36 To solve this problem, we directly determined the interaction of a given RNA and its different types of off-target sites using the engineered reporter gene tools in living cells, which was not affected by the cell cycle or the complex chromatin structures.37,38 The reporter like fluorescent proteins and luciferase reporter have been previously used to determine the on-target activity of Cas9 but seldom used to detect the off-target effect.39–42
In this work, we here designed a target DNA mutagenesis-based fluorescence assay with reengineered dual-luciferase reporter toolkits (Fig. 1). A series of different off-target DNA cassettes (PAM variants, mono-nucleotide, di-nucleotide, tri-nucleotide and multi-nucleotide mismatches to the guide RNA) were inserted into the region upstream of the start codon ATG of the firefly luciferase gene. The cleavage activity of Cas9 at off-target sites was quantified by the relative ratio of firefly luciferase activity and Renilla luciferase activity normalized to the untreated group. This work would directly exhibit the region effect of off-target DNA sequence on gene cleavage in living cells and provide the off-target prediction guidance to minimize off-target effect to enable safer genome engineering.
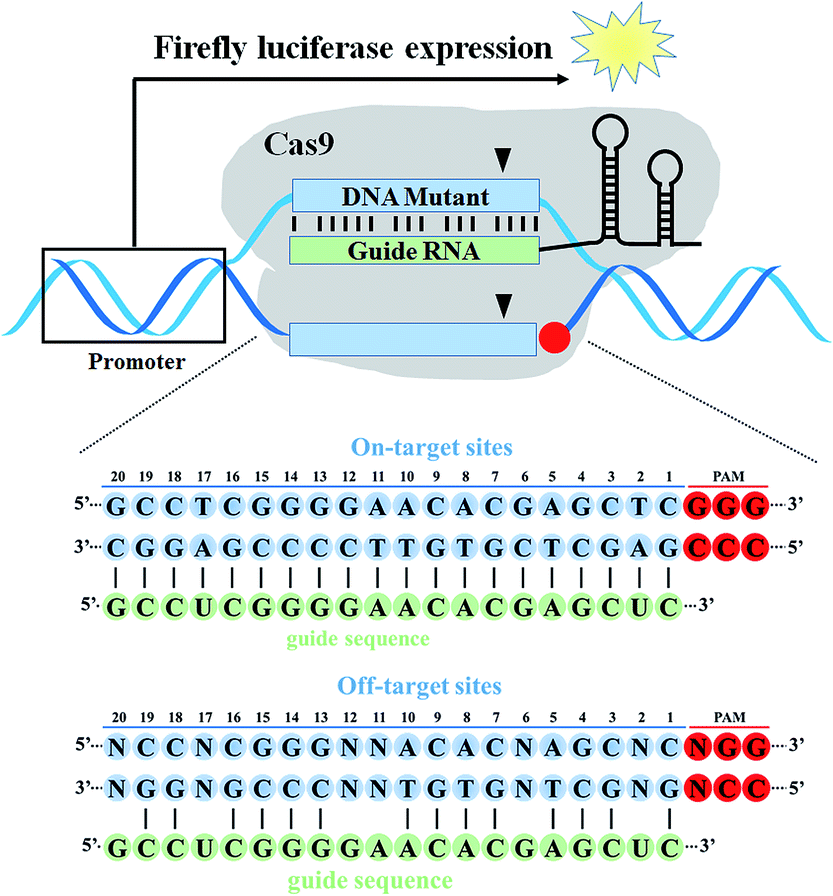 | ||
| Fig. 1 The illustration of a target DNA mutagenesis-based fluorescence assay with reengineered dual-luciferase reporter toolkits. The on-target DNA cassette or off-target DNA cassettes containing different mismatches with the guide RNA sequence were inserted into the region upstream of the start codon ATG of the firefly luciferase gene. Cas9 cleavage activity at the on-target and off-target sites was quantified by the relative ratio of firefly luciferase activity and Renilla luciferase activity. | ||
Results and discussion
The construction of the target DNA mutagenesis-based fluorescence reporter
To study the base-pairing interaction of a given sgRNA with off-target DNA in living cells, we engineered the firefly luciferase-expressing plasmid (pGL3-Fluc) as a fluorescence reporter indicating gene-specific cleavage.43 The on-target or off-target DNA cassettes were inserted upstream of the start site ATG of the firefly luciferase gene without affecting the fluorescence intensity (Fig. S1†). When a given sgRNA was produced, the firefly luciferase activity would be reduced for the site-specific cleavage in the on-target or off-target DNA cassettes (Fig. 2).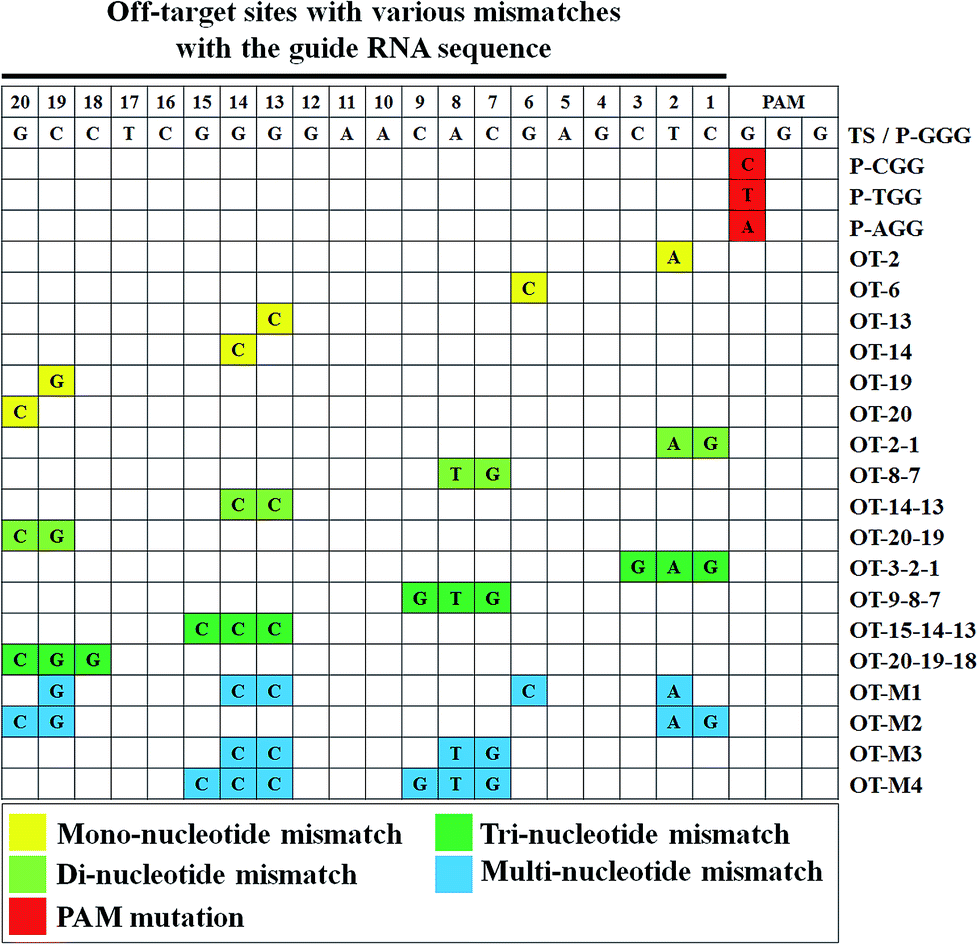 | ||
| Fig. 2 The types of off-target sites containing different mismatches with the guide sequence of sgRNA. Positions of the mismatch nucleotide are highlighted for PAM mutation (red), mono-nucleotide mismatch (yellow), di-nucleotide mismatch (light green), tri-nucleotide mismatch (dark green) and multi-nucleotide mismatch (blue) in the grid. | ||
Firstly, we used this fluorescence reporter to test the on-target activity of 11 designed sgRNAs targeting different sites of EMX1 gene in HEK293T cells. This fluorescence reporter showed a sensitive discrimination between good on-target activity and bad on-target activity. Several highly-potent sgRNAs like EMX1-345, EMX1-429 and EMX1-771 all exhibited more than 75% inhibition of luciferase activity (Fig. S2†). Based on the results of T7EI assay and sequencing, those highly-potent sgRNAs could also induce a high rate of insertion and deletion mutations (InDel) (Fig. S3†).
We then selected EMX1-345 as the following sgRNA to study the off-target effect. All the off-target sequences were derived from the EMX1-345 target sequence. To study the effect of different PAM sequences on the on-target activity, we tested four PAM variants (TGG, AGG, CGG, GGG). The dual luciferase reporter-based results showed that Cas9 nuclease exhibited less activity on CGG PAM than all other PAM variants (Fig. 3). The in vitro gel-based DNA cleavage assay also validated this result (Fig. S4 and S5†). Considering our results and literature reports,19,27 it seemed that Cas9 was less sensitive to CGG PAM than other three PAMs. Since the high-fidelity Cas9 (HF-Cas9) was reported to decrease the off-target effect by reducing the non-specific interactions with target DNA, HF-Cas9 was used to detect its sensitivity to those four PAM variants.22 The results showed that the on-target activity of HF-Cas9 to all PAMs was all reduced up to 2–3 fold compared with WT-Cas9 (Fig. S6, S7,† and 3), indicating a tradeoff between Cas9 on-target activity and non-specific interactions. Hence, these results demonstrated that this fluorescence reporter-based detection of on-target activity was fast, feasible and more accurate in quantification.
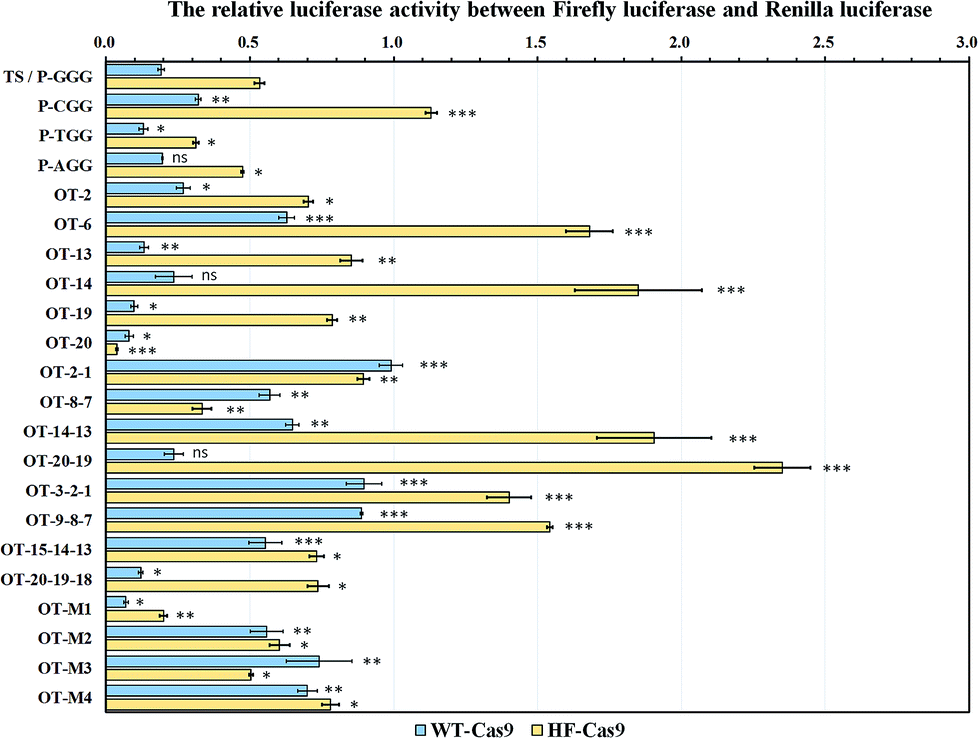 | ||
| Fig. 3 Off-target site evaluation based on the relative luciferase activity between firefly luciferase and Renilla luciferase. The pGL3-Fluc plasmids containing 21 off-target sites were co-transfected into HEK293T cells with PX458 plasmids expressing the wild-type Cas9 (WT-Cas9) or high-fidelity Cas9 (HF-Cas9). After incubation at 37 °C for 48 h, the relative luciferase activity was determined to compare the gene cleavage efficiency. For all of the tests, ns showed no statistical significance, while P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***) was considered to be statistically significant compared with the target site (TS/P-GGG). | ||
Cas9 had a wide-range region tolerance to multiple nucleotide mismatches in the off-target sites
As the computer-based prediction of potential off-target sites including nucleotide mismatches occurred at a high frequency in genomic DNA, we mimicked the natural mismatches in the off-target sites to study the base-pairing interaction of a given sgRNA with off-target sites in living cells.To find the site determinants in off-target sequences, we firstly chose 6 off-target sites with the mono-nucleotide mismatch throughout the EMX1-345 site (from 1st to 20th). The results showed that WT-Cas9 cleaved OT-19 and OT-20 more efficiently than other sites (OT-2, OT-6, OT-13, OT-14) and even the perfectly-matched on-target site (TS) (Fig. 3). This difference of gene cleavage activity was not easily observed when in vitro DNA cleavage assay was performed (Fig. S4 and S5†). In contrast, HF-Cas9 showed more strict selectivity to the single mismatch. Except the 20th mismatch, HF-Cas9 exhibited the much lower activity on all other off-target sites than TS, which was also confirmed by in vitro DNA assay (Fig. S8†).
It has been reported that truncated sgRNAs with at least 17 nucleotides of complementarity at the 3′ end induced gene cleavage at the on-target sites with improved specificity and efficiency compared with normal sgRNAs (20 nucleotides of complementarity).44,45 It suggested that some sgRNAs with shorter complementarity to target DNA might also showed higher activity. Similarly, a number of mismatches at the PAM-distal sites of their target DNA sequence like OT-20 and OT-19 might show the stronger on-target cleavage activity than TS. Hence, WT-Cas9 could exhibit a certain tolerance to those single mismatches in the PAM-distal sites.
We subsequently introduced four kinds of the di-nucleotide mismatches into the EMX1-345 site. Consistent with OT-20 and OT-19, WT-Cas9 also cleaved OT-20-19 more efficiently than other three di-nucleotide mismatch sites. WT-Cas9 showed no significant difference in gene cleavage on OT-20-19 and TS, indicating that the progressive two nucleotide mismatches at the 5′ end did not affect the binding of sgRNA.44,45 In contrast, the progressive 1st and 2nd di-nucleotide mismatch at the off-target site could effectively resist the cleavage of WT-Cas9 and HF-Cas9, which was consistent with sgRNA variants targeted to the genomic loci containing mono-nucleotide DNA bulges at the 1st and 2nd site.21 Notably, WT-Cas9 could still cleave other off-target sites like OT-14-13 at the cleavage efficiency of 35% whereas HF-Cas9 could not cleave OT-14-13 (Fig. 3). This revealed that the combination of N497A, R661A, Q695A, and Q926A substitutions rendered Cas9 a high selectivity on the middle region in the target site.
As WT-Cas9 hardly cleaved OT-2-1, we further detected whether the tri-nucleotide mismatch at progressive 3rd, 2nd and 1st site could still resist Cas9 cleavage. Consistent with the above results, WT-Cas9 and HF-Cas9 still retained a very low activity on the off-target sites like OT-9-8-7 and OT-3-2-1 while WT-Cas9 still exhibited a high activity on OT-20-19-18 (Fig. 3), which was consistent with the above result of OT-20-19. The progressive tri-nucleotide mismatch at the 5′ end (OT-20-19-18) made the full sgRNA like truncated sgRNAs with 17 nucleotides of complementarity at the 3′ end, which has been proved to be more efficient than normal sgRNAs.44,45 Those results further indicated that the progressive tri-nucleotide mismatch in the seed region (1–9 bp) in the off-target site was resistant to Cas9 cleavage and three progressive nucleotide mismatches at the 5′ end might be negligible for full sgRNAs.
Because many off-target sites in genomic DNA contained multiple mismatches, those complex off-target sites were very hard to detect by gel-based cleavage assay or computer-based prediction. Using this fluorescence assay, we introduced multiple nucleotide mismatches at diverse sites to mimic the actual off-target sites. Although OT-M1 included 5 evenly-distributed site mismatches (19th, 14th, 13th, 6th, 2nd), WT-Cas9 and HF-Cas9 both had much better activity on OT-M1 than the target site (TS). For other three off-target sites, OT-M2 (20th, 19th, 2nd, 1st) included four site mismatches in the two ends of the sequence while OT-M3 and OT-M4 contained the mismatches in the middle region of the sequence (Fig. 3). WT-Cas9 and HF-Cas9 both exhibited much lower activity on those off-target sites than the target site (Fig. 3). This revealed that Cas9 had a wide-range region tolerance to multiple nucleotide mismatches evenly-distributed in the off-target sites.
CRISTA-assisted fluorescence assay provided more accurate off-targeting predictions
To date, based on different attributes including the effect of DNA and RNA bulges, base-paring matching, sequence similarity and the thermodynamics of the sgRNA, a method called CRISPR Target Assessment (CRISTA) provided more accurate off-target predictions.34 It mainly relied on the machine learning method to determine the propensity of a potential genomic DNA site to be cleaved by a given sgRNA. However, no direct experimental test method was currently developed to prove those feasibility of those computer-based predictions. To validate the predictions by CRISTA, we hence compared the difference between CRISTA scores and calculated CRISPR-off scores at those off-target sites. The CRISPR-off score was obtained from the multiplication of the tested off-target score and CRISTA score, and the tested off-target score was calculated as the gene cleavage efficiency from the formulation (off-target score = 1 − the relative luciferase activity).The comparison of fluorescence-based cell assay and CRISTA-based prediction assay showed that the tested off-target activity was well consistent with CRISTA predictions in off-target sites with the mono-nucleotide mismatch while the fluorescence-based cell assay provided more consistent evaluations with previously reported results for off-target sites with the di-nucleotide, tri-nucleotide and multi-nucleotide mismatches (Fig. 4A and Table S1†).19,27 The overall correlation coefficient between predicted CRISTA score and measured CRISPR-off score was 0.62 (Fig. 4B). As we see, the two progressive nucleotide mismatches like OT-2-1 commonly used as the HDR repair template was reported to be highly resistant to cleavage by a given sgRNA, while CRISTA predictions gave it a high score.21,53 The CRISTA score of OT-2-1 (0.8025) was much higher than the calculated CRISPR-off score (0.0078), indicating the complexity of off-target predictions. The combination of fluorescence-based cell assay and CRISTA-based prediction assay helped to set up a more accurate and actual off-target prediction method for diverse genomic DNA. The average CRISPR-off score greatly reflected the overall gene intervention at the off-target site by a given sgRNA, which would greatly facilitate the accurate discrimination of real off-target sites in cells.
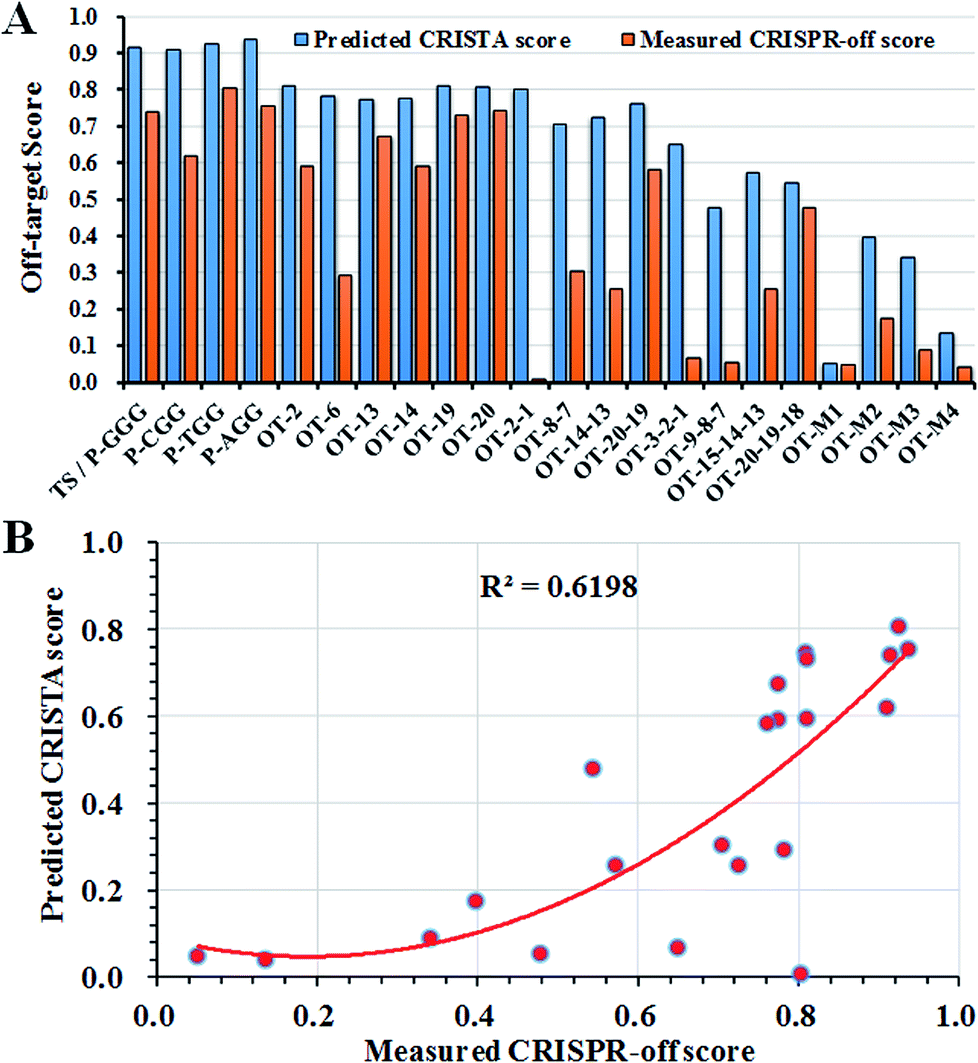 | ||
| Fig. 4 The comparison of fluorescence-based cell assay and CRISTA-based prediction assay. (A) The off-target scores were compared between CRISTA-based off-target prediction and fluorescence-based off-target detection for all the off-target sites. (B) The correlation assay of predicted CRISTA score and measured CRISPR-off score. The score scope ranged from 0 to 1, indicating that the lowest value represented the inefficient cleavage on the DNA while the highest value represented the highly-efficient cleavage on the DNA. | ||
Conclusions
Since the development of CRISPR-Cas9 components as therapeutics, reducing the off-target effect arouse great attention. How to predict and detect the off-target sites in whole genome was the key problem. Although many sequencing-based off-target detection methods had been widely used, it was still very hard to predict the off-target site. It was still necessary to develop a fast and accurate evaluation platform to detect the off-target sites. The existing detection method mainly relied on sequencing to find out the final mutations induced by off-target cleavage in cells. If we could detect the off-target activity before the application, gene editing would be very time-saving and much safer.As usual, the dual-luciferase reporter toolkits have been artificially modified and transfected into cells of interest for the rapid assessment of gene delivery, gene expression, gene silencing and gene cleavage.46–49 However, this dual-luciferase reporter-based gene assay was rarely used to directly determine the off-target activity. In this study, we have demonstrated this tool's capacity of quantitative evaluation about the off-target activity of Cas9 without considering chromatin structures or cell cycles. This target DNA mutagenesis-based fluorescence assay could provide a comprehensive analysis about the off-target sites containing mono-nucleotide, di-nucleotide, tri-nucleotide and multi-nucleotide mismatches. It would reveal the role of each nucleotide in the target sites in Cas9 cleavage.
Due to many unknown off-target sequences in genomic DNA, this dual-luciferase reporter-based gene assay still had its weakness. It was in fact not an unbiased evaluation method because it also need the designed or intended off-target site sequences for the test in advance. The bona-fide off-targets might be lost by this method. In contrast, the current unbiased off-target detection tools have been widely developed to find unintended cleavage sites in living cells such as in vitro detection methods (Digenome-seq,31 CIRCLE-seq,32 SITE-seq33) and in vivo detection methods (GUIDE-seq,50 HTGTS,51 BLESS/BLISS,52 IDLV capture,53 ChIP-seq54). They all relied on cut-edge or gene repair-based tagging enrichment and high-throughput sequencing in the whole genome for high sensitivity, which could be not reached by the dual-luciferase reporter-based gene assay. Despite its weakness in high-throughput profiling of all possible off-target sites in the whole genome, this dual-luciferase reporter-based gene assay provided a fast, simple and accurate approach to evaluate both on-target and off-target site sequences in any cells of interest, which could be also compatible with current sequencing-based detection methods and the computer prediction platform.
To sum, we established a target DNA mutagenesis-based fluorescence assessment method to evaluate both the on-target and off-target activity of CRISPR-Cas9 system. The experimental results were well coordinated with computer-based predictions. The combination of computer-based predictions and this target DNA mutagenesis-based fluorescence assay could further provide the accurate guidance of how to reduce the off-target effect by engineering Cas9 or sgRNA variants. Hence, this target DNA mutagenesis-based fluorescence assay could be further used to search the matching pair of high-efficient sgRNAs and high-fidelity Cas9 variants with the fewest kinds of off-target mutations.
Experimental
Plasmid construction
The firefly luciferase reporter pGL3-Fluc plasmids (Promega) were re-engineered to insert an extra coding frame upstream of the initiation codon ATG of the firefly luciferase gene, which did not affect the normal translation of firefly luciferase.43 The inserted coding frame included two restriction enzyme sites Apa I and Bgl II, followed by a protospacer adjacent motif (PAM) as GGG. For different target DNA variants, we synthesized different ssDNA guides with many nucleotide modifications such as PAM variants, mono-nucleotide, di-nucleotide, tri-nucleotide and multi-nucleotide mismatches to the guide RNA. ssDNA guides (10 μM) were firstly phosphorylated by T4 PNK (10 U) in 1× T4 ligation buffer (New England Biolabs, NEB) for 30 min at 37 °C and then annealed into DNA duplexes with four base overhangs according to the following procedure: 99 °C for 3 min, 2 cycles of 60 °C for 5 min and 37 °C for 15 min, and finally holding at 16 °C for 1 h. The annealed dsDNA could be then effectively ligated into the Apa I and Bgl II linearized pGL3-Fluc plasmids (50 ng) and then transformed into DH5α competent cells to pick the right pGL3-Fluc plasmids containing different guide sequences, which were verified by Sanger sequencing (Sangon biotechnology co., LTD, Shanghai, China).pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid #48138).55 According to the above-mentioned instructions, annealed DNA duplexes with four overhangs could be ligated into Bbs I-linearized PX458 plasmids for the sgRNA transcription under the human U6 promoter (hU6). The engineered PX458 plasmids containing different sgRNA cassettes were also confirmed by Sanger sequencing (Sangon biotechnology co., LTD, Shanghai, China).
A reported high-fidelity Cas9 variant (HF-Cas9) including four mutations (N497A/R661A/Q695A/Q926A) was constructed according to the Golden gate assembly method.22 The primers containing the mutation close to the Bsa I-digested four sites at the 5′-end was used to amplify different parts of Cas9 gene, respectively. PX458 plasmid (50 ng) was mixed with primers and Q5 DNA polymerase (NEB) for PCR amplification according to the following procedure: 98 °C 30 s, 30 cycles of 98 °C 10 s, 60 °C 30 s, 72 °C 60 s, 72 °C 5 min, and finally 16 °C for 30 min. The five fragments were then incubated with Age I and EcoR I-linearized PX458 vector backbones (50 ng) in the Bsa I and T4 ligase mixture (NEB) according to the standard procedure: 25 cycles of 37 °C 3 min, 16 °C 4 min, followed by 50 °C 5 min and 80 °C 5 min. The reaction product could be transformed into DH5α competent cells to select the right HF-Cas9-expressing plasmid PX458-HF-Cas9, which was confirmed by Sanger sequencing (Sangon biotechnology co., LTD, Shanghai, China).
pET21 vectors expressing the wildtype Cas9 nuclease (WT-Cas9) and high-fidelity Cas9 variant (HF-Cas9) were constructed according to Gibson cloning assembly. The primers containing the homology sequence of pET21 vectors was used for the amplification of SpCas9 gene and HF-Cas9 gene from the recombinant PX458 plasmids using Q5 DNA polymerase (NEB). The PCR amplification was performed according to the following procedure: 98 °C 30 s, 30 cycles of 98 °C 10 s, 60 °C 30 s, 72 °C 2.5 min, 72 °C 5 min, and finally 16 °C for 30 min. After PCR amplification and gel extraction, the purified DNA was incubated with the pET21 vector backbone DNA for 15 min at 50 °C. The reaction product was transformed into DH5α competent cells to pick the colony. All pET21 vectors expressing SpCas9 nuclease and HF-Cas9 nuclease were confirmed by Sanger sequencing (Sangon biotechnology co., LTD, Shanghai, China).
Purification of WT-Cas9 and HF-Cas9 nuclease
pET21 vectors expressing WT-Cas9 and HF-Cas9 nuclease were transformed into BL21 (DE3) cells. A colony was picked into 10 mL LB liquid medium for overnight culture. When 10 mL bacterial culture was inoculated into 1 L LB liquid medium for 3 h (OD600 = 0.6), isopropyl-β-D-thio-galactoside (IPTG, 0.5 mM final) was then supplemented. The target protein was induced for 16 h at 18 °C. Cell pellet was suspended in 20 mL lysis buffer (20 mM Tris–HCl, 500 mM NaCl, 20 mM imidazole, pH 7.5) with the addition of one tablet of protease inhibitor cocktails (Roche cOmplete, EDTA-free) and lysozyme (l mg mL−1, Sigma). After incubated on ice for 15 min, the suspension was sonicated for cell lysis and centrifuged at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C for 30 min. The supernatant was transferred into the column containing the Ni-NTA agarose (Qiagen) and placed for 2 h at 4 °C. After washing with lysis buffer for three times and subsequent elution with lysis buffer containing 250 mM imidazole for five times, the target proteins were collected. The proteins were concentrated with ultra-15 centrifugal filter unit with 100 kDa cutoff (Millipore) and further buffer exchanged with storage buffer (40 mM Tris–HCl, 300 mM KCl, 2 mM DTT, 0.2 mM EDTA, pH 7.5). The SpCas9 nuclease and SpCas9 variants were finally mixed with an equal volume of pure glycerol and stored at −80 °C for in vitro DNA cleavage assay.
000 rpm at 4 °C for 30 min. The supernatant was transferred into the column containing the Ni-NTA agarose (Qiagen) and placed for 2 h at 4 °C. After washing with lysis buffer for three times and subsequent elution with lysis buffer containing 250 mM imidazole for five times, the target proteins were collected. The proteins were concentrated with ultra-15 centrifugal filter unit with 100 kDa cutoff (Millipore) and further buffer exchanged with storage buffer (40 mM Tris–HCl, 300 mM KCl, 2 mM DTT, 0.2 mM EDTA, pH 7.5). The SpCas9 nuclease and SpCas9 variants were finally mixed with an equal volume of pure glycerol and stored at −80 °C for in vitro DNA cleavage assay.
Cell culture and plasmid transfection
Human embryo kidney cells (HEK293T) were kindly provided by Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). HEK293T cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM, GIBCO) with high glucose (25 mM) and L-glutamine (4 mM) under the culture condition of 37 °C and 5% CO2 in the incubator. The culture medium was also supplemented with 10% fetal bovine serum (FBS, GIBCO), 100 U per mL penicillin and 100 μg mL−1 streptomycin (GIBCO). When kept in the stage of exponential growth, cells were seeded into 24-well plates (∼30000 cells per well) to reach about 80% confluence for subsequent transfection. Before transfection, the old culture medium was then removed and replaced with serum-free Opti-MEM (0.5 mL per well, GIBCO). The cells were further co-transfected with dual-luciferase reporter plasmids (100 ng pGL3-Fluc/50 ng pRL-TK per well) and Cas9-expressing plasmids (400 ng per well) with the help of Lipofectamine 2000 (Invitrogen, USA). The amount of Cas9-expressing plasmids could be varied according to the experiment intention. After four hours of cell transfection, each well was then supplemented with 1 mL DMEM and maintained for 48 hours.
Dual-luciferase reporter assay
At 48 hours of post-transfection, cells were harvested and lysed with 100 μL passive lysis buffer (PLB) to release the luciferase proteins, which could be then quantitatively assayed according to the procedure of the dual-luciferase reporter assay kit (Promega). After centrifugation at 13000 rpm, the supernatant (80 μL) was transferred into a new tube. 20 μL cell lysate was pipetted into the 96-well plate to measure the relative firefly luciferase activity. Firefly luciferase assay reagent I (100 μL) and Renilla luciferase assay reagent II (100 μL) were in stepwise transferred into 96-well to measure the respective fluorescence intensity for firefly luciferase and Renilla luciferase on Safire Microplate Reader (Tecan). The relative luciferase unit (RLU) was calculated as the P/N according to the following formula:
P/N (ratio) = [D (firefly)/D (Renilla) + C (firefly)/C (Renilla)]/[B (firefly)/B (Renilla) + A (firefly)/A (Renilla)]. A and B denoted two replicates transfected with only the dual-luciferase reporter plasmids, while C and D showed two replicates transfected with Cas9-expressing plasmids and the dual-luciferase reporter plasmids.
T7 endonuclease I (T7EI) assay
The HEK293T cells were transfected with Cas9-expressing plasmids (400 ng per well). Cells were incubated at 37 °C for 48 hours before genomic DNA extraction. Genomic DNA was extracted using the DNALyse Amplification Kit (CWBIO). After genomic DNA purification, gene-specific primers flanking the cleavage site for each coding region were used to amplify a 600–800 bp product with Q5 DNA polymerase (New England Biolabs, NEB). The PCR amplification was performed according to the following procedure: 98 °C 30 s, 30 cycles of 98 °C 10 s, 60 °C 30 s, 72 °C 60 s, 72 °C 5 min, and finally 16 °C for 30 min. PCR products were then purified using GeneJET PCR Purification Kit (Thermo Scientific).The purified PCR products (500 ng) were added in 13 μL 1× NEB buffer 2.1 to form the heterodimer according to the procedure: 95 °C for 10 min, ramping down from 95 °C to 85 °C at 2°C s−1, 85 °C to 75 °C at 0.3°C s−1, 75 °C to 65 °C at 0.3°C s−1, 65 °C to 55 °C at 0.3°C s−1, 55 °C to 45 °C at 0.3 °C s−1, 45 °C to 35 °C at 0.3°C s−1, 35 °C to 25 °C at 0.3 °C s−1, and finally holding 25 °C for 1 h. The re-annealed PCR products were incubated with 10 U T7 endonuclease I (T7EI, NEB) at 37 °C for 30 min. The T7EI-treated PCR products were analyzed on 2% agarose. Gels were imaged with a Gel Doc gel imaging system (Bio-rad). InDel percentage was calculated based on relative band intensities according to the following formula:
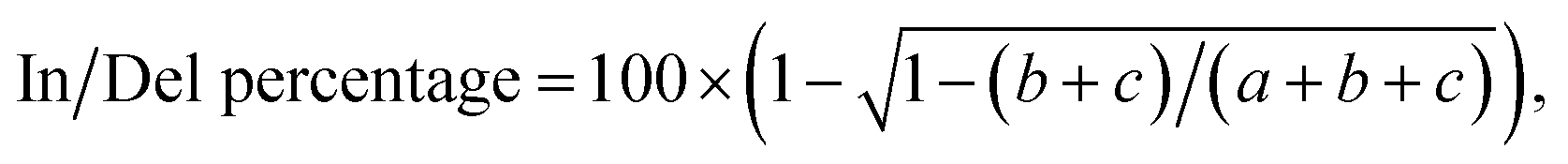 where a was the light intensity of the normal PCR product, while b and c were the light intensity of two respective cleaved products.55
where a was the light intensity of the normal PCR product, while b and c were the light intensity of two respective cleaved products.55
InDel assay by blunt ligation-based sequencing
PCR products were gel purified by QIAquick Gel Extraction Kit (Qiagen) and then ligated into pUC19 plasmid with the pEASY®-Blunt Cloning Vector (Transgen Biotech). The ligation mix (1 μL) was subsequently transformed into DH5α competent cells for colony selection. 20 colonies were randomly picked for sequencing with M13F primer. The sequences of 20 colonies were alignmented with DNAMAN 7 software to find out the insertion or deletions in the modification site induced by gene cleavage.In vitro transcription of EMX1-345 sgRNA
Before transcription, two oligonucleotides (10 μM) of sgRNA backbone (sgRNA-BB) and FS-EMX1-345 were treated with Q5 DNA polymerase (New England Biolabs, NEB). The extension reaction was performed according to the following procedure: 98 °C 30 s, 25 cycles of 98 °C 10 s, 60 °C 30 s, 72 °C 30 s, 72 °C 5 min, and finally 16 °C for 30 min. The product was then purified with Oligo Clean & Concentrator™ (ZYMO). EMX1-345 sgRNA was transcribed using TranscriptAid T7 High Yield transcription Kit (Thermo Scientific) in 20 μL reaction, which was incubated at 37 °C for 2 h and then treated to degrade all the DNA templates with 1U DNase I for 15 min at 37 °C. EMX1-345 sgRNA transcripts were purified with Oligo Clean & Concentrator™ (ZYMO) and eluted with RNase-free water for the following DNA cleavage assay.In vitro DNA cleavage assay
The in vitro DNA cleavage assay was performed according to the previously reported method.56 The pGL3-FLuc plasmids and linear DNAs containing the sequences perfectly or partially complementary to the sgRNA guide sequence were chosen as the target DNA. The linear target DNA was amplified with primers (pGL3-F529 and pGL3-R1029) from pGL3-FLuc plasmid using Q5 DNA polymerase (New England Biolabs, NEB). The PCR amplification was performed according to the following procedure: 98 °C 30 s, 30 cycles of 98 °C 10 s, 60 °C 30 s, 72 °C 30 s, 72 °C 5 min, and finally 16 °C for 30 min. After PCR amplification, PCR products were gel purified by QIAquick Gel Extraction Kit (Qiagen). In vitro DNA cleavage reactions were performed in CutSmart buffer (5 mM DTT, NEB) with a molar ratio of 10:10:1 (Cas9/sgRNA/target DNA) in 20 μL reactions. After the pre-incubation of Cas9 and sgRNA at 37 °C for 10 min, each pGL3-FLuc plasmid (600 ng) or linear DNA (100 ng) was then supplemented and incubated for 60 min or 12 h, respectively. Enzyme reaction was stopped with 10× DNA loading dye (Takara) and were resolved on 0.7% agarose gel (for plasmid DNA) or 2% agarose gel (for linear DNA). The gel bands were visualized with Quantity One software and analyzed with Image J software.
Statistical analysis
The cell test was performed in four independent repeats. Student's t-test and analysis of variance were used to compare the P value with two different groups. For all of the tests, P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***) was considered to be statistically significant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key R&D Program of China (2017YFD0200500), NSFC (21332004, 21472101, 21672118, 21740002).Notes and references
- S. J. Brouns, M. M. Jore, M. Lundgren, E. R. Westra, R. J. Slijkhuis, A. P. Snijders, M. J. Dickman, K. S. Makarova, E. V. Koonin and J. V. Oost, Science, 2008, 321, 960–964 CrossRef CAS PubMed.
- R. Sorek, V. Kunin and P. Hugenholtz, Nat. Rev. Microbiol., 2008, 6, 181–186 CrossRef CAS PubMed.
- F. Hille, H. Richter, S. P. Wong, M. Bratovič, S. Ressel and E. Char-pentier, Cell, 2018, 172, 1239–1259 CrossRef CAS PubMed.
- G. Gasiunas, R. Barrangou, P. Horvath and V. Siksnys, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E2579–E2586 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- P. M. Li, L. Yang, K. M. Esvelt, J. Aach, M. Guell, J. E. DiCarlo, J. E. Norville and G. M. Church, Science, 2013, 339, 823–826 CrossRef PubMed.
- L. Cong, R. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- T. Shah, T. Andleeb, S. Lateef and M. A. Noor, Plant Physiol. Biochem., 2018, 131, 12–21 CrossRef CAS PubMed.
- G. R. McFarlane, C. B. A. Whitelaw and S. G. Lillico, Trends Biotechnol., 2018, 36, 130–133 CrossRef CAS PubMed.
- G. J. Knott and J. A. Doudna, Science, 2018, 361, 866–869 CrossRef CAS PubMed.
- P. D. Donohoue, R. Barrangou and A. P. May, Trends Biotechnol., 2018, 36, 134–146 CrossRef CAS PubMed.
- H. Mollanoori and S. Teimourian, Biotechnol. Lett., 2018, 40, 907–914 CrossRef CAS PubMed.
- S. H. Sternberg, S. Redding, M. Jinek, E. C. Greene and J. A. Doudna, Nature, 2014, 507, 62–67 CrossRef CAS PubMed.
- P. D. Hsu, D. A. Scott, J. A. Weinstein, F. A. Ran, S. Konermann, V. Agarwala, Y. Li, E. J. Fine, X. Wu, O. Shalem, T. J. Cradick, L. A. Marraffini, G. Bao and F. Zhang, Nat. Biotechnol., 2013, 31, 827–832 CrossRef CAS PubMed.
- X. H. Zhang, L. Y. Tee, X. G. Wang, Q. S. Huang and S. H. Yang, Mol. Ther.--Nucleic Acids, 2015, 4, e264 CrossRef CAS PubMed.
- S. W. Cho, S. Kim, Y. Kim, J. Kweon, H. S. Kim, S. Bae and J. S. Kim, Genome Res., 2014, 24, 132–141 CrossRef CAS PubMed.
- E. A. Josephs, D. D. Kocak, C. J. Fitzgibbon, J. McMenemy, C. A. Gersbach and P. E. Marszalek, Nucleic Acids Res., 2015, 43, 8924–8941 CrossRef CAS PubMed.
- C. Kuscu, S. Arslan, R. Singh, J. Thorpe and M. Adli, Nat. Biotechnol., 2014, 32, 677–683 CrossRef CAS PubMed.
- Y. L. Zhang, X. L. Ge, F. Y. Yang, L. P. Zhang, J. Y. Zheng, X. F. Tan, Z. B. Jin, J. Qu and F. Gu, Sci. Rep., 2014, 4, 5405 CrossRef PubMed.
- Q. Zhang, H. L. Xing, Z. P. Wang, H. Y. Zhang, F. Yang, X. C. Wang and Q. J. Chen, Plant Mol. Biol., 2018, 96, 445–456 CrossRef CAS PubMed.
- Y. Lin, T. J. Cradick, M. T. Brown, H. Deshmukh, P. J. Ran, N. Sarode, B. M. Wile, P. M. Vertino, F. J. Stewart and G. Bao, Nucleic Acids Res., 2014, 42, 7473–7485 CrossRef CAS PubMed.
- B. P. Kleinstiver, V. Pattanayak, M. S. Prew, S. Q. Tsai, N. T. Nguyen, Z. Zheng and J. K. Joung, Nature, 2016, 529, 490–495 CrossRef CAS PubMed.
- Y. Fu, J. D. Sander, D. Reyon, V. M. Cascio and J. K. Joung, Nat. Biotechnol., 2014, 32, 279–284 CrossRef CAS PubMed.
- J. S. Chen, Y. S. Dagdas, B. P. Kleinstiver, M. M. Welch, A. A. Sousa, L. B. Harrington, S. H. Sternberg, J. K. Joung, A. Yildiz and J. A. Doudna, Nature, 2017, 550, 407–410 CrossRef CAS.
- M. Jama, A. Ullah, M. Ahsan, R. Tyagi, Z. Habib and K. Rehman, Curr. Issues Mol. Biol., 2018, 26, 65–80 CrossRef.
- I. M. Slaymaker, L. Gao, B. Zetsche, D. A. Scott, W. X. Yan and F. Zhang, Science, 2016, 351, 84–88 CrossRef CAS PubMed.
- T. Zheng, Y. Z. Hou, P. J. Zhang, Z. X. Zhang, Y. Xu, L. T. Zhang, L. L. Niu, Y. Yang, D. Liang, F. Yi, W. Peng, W. J. Feng, Y. Yang, J. X. Chen, Y. Y. Y. Zhu, L. H. Zhang and Q. Du, Sci. Rep., 2017, 7, 40638 CrossRef CAS PubMed.
- R. J. Mean, A. Pierides, C. C. Deltas and M. Koptides, BioTechniques, 2004, 36, 758–760 CrossRef CAS PubMed.
- H. O'Geen, I. M. Henry, M. S. Bhakta, J. F. Meckler and D. J. Segal, Nucleic Acids Res., 2015, 43, 3389–3404 CrossRef PubMed.
- T. Koo, J. Lee and J. S. Kim, Mol. Cells, 2015, 38, 475–481 CrossRef CAS PubMed.
- D. Kim, S. Bae, J. Park, E. Kim, S. Kim, H. R. Yu, J. H. Wang, J. I. Kim and J. S. Kim, Nat. Methods, 2015, 12, 237–243 CrossRef CAS PubMed.
- S. Q. Tsai, N. T. Nguyen, L. J. Malagon, V. V. Topkar, M. J. Aryee and J. K. Joung, Nat. Methods, 2017, 14, 607–614 CrossRef CAS PubMed.
- P. Cameron, C. K. Fuller, P. D. Donohoue, B. N. Jones, M. S. Thomp son, M. M. Carter, S. Gradia, B. Vidal, E. Garner, E. M. Slorach, E. Lau, L. M. Banh, A. M. Lied, L. S. Edwards, A. H. Settle, D. Capurso, V. Llaca, S. Deschamps, M. Cigan, J. K. Young and A. P. May, Nat. Methods, 2017, 14, 600–606 CrossRef CAS PubMed.
- S. Abadi, W. X. Yan, D. Amar and I. Mayrose, PLoS Comput. Biol., 2017, 13, e1005807 CrossRef PubMed.
- M. Stemmer, T. Thumberger, J. Wittbrodt and J. L. Mateo, PLoS One, 2015, 10, e0124633 CrossRef PubMed.
- F. Alkan, A. Wenzel, C. Anthon, J. H. Havgaard and J. Gorodkin, Genome Biol., 2018, 19, 177 CrossRef PubMed.
- S. Lin, B. T. Staahl, R. K. Alla and J. A. Doudna, eLife, 2014, 3, e04766 CrossRef PubMed.
- R. M. Yarrington, S. Verma, S. Schwartz, J. K. Trautman and D. Carroll, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9351–9358 CrossRef CAS PubMed.
- H. Zhang, X. Zhang, C. Fan, Q. Xie, C. Xu, Q. Zhao, Y. Liu, X. Wu and H. Zhang, Biochem. Biophys. Res. Commun., 2016, 471, 528–532 CrossRef CAS PubMed.
- R. A. Johnson, V. Gurevich, S. Filler, A. Samach and A. A. Levy, Plant Mol. Biol., 2015, 87, 143–156 CrossRef CAS PubMed.
- C. W. White, H. K. Vanyai, H. B. See, E. K. M. Johnstone and K. D. G. Pfleger, Sci. Rep., 2017, 7, 3187 CrossRef PubMed.
- A. Glaser, C. McColl and J. Vadolas, Mol. Ther.--Nucleic Acids, 2016, 5, e334 CrossRef CAS PubMed.
- Q. Du, H. Thonberg, H. Y. Zhang, C. Wahlestedt and Z. Liang, Biochem. Biophys. Res. Commun., 2004, 325, 243–249 CrossRef CAS PubMed.
- J. P. Zhang, X. L. Li, A. Neises, W. Q. Chen, L. P. Hu, G. Z. Ji, J. Y. Yu, J. Xu, W. P. Yuan, T. Cheng and X. B. Zhang, Sci. Rep., 2016, 6, 28566 CrossRef CAS PubMed.
- Y. F. Fu, J. D. Sander, D. Reyon, V. M. Cascio and J. K. Joung, Nat. Biotechnol., 2014, 32, 279–284 CrossRef CAS PubMed.
- R. L. Sheng, F. F. An, Z. Wang, M. R. Li and A. M. Cao, RSC Adv., 2015, 5, 12338–12345 RSC.
- J. Y. Lee, S. Kim, D. H. Wang, J. M. Jeong, J. K. Chung, M. C. Lee and D. S. Lee, J. Nucl. Med., 2008, 49, 285–294 CrossRef CAS PubMed.
- G. Huang, Q. Gao, Y. Zhao, Z. Dong, T. Li, X. Guan and J. Jiang, Gene, 2015, 558, 278–286 CrossRef CAS PubMed.
- N. Smart, P. J. Scambler and P. R. Riley, Biol. Proced. Online, 2005, 7, 1–7 CrossRef CAS PubMed.
- S. Q. Tsai, Z. Zheng, N. T. Nguyen, M. Liebers, V. V. Topkar, V. Thapar, N. Wyvekens, C. Khayter, A. J. Iafrate, L. P. Le, M. J. Aryee and J. K. Joung, Nat. Biotechnol., 2015, 33, 187–197 CrossRef CAS PubMed.
- R. L. Frock, J. Hu, R. M. Meyers, Y. J. Ho, E. Kii and F. W. Alt, Nat. Biotechnol., 2015, 33, 179–186 CrossRef CAS PubMed.
- R. Mirzazadeh, T. Kallas, M. Bienko and N. Crosetto, Methods Mol. Biol., 2018, 1672, 167–194 CrossRef CAS PubMed.
- T. Koo, J. Lee and J. S. Kim, Mol. Cell, 2015, 38, 475–481 CrossRef CAS PubMed.
- L. Khair, R. E. Baker, E. K. Linehan, C. E. Schrader and J. Stavnezer, PLoS Genet., 2015, 11, e1005438 CrossRef PubMed.
- F. A. Ran, P. D. Hsu, J. Wright, V. Agarwala, D. A. Scott and F. Zhang, Nat. Protoc., 2013, 8, 2281–2308 CrossRef CAS PubMed.
- D. Wang, D. Ma, J. Han, L. Kong, L. Y. Li and Z. Xi, ChemBioChem, 2018, 19, 2195–2205 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10017a |
| This journal is © The Royal Society of Chemistry 2019 |