
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Nanocomposite functionalized membranes based on silica nanoparticles cross-linked to electrospun nanofibrous support for arsenic(V) adsorption from contaminated underground water†
L. Yohai *a,
H. Giraldo Mejíaa,
R. Procaccinia,
S. Pellicea,
K. Laxman Kunjalib,
J. Duttab and
A. Uheida*b
*a,
H. Giraldo Mejíaa,
R. Procaccinia,
S. Pellicea,
K. Laxman Kunjalib,
J. Duttab and
A. Uheida*b
aDivisión Cerámicos, INTEMA, CONICET, UNMdP, B7608FDQ Mar del Plata, Argentina. E-mail: yohai@fi.mdp.edu.ar
bFunctional Materials Group, Department of Applied Physics, KTH Royal Institute of Technology, 16440 Kista, Stockholm, Sweden. E-mail: salam@kth.se
First published on 12th March 2019
Abstract
Nanocomposite functionalized membranes were synthesized using surface functionalized mesoporous silica nanoparticles (MCM-NH2 or MCM-PEI) cross-linked to a modified polyacrylonitrile (mPAN) nanofibrous substrate for the removal of 1 mg L−1 of As(V); a concentration much higher than what has been reported for underground water in Argentina. Adsorption studies were carried out in batch mode at pH 8 with nanoparticles in colloidal form, as well as the nanoparticles supported on the modified PAN membranes (mPAN/MCM-NH2 and mPAN/MCM-PEI). Results indicate a twenty-fold improvement in As(V) adsorption with supported nanoparticles (nanocomposite membranes) as opposed to their colloidal form. The adsorption efficiency could be further enhanced by modifying the nanocomposite membrane surface with Fe3+ (mPAN/MCM-NH2-Fe3+ and mPAN/MCM-PEI-Fe3+) which resulted in more than 95% arsenic being removed within the first 15 minutes and a specific arsenic adsorption capacity of 4.61 mg g−1 and 5.89 mg g−1 for mPAN/MCM-NH2-Fe3+ and mPAN/MCM-PEI-Fe3+ nanocomposite membranes, respectively. The adsorption characteristics were observed to follow a pseudo-first order behavior. The results suggest that the synthesized materials are excellent for quick and efficient reduction of As(V) concentrations below the WHO guidelines and show promise for future applications.
1. Introduction
Arsenic (As) is an abundant element on earth, widely distributed in water, air, rocks and soil. The presence of arsenic in the environment occurs because of both natural and anthropogenic activities.1 Arsenic exists in its inorganic and organic forms, however, dissolved species of As in water are mainly inorganic, arsenite being the dominant form under reducing conditions and arsenate the most stable species in oxygenated environments.2 Organic forms of arsenic, mostly found in fishes and crustaceans, are believed to be non-toxic. Exposure of the general population to As primarily occurs through groundwater consumption and, in addition, there is a risk of exposure in human health due to the introduction of arsenic into the food chain through crops, rice, milk, meat and others.3,4 The most affected countries are India, Bangladesh, China, Japan, Taiwan, Canada, USA, Mexico, Chile and Argentina. Inorganic arsenic is classified as a carcinogenic and neurotoxic contaminant. Long-term arsenic exposures can cause skin, kidney, liver and bladder cancers. Arsenic exposure can also manifest in other outcomes such as hypertension, diabetes and heart diseases.3 In 2011, the World Health Organization (WHO) and the United States Environmental Protection Agency (US EPA) recommended a maximum tolerable limit of 0.01 mg L−1 of As in drinking water.2,5,6 In Argentina, it is common to find many citizens affected by chronic endemic regional hydroarsenism (H.A.C.R.E., letters due to words in Spanish) characterized by dermal lesions on palms and feet.7 This kind of disease is related to the consumption of water contaminated with an As concentration exceeding WHO's recommended limit. More than 4 million Argentinian citizens are exposed to water and groundwater contaminated with As. The most affected regions in Argentina are the provinces of La Pampa, Tucumán, Córdoba, Santa Fe, Chaco, Santiago del Estero and Salta.8–11 In Buenos Aires province, 87% of water and groundwater analysis have shown arsenic concentrations far beyond the concentrations recommended for drinking water.12 In the rural areas, for example the city of Rafaela, which is the main milk producing region of Santa Fe, arsenic concentrations in water between 0.18 and 0.32 mg L−1 have been found.13Several technologies are used to reduce arsenic concentrations to safe limits, such as co-precipitation, oxidation/filtration, selective adsorption, reverse osmosis, ion exchange, activated alumina, coagulation/filtration, photocatalysis and nanofiltration.6 Adsorption has been widely used being simple, inexpensive and efficient technology for treatment of ground and wastewater, air emissions or for removing a series of toxic chemicals and heavy metals.14 Adsorbents such as carbon nanospheres,15 metal oxide nanoparticles,16–19 carbon nanotubes (CNTs),20–22 Fe species such as Fe, Fe2O3, Fe3O4 (ref. 23–26) and mesoporous silica nanoparticles23,27–29 have been employed to remove arsenic from contaminated water.
Mesoporous silica nanoparticles have caught attention due to its unique properties like large surface area, high pore volume, ordered structure, chemical versatility and good mechanical and thermal stability. In addition, it is possible to functionalize silanol groups present in the structure with amine groups.24 In particular, MCM-41 is an ordered mesoporous silica characterized by a two dimensional hexagonal array of cylindrical pores.28,30 MCM-41, with high density of amino groups and well-defined mesochannels, can enhance the accessibility of molecules capable of adsorb cations, such as cobalt, copper, zinc and arsenic. The adsorbents, however, tend to aggregate into larger particles, leading to lower adsorption capacity due to reduction of surface area. The use of supporting materials, such as nanofibers and carbon nanotubes, can improve the adsorptive performance of nanoparticles due to the distribution of these particles on its surfaces.31 Nanofibrous membranes have small fiber diameters that lead to large specific surface area and high flexibility for chemical/physical surface functionalization.32 The use of nanofibrous membranes is favored over other technologies for water treatment due to lower energy consumption, high selectivity and continuous mode operation possibilities.33 Polyacrylonitrile (PAN) nanofibers are popular due to its excellent mechanical properties, commercial availability, environmental stability and chemical versatility. Nitrile groups on PAN surface can be modified through simple chemical reactions to be easily cross-linked with polyamines.5 Recently, the use of nanocomposite materials has become a novel and promising technology for water remediation. Particularly, one successful technique is to add nanoparticles to polymeric or ceramic membranes during membrane synthesis.34
In this study, we developed nanocomposite membranes for efficient removal of As(V). For this purpose, As(V) adsorption studies were carried out with amino functionalized nanoparticles (NPs) cross-linked to a support of modified polyacrylonitrile nanofibers (mPAN). The attachment of Fe3+ to the nanocomposite nanofibers membranes was also evaluated for arsenic removal efficiency. Experiments were performed in the batch mode at pH 8 with constant initial arsenic concentration, 1 mg L−1, higher concentration than what is found in contaminated Argentinian groundwater. Kinetic behavior was studied to evaluate the adsorption process. Techniques such as High Resolution Transmission Electron Spectroscopy (HR-TEM), Scanning Electron Microscopy (SEM), Fourier Transform Infrared Spectroscopy (FTIR) and Zeta Potential (ZP) were used for characterization. Inductively coupled plasma-atomic emission spectroscopy (ICP-OES) was used to quantify As(V) concentration.
2. Materials and methods
2.1. Chemical and materials
Polyacrylonitrile powder (PAN, MW = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), N,N-dimethylformamide (DMF), tetraethylorthosilicate (TEOS), aminopropyltriethoxysilane (APTES), HCl 37% wt, cetyltrimethylammonium bromide (CTAB), hydroxylamine hydrochloride (NH2OH·HCl), NH4OH 25% wt, Fe(NO3)3·9H2O, Na2HAsO4·7H2O, Na2CO3, absolute ethanol, toluene and glutaraldehyde (GA) 50%, were purchased from Sigma-Aldrich. Branched polyethylenimine (PEI, MW = 10000 g mol−1) was purchased from Alfa-Aesar. All chemicals were analytical grade and were directly used without further purification. High purity water with a resistivity of 15 MΩ cm−1 was used in all the experiments.
000 g mol−1), N,N-dimethylformamide (DMF), tetraethylorthosilicate (TEOS), aminopropyltriethoxysilane (APTES), HCl 37% wt, cetyltrimethylammonium bromide (CTAB), hydroxylamine hydrochloride (NH2OH·HCl), NH4OH 25% wt, Fe(NO3)3·9H2O, Na2HAsO4·7H2O, Na2CO3, absolute ethanol, toluene and glutaraldehyde (GA) 50%, were purchased from Sigma-Aldrich. Branched polyethylenimine (PEI, MW = 10000 g mol−1) was purchased from Alfa-Aesar. All chemicals were analytical grade and were directly used without further purification. High purity water with a resistivity of 15 MΩ cm−1 was used in all the experiments.
2.2. Synthesis of the adsorbents
:3) solution. Particles were recovered by centrifugation, washed several times with DI water and dried at 80 °C overnight. Mesoporous silica nanoparticles were then calcined at 550 °C for 5 h to remove the surfactant with a ramp of 5 °C min−1 during heating or cooling.Two types of amino precursors were used for surface functionalization: APTES and branched PEI.
APTES functionalization. Activated NPs were refluxed in 100 mL of dry toluene containing 0.85 mL of APTES during 4 h at 100 °C. These nanoparticles were labeled as MCM-NH2.
PEI functionalization. Activated NPs were functionalized with PEI to enhance amino groups in the adsorbent. Two steps were followed. First, 0.1 g of MCM-NH2 nanoparticles were mixed with 0.32 g of GA solution. DI water was added to reach 10 g of total solution. After mixing overnight at room temperature, NPs were centrifuged and washed several times with DI water to remove any excess of unreacted GA. After air-drying, 0.075 g of these NPs were mixed with 6.5 g of branched PEI and 20 mL of DI water and stirred overnight at 50 °C. After washing several times with DI water, NPs were vacuum-dried at 60 °C. These NPs were labeled as MCM-PEI.
Incorporation of Fe3+ was done by immersion of the nanocomposites in a solution of 2 g of Fe(NO3)3·9H2O in 40 mL of ethanol. This procedure was performed at room temperature, in a hermetically closed recipient by orbital shaking overnight. Subsequently, nanocomposites were washed several times with DI water in order to remove residual iron(III), and labeled as mPAN/MCM-NH2-Fe3+ or mPAN/MCM-PEI-Fe3+.
2.3. Characterization
Images of MCM-41 nanoparticles were taken using a High Resolution Transmission Electron Microscope (HR-TEM, JEOL 2100 FEG-TEM). Raw electrospun nanofibers and nanocomposite membranes were imaged with a Scanning Electron Microscope (Zeiss Ultra 55 SEM instrument). Zeta Potential for NPs was determined by Dynamic Light Scattering (DLS) using a Beckman Coulter™ Delsa Nano C particle analyzer, (Brea, CA, USA). Fourier Transform Infrared Spectroscopy (FTIR Nicolet Instruments model Avatar-100 equipped with ATR diamond at 303 K, Madison, WI, USA) was used to follow, step by step, the synthesis of nanoparticles, nanofibers and the cross-linking procedures.2.4. Batch adsorption studies
Different arsenic solutions were prepared from a 1000 mg L−1 of As(V) standard stock solution obtained by the dilution of Na2HAsO4·7H2O salt. All arsenic solutions were kept at pH 8. Batch adsorption experiments were carried out at room temperature and orbital shaking. Induced Coupled Plasma (ICP) technique was used to determine arsenic concentration (ICP-OES, iCAP 6500 Thermo Scientific). As(V) adsorption was tested in three different ways: (a) modified nanoparticles (MCM-NH2 or MCM-PEI) suspension in 10 mL water was exposed to 1 mg L−1 of As(V). After 90 min, particles were separated by centrifugation at 12000 rpm for 15 minutes and the remaining concentration of As in water was analyzed; (b) membranes (mPAN/MCM-NH2 and mPAN/MCM-PEI) were immersed in a 100 mL solution of 1 mg L−1 As(V). After 90 min, a water sample was collected and analyzed; (c) membranes with incorporated Fe3+ (mPAN/MCM-NH2-Fe3+ and mPAN/MCM-PEI-Fe3+) were immersed in 100 mL solution of 1 mg L−1 As(V). Water samples were collected every 5 minutes during the adsorption process over 90 minutes and analyzed. The amount of modified nanoparticles (20 ± 2 mg) was kept constant for all experiments reported here.
3. Results and discussion
3.1. Nanocomposite characteristics
The crosslinking reaction scheme of the nanocomposites membranes is schematically represented in Scheme 1. To follow the incorporation of the functional groups, FTIR spectra were collected for the synthesized NPs (Fig. 1(a)). IR bands for MCM-41 are typical of mesoporous silica26,28 with a wide band at 3380 cm−1 corresponding to O–H stretching. The adsorption at 1067 cm−1 is attributed to the transversal optic (TO) mode of the asymmetric vibration of Si–O–Si and at 960 cm−1 to Si–OH vibration. Upon functionalization, the Si–O–Si shifts to 1041 and 1050 cm−1 for MCM-NH2 and MCM-PEI, respectively. Moreover, the absorption at 960 cm−1 is less intense due to the reaction of silanol groups with amino precursors. N–H stretching is found in the 3200–3500 cm−1 region, where it overlaps with O–H stretching. Amino groups appear between 1650–1465 cm−1 due to –NH2 and N–H bending. More intense bands in MCM-PEI are due to higher primary and secondary amino groups present in PEI structure. Typical bands for –CH2 and C–H stretching are observed between 2980 and 2830 cm−1.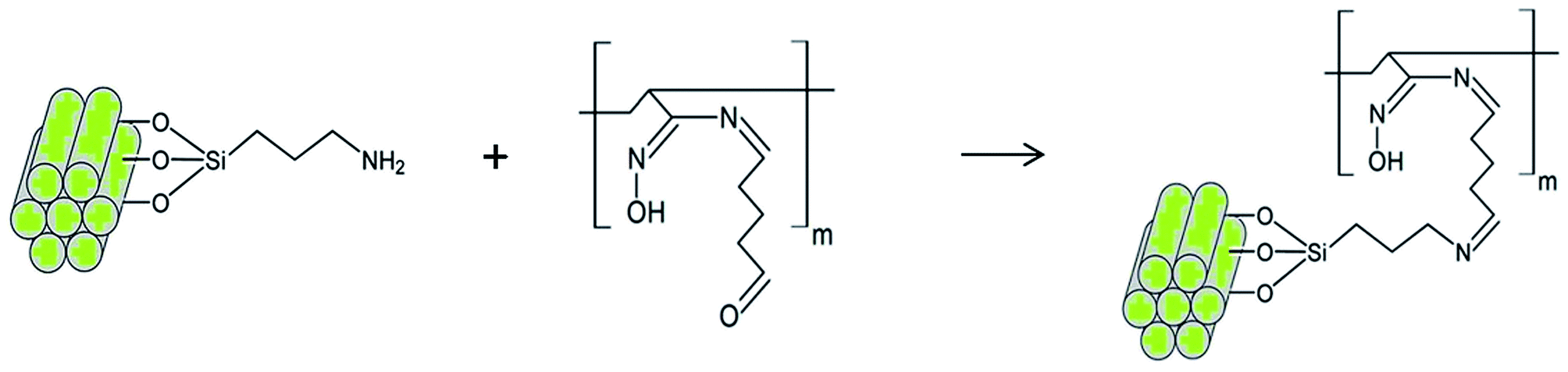 | ||
| Scheme 1 Schematic representation of the cross-linking reaction between MCM-NH2 and modified nanofibers (mPAN). | ||
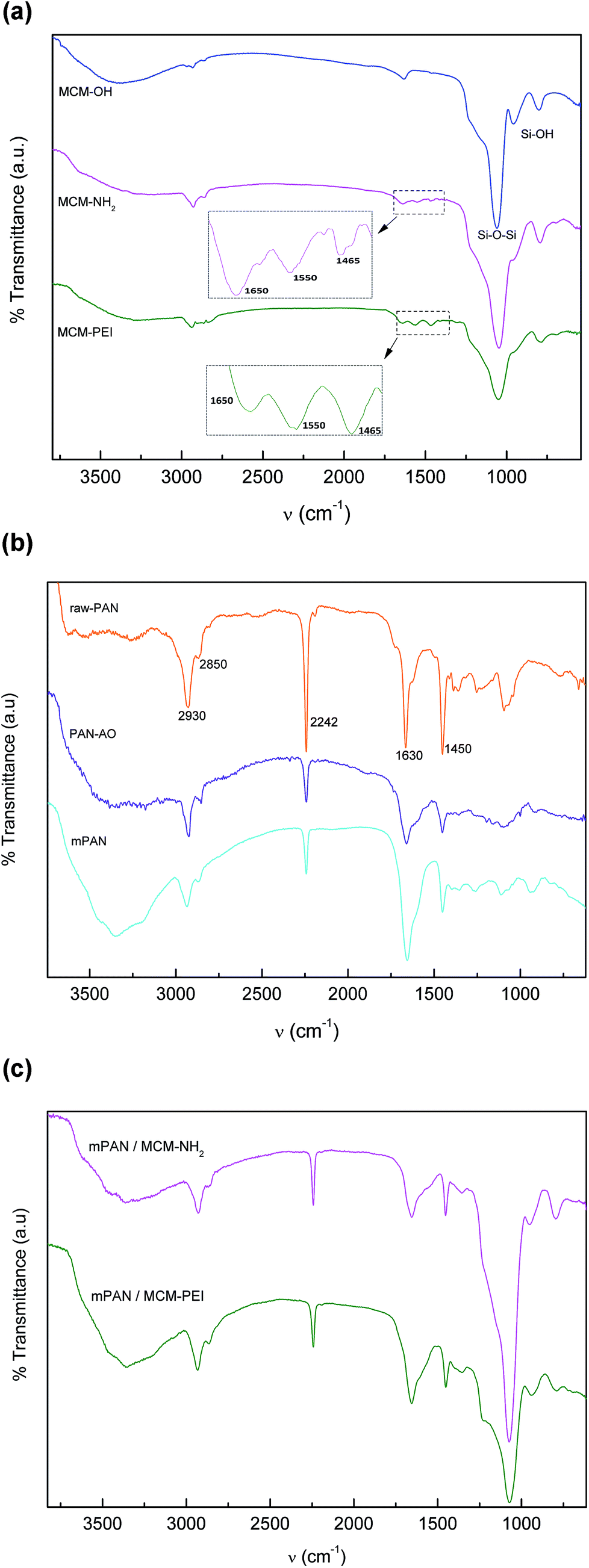 | ||
Fig. 1 (a) FTIR spectra of MCM-OH ( ), MCM-NH2 ( ), MCM-NH2 ( ) and MCM-PEI ( ) and MCM-PEI ( ). (b) FTIR spectra for raw-PAN ( ). (b) FTIR spectra for raw-PAN ( ), PAN-AO ( ), PAN-AO ( ) and mPAN ( ) and mPAN ( ). (c) FTIR spectra for mPAN/MCM-NH2 ( ). (c) FTIR spectra for mPAN/MCM-NH2 ( ) and mPAN/MCM-PEI ( ) and mPAN/MCM-PEI ( ). ). | ||
Fig. 1(b) shows PAN spectra during the synthesis steps. In electrospun raw nanofibers (raw-PAN), an intense band at 2242 cm−1 is observed that arise from C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching.35 Peaks at 2933 cm−1 and 2850 cm−1 corresponding to –CH2– stretching and the one at 1455 cm−1 to bending vibration of C–H in –CH2– groups are also observed. Absorption band at 1630 cm−1 is possibly the vibration of the C
N stretching.35 Peaks at 2933 cm−1 and 2850 cm−1 corresponding to –CH2– stretching and the one at 1455 cm−1 to bending vibration of C–H in –CH2– groups are also observed. Absorption band at 1630 cm−1 is possibly the vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds due to residual DMF solvent in the matrix.35 After amidoxime functionalization (PAN-AO), the nitrite absorption at 2242 cm−1 becomes less intense suggesting that these groups are partially functionalized to amidoxime groups. In addition, a wide band appears around 3360 cm−1 due to overlapped N–H and O–H stretching vibrations. The wide band at 1655 cm−1 can be attributed to overlapped CN and –NH2 vibrations. In mPAN spectrum increased peak at 1655 cm−1 is due to contribution of both CN and CO stretching.
O bonds due to residual DMF solvent in the matrix.35 After amidoxime functionalization (PAN-AO), the nitrite absorption at 2242 cm−1 becomes less intense suggesting that these groups are partially functionalized to amidoxime groups. In addition, a wide band appears around 3360 cm−1 due to overlapped N–H and O–H stretching vibrations. The wide band at 1655 cm−1 can be attributed to overlapped CN and –NH2 vibrations. In mPAN spectrum increased peak at 1655 cm−1 is due to contribution of both CN and CO stretching.
Fig. 1(c) shows spectra for mPAN/MCM-NH2 and mPAN/MCM-PEI membranes. As it can be seen, cross-linking has been successfully achieved; in both cases, peak at 1655 cm−1 is less intense due to reaction of CO of the modified nanofibers with –NH2 groups from MCM-NH2 or MCM-PEI nanoparticles. Moreover, the wide band between 3600–3000 cm−1 becomes less intense due to the lowering of N–H stretching groups. Absorption band at 1070 cm−1 is typically from Si–O–Si vibrations of silica nanoparticles.
Fig. 2 shows (a) HRTEM image of MCM-NH2 and (b) HRTEM image of MCM-PEI. Rod-like functionalized nanoparticles with regular hexagonal structure are observed. Fig. 2(c), (d) and (e) presents SEM image of mPAN, mPAN/MCM-NH2 and mPAN/MCM-PEI, respectively. Several authors had demonstrated that functionalization does not change mesoporous structure so no comparison has been carried out between as-synthesized and functionalized NPs.26 SEM image shows the nanoparticles linked to nanofibers after the cross-linking step, due to covalent bonding as evidenced from FTIR analysis.
 | ||
| Fig. 2 HRTEM image for (a) MCM-NH2, and (b) MCM-PEI; and SEM images for (c) mPAN, (d) mPAN/MCM-NH2 and (e) mPAN/MCM-PEI nanocomposites. | ||
Zeta potential analyses of the different synthesized NPs were determined to confirm the functionalization with APTES and PEI precursors and to explain how the types of functionalization may be crucial for arsenic adsorption. ZP measurements were performed in water as a function of pH as shown in Fig. 3(a). For MCM-OH, surface charge is positive in acidic solution changing to negative in basic solution. The point of zero charge (pzc), or isoelectric point, corresponds to the condition where the electrical charge density on a surface is zero. In agreement with other reports, the point of zero charge (pHpzc) was found at 5.8 for MCM-OH.28 For amino functionalized NPs, the pHpzc shifted towards basic region. pHpzc was found to be 8.06 for MCM-NH2 and 11.3 for MCM-PEI, similar to what has been reported in the literature.36 The different pHpzc values showed that functionalization has effects on the surface charge and that pHpzc varies with the quantity of amine groups on the surface. Higher pHpzc found for MCM-PEI than for MCM-NH2 is consistent with the more amine groups present in PEI in comparison to APTES precursor. In acidic solutions, PEI is a polycation. According to Demadis et al.,37 PEI bears primary (36%), secondary (27%) and tertiary (36%) amine groups, with pKa values close to 4.5, 6.7 and 11.6, respectively. These results further justify the observations made during the FTIR studies.
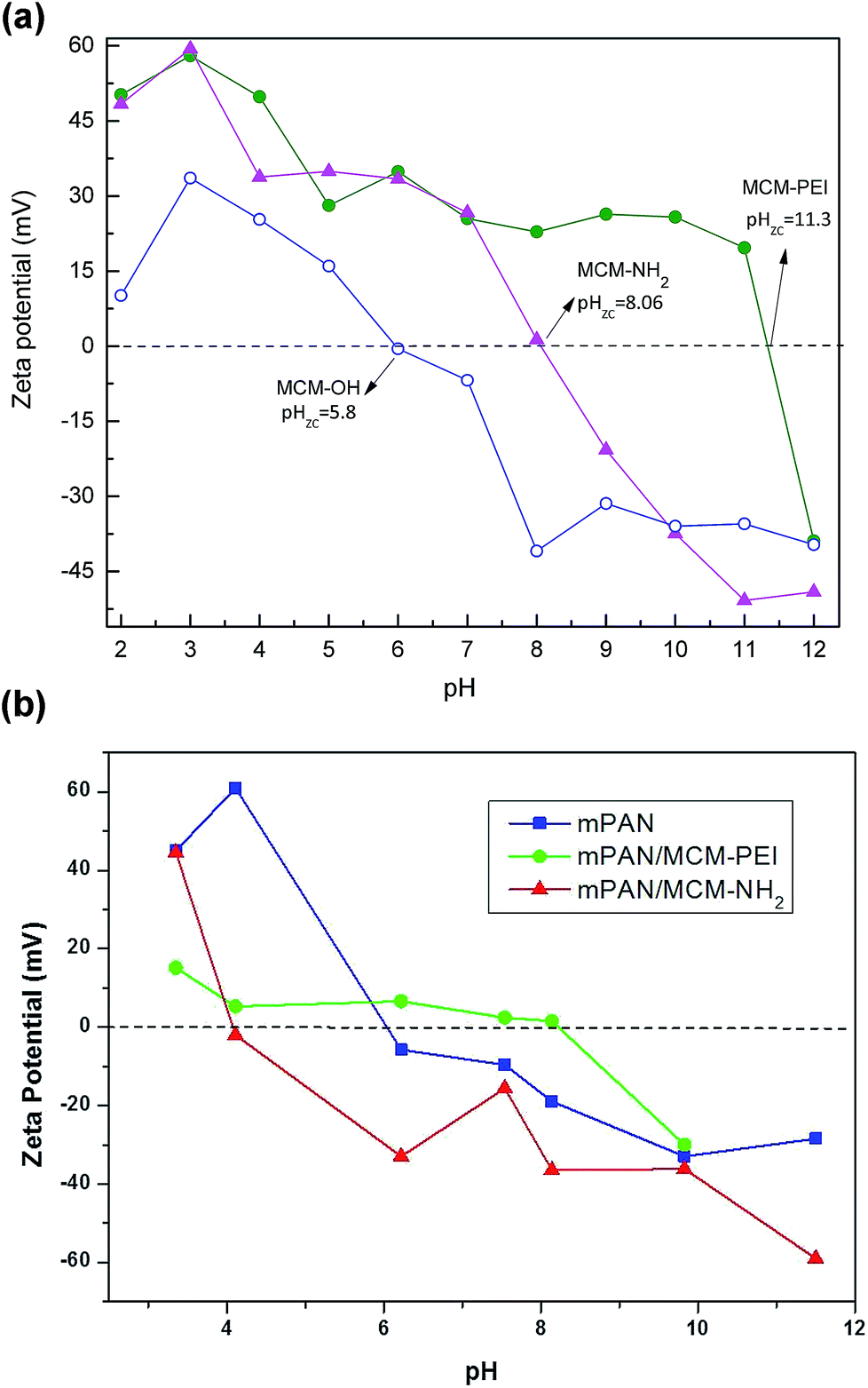 | ||
Fig. 3 Zeta potential and point-of-zero charge for (a) MCM-OH ( ), MCM-NH2 ( ), MCM-NH2 ( ) and MCM-PEI ( ) and MCM-PEI ( ); (b) mPAN ( ); (b) mPAN ( ), mPAN/MCM-NH2 ( ), mPAN/MCM-NH2 ( ) and mPAN/MCM-PEI ( ) and mPAN/MCM-PEI ( ). ). | ||
Zeta potential was also investigated for nanocomposites as shown in Fig. 3(b). As can be observed from the data, the mPAN surface has a PZC at approximately pH 6.0, making it negatively charged (−19 mV) and hence ineffective as an arsenic adsorbent at pH 8. On the contrary, mPAN/MCM-PEI has a low positive charge at pH 8.0 (+1.52 mV), which correlates well and explains its higher relative adsorption as compared to mPAN/MCM-NH2, which has a point-of-zero-charge at pH 4.2 and negative surface (−35 mV) at pH 8.0. The results here indicate that there is a moderate shift in the pHpzc towards the acidic side for both mPAN/MCM-PEI and mPAN/MCM-NH2 wherein the higher number of amine groups in PEI help in maintaining a more basic pHpzc. Effectively, it appears that the mPAN surface charge screens some of the negative MCM particle charges, reducing the net adsorption capacity of the As(V) species. Nonetheless what is interesting to note here is that when the functionalized MCM particles are supported on mPAN membranes, their specific adsorption capacity is considerably enhanced as compared to their colloidal form, indicating that not all free surface functional groups contribute to the adsorption process in the colloidal solution.
3.2. Adsorption studies
Adsorption is a surface-based process and a consequence of surface energy. The exact nature of the bonding depends on the involved species, but the adsorption process is generally classified as physisorption (characteristic of weak van der Waals forces), chemisorption (characteristic of covalent bonding) or electrostatic attraction between the adsorbate and the adsorbent. According to Benhamou et al.,28 large adsorption capacity of aminated materials may be explained by a local chemisorption mechanism between amines groups and As(V) species. Besides, pH of the solution is a critical parameter that rules arsenic removal since it may affect sorbent charge and arsenic speciation in solution. Arsenic is a triprotic acid that dissociates according to the following equations:28| H3AsO4 + H2O ⇆ H2AsO4− + H3O+, pKa1 = 2.2 | (1) |
| H2AsO4− + H2O ⇆ HAsO42− + H3O+, pKa2 = 6.9 | (2) |
| HAsO42− + H2O ⇆ AsO43− + H3O+, pKa3 = 11.5 | (3) |
Fig. 4 shows distribution of arsenic(V) species as a function of pH. Calculations were performed with Hydra/Medusa computer software38 with an initial concentration of 1.33 10−5 mol L−1 (1 mg L−1) of As(V). In the present work, arsenic solutions are studied at pH 8, simulating typical pH found in Argentinian groundwater,13 where arsenic is mostly present in anionic form, HAsO42−. Taking into account zeta potential values at pH 8, it is expected a neutral charge for MCM-NH2 and a positive charge for MCM-PEI.
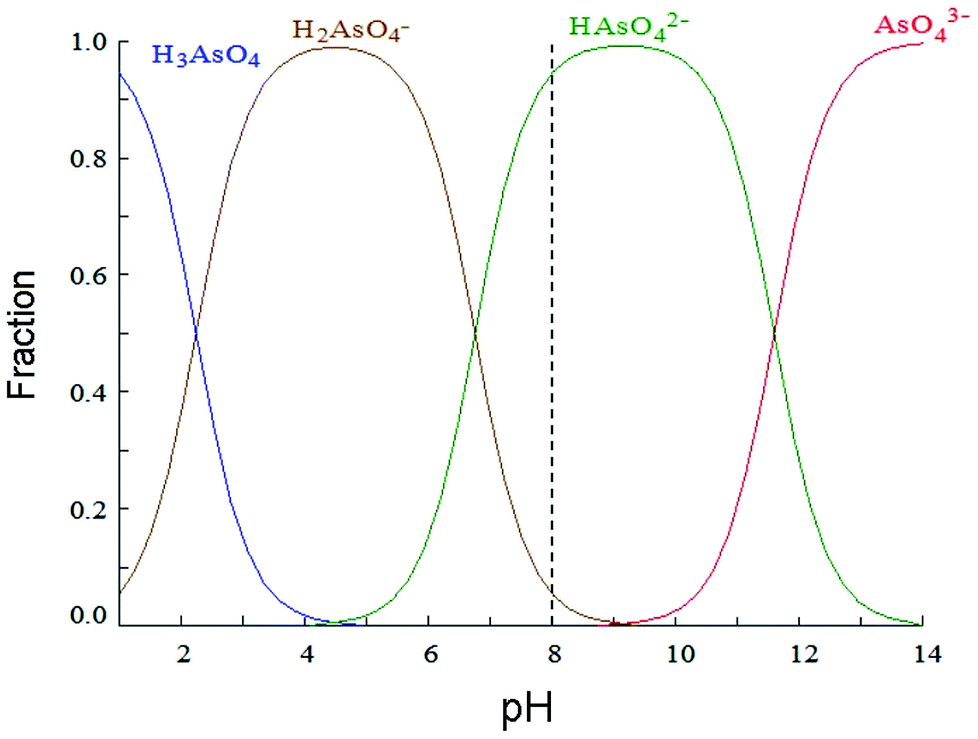 | ||
| Fig. 4 Speciation of As(V) according to solution pH as determined using Medusa software. | ||
Adsorption capacity (q) was calculated using the following equation:
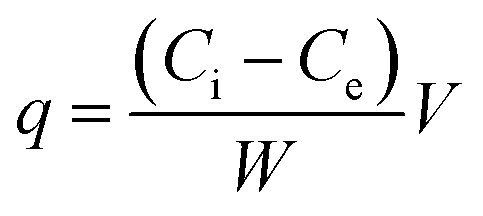 | (4) |
Adsorption tests performed with suspended MCM-NH2 and MCM-PEI nanoparticles in As(V) solution at pH = 8 revealed a capacity concentration at equilibrium, qe, of 0.003 mg g−1 with MCM-NH2 and 0.053 mg g−1 with MCM-PEI within 90 min of adsorption. In this case, considering qe and the charge of nanoparticles at pH 8, the higher and effective adsorption of arsenate species with MCM-PEI might be due to the negative charge of HAsO42− interacting with the positive charge of amino groups present in MCM-PEI nanoparticles. Contrary to the observations made by Benhamou et al.,28 the positively charged surface of MCM-PEI nanoparticles are expected to provide better adsorption for negative arsenate ions (HAsO42−) via electrostatic attraction.
It is important to consider that the adsorption of arsenic species with nanoparticles suspended in solution needs a further separation step, either centrifugation or filtration. This is inconvenient for applications in continuous flow reactors and is impractical since it would lead to higher complexities in operations. Moreover, suspended NPs tend to agglomerate in solution that has led to the studies of many immobilization matrices.39 In this work, nanoparticles were cross-linked with functionalized nanofibers. Adsorption tests with these membranes were carried out in batch mode operation using orbital shaking as it has been described in Section 1.4. As stated before, the amount of nanoparticles linked to the nanofibers was 20 ± 2 mg in each case, which represents a 10% in weight of the adsorbent. Results of adsorption characteristics showed qe = 0.05 mg g−1 for mPAN/MCM-NH2 and qe = 1 mg g−1 for mPAN/MCM-PEI. These values represent an increment of nearly twenty times of adsorbed arsenic compared to suspended nanoparticles, indicating that dispersed NPs on mPAN surface have higher efficiency in the adsorption of As(V). However, particles attached to the nanofibrous support lead to a reduction in the net positive charge which reduces HAsO42− adsorption.
To enhance the adsorption capacity further, membranes were treated with a Fe3+ solution. Fe3+ coordinated to amino ligands has been studied before by Yokoi et al.24 where it was reported that Fe3+ acts as a strong adsorbent due to its high selectivity to arsenic(V). Fe3+ in solution is easily captured by amino ligands which work effectively as adsorption sites for metal cations.40 Incorporation of Fe3+ has been studied by other authors as well.30,41 In order to increase the percentage of arsenic adsorption, nanocomposites were treated with an excess amount of Fe3+ solution. Adsorption tests performed using mPAN/MCM-NH2-Fe3+ and mPAN/MCM-PEI-Fe3+ membranes show substantial improvements. For comparison, Fig. 5(a) shows arsenic adsorption capacity obtained in each case studied after 90 minutes of exposure, where it can be seen the increase in the adsorption capacity of the nanocomposite membranes when treated with Fe3+. These results showed that the presence of amino groups on surface are responsible for increasing density of Fe3+ which plays the main role for arsenic adsorption. Fig. 5(b) shows arsenic adsorption capacity as a function of time. For mPAN/MCM-NH2-Fe3+ arsenic removal reached 87% in 15 minutes and 97% after 30 minutes. In the case of mPAN/MCM-PEI-Fe3+, 95% removal was observed during the first 15 minutes and after 30 minutes practically all the arsenic was removed. The difference in the percentage of adsorption between mPAN/MCM-PEI-Fe3+ and mPAN/MCM-NH2-Fe3+ shows that the presence of more amino groups on surface are responsible for increasing density of Fe3+ on surface, leading in an increment of the arsenic adsorbed.
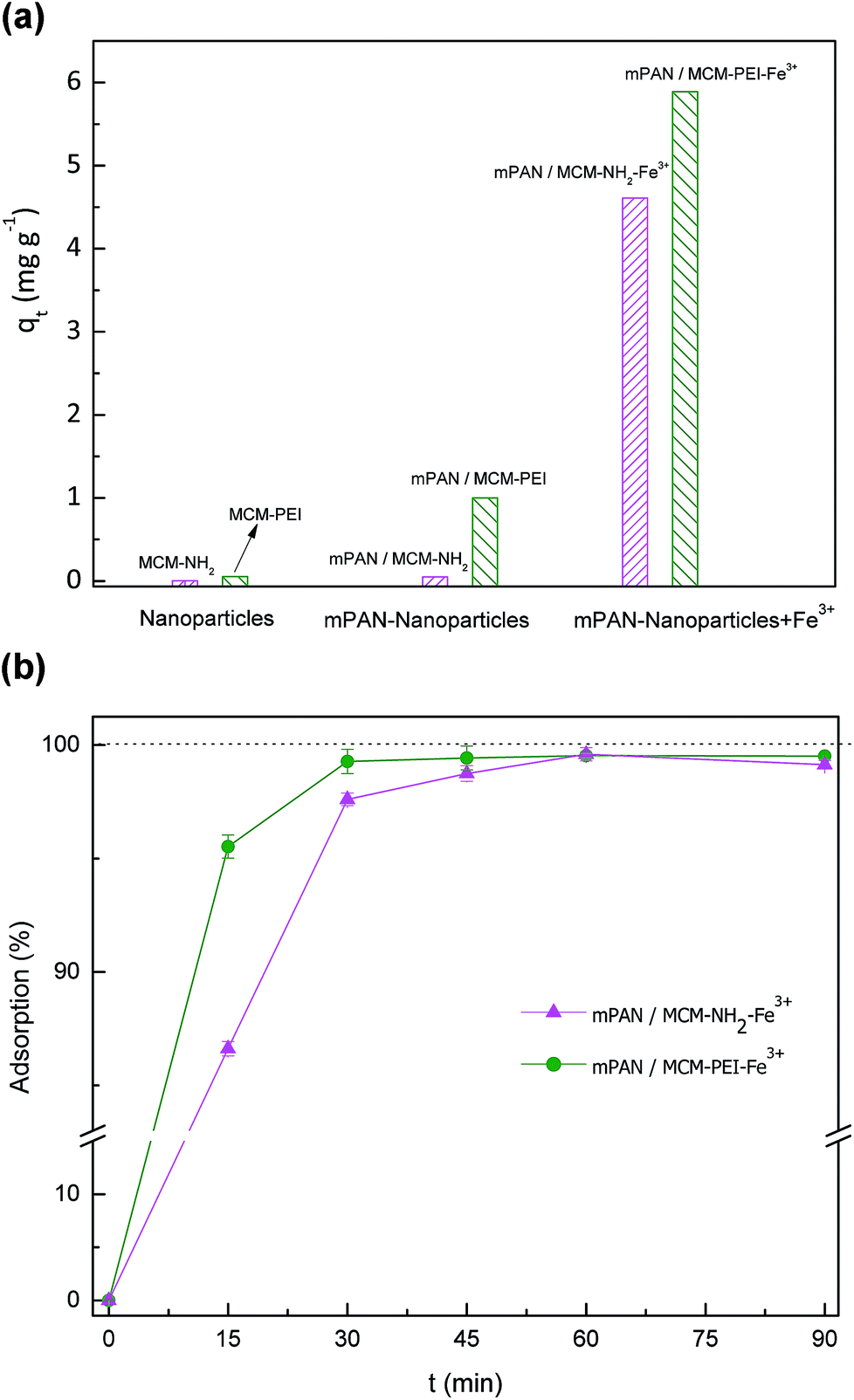 | ||
Fig. 5 (a) Adsorption percentage for comparison between nanoparticles, nanoparticles cross-linked to mPAN and with the incorporation of Fe3+; and (b) adsorption percentage for mPAN/MCM-NH2-Fe3+ ( ) and mPAN/MCM-PEI-Fe3+ ( ) and mPAN/MCM-PEI-Fe3+ ( ) as a function of time. ) as a function of time. | ||
3.3. Adsorption kinetics
Adsorption kinetics of As(V) was investigated in order to understand adsorption behavior. In Fig. 6 and 7, adsorption capacities (q) versus contact time are respectively shown for mPAN/MCM-NH2-Fe3+ and mPAN/MCM-PEI-Fe3+ nanocomposites membranes. The adsorption capacity for As(V) removal rapidly reached 4.04 mg g−1 and 5.66 mg g−1 within the first 15 min for mPAN/MCM-NH2-Fe3+ and mPAN/MCM-PEI-Fe3+, respectively. It can be noticed that for mPAN/MCM-NH2-Fe3+, adsorption reached equilibrium after 45 min with a removal efficiency of 98.98% while for mPAN/MCM-PEI-Fe3+ the equilibrium was reached after 30 min with a removal efficiency of 99.6%. After 90 min of exposure, the amount of adsorbed arsenic was 4.61 mg g−1 for mPAN/MCM-NH2-Fe3+ and 5.89 mg g−1 for mPAN/MCM-PEI-Fe3+.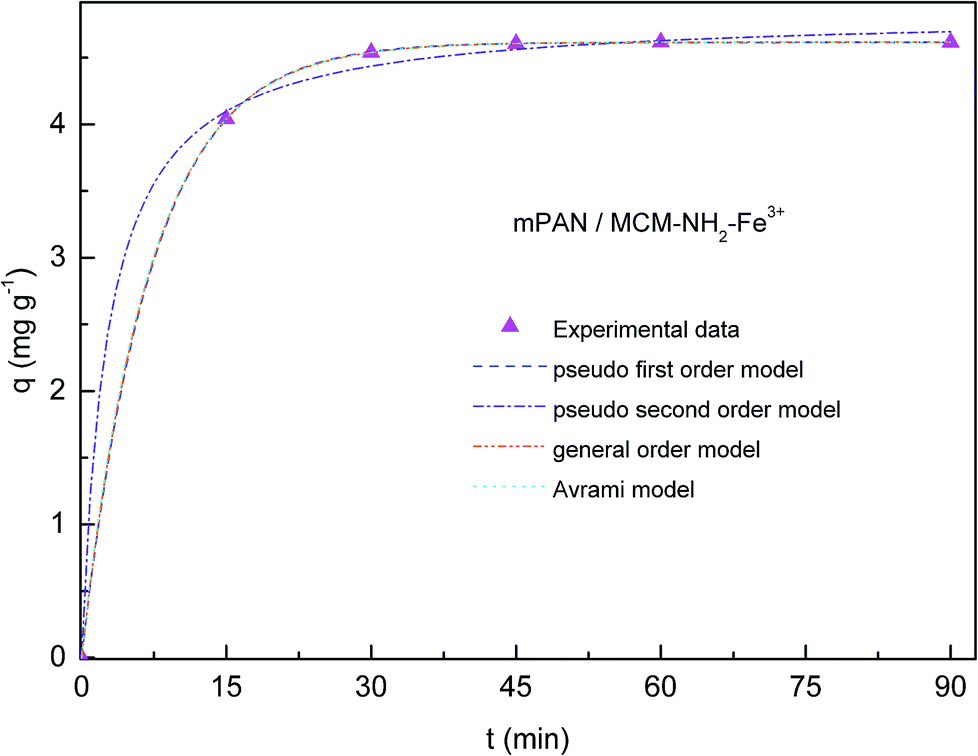 | ||
Fig. 6 Adsorption capacity for mPAN/MCM-NH2-Fe3+. Experimental data is represented by ( ). Dashed and dot lines represents kinetic models used for fitting. Experimental conditions: Initial concentration = 1 mg L−1, adsorbent amount = 20 mg, pH = 8. ). Dashed and dot lines represents kinetic models used for fitting. Experimental conditions: Initial concentration = 1 mg L−1, adsorbent amount = 20 mg, pH = 8. | ||
 | ||
Fig. 7 Adsorption capacity for mPAN/MCM-PEI-Fe3+. Experimental data is represented by ( ). Dashed and dot lines represents kinetic models used for fitting. Experimental conditions: initial concentration = 1 mg L−1, adsorbent amount = 20 mg, pH = 8. ). Dashed and dot lines represents kinetic models used for fitting. Experimental conditions: initial concentration = 1 mg L−1, adsorbent amount = 20 mg, pH = 8. | ||
The obtained results show that the synthesized nanocomposites have a high adsorption efficiency and this characteristic is typical for porous adsorbents with high surface area and mesopore sizes that allow easy diffusion of arsenic species into the mesoporous channels.42 High adsorption of As(V) on the surface of mPAN/MCM-NH2-Fe3+ and mPAN/MCM-PEI-Fe3+ within a short time may be attributed to the availability of a large number of active binding sites (Fe3+) on the surface of the nanoparticles attached to the nanofibrous support. The density of adsorbed Fe3+ increases with an increase in the surface density of the amino groups. These results are in agreement with a previously reported work.24
The experimental data was fitted using the nonlinear regression of pseudo-first order (eqn (5)), pseudo-second order (eqn (6)), general-order (eqn (7)) and the Avrami (eqn (8)) kinetic models:
| qt = qe,cal(1 − exp−k1t) | (5) |
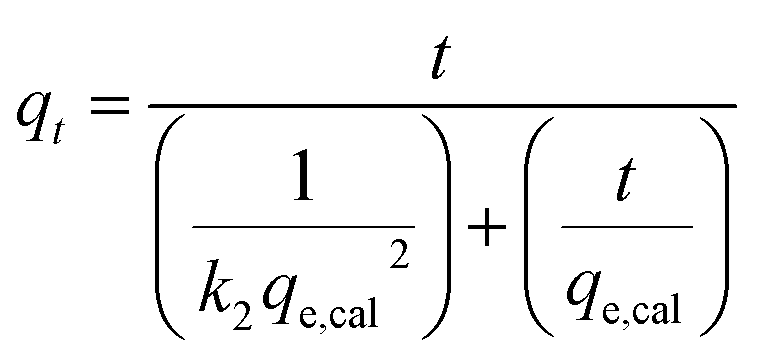 | (6) |
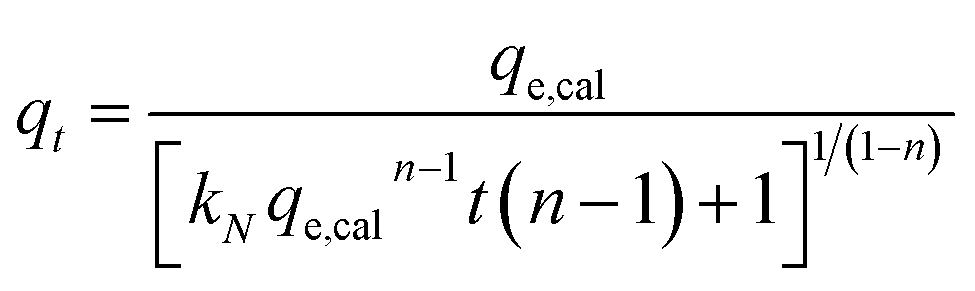 | (7) |
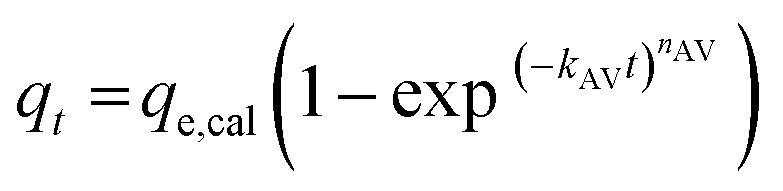 | (8) |
The coefficient of determination (R2), adjusted coefficient of determination (Radj2), and the standard deviation (SD) was used to test the best fitting of the kinetic model to the experimental data:
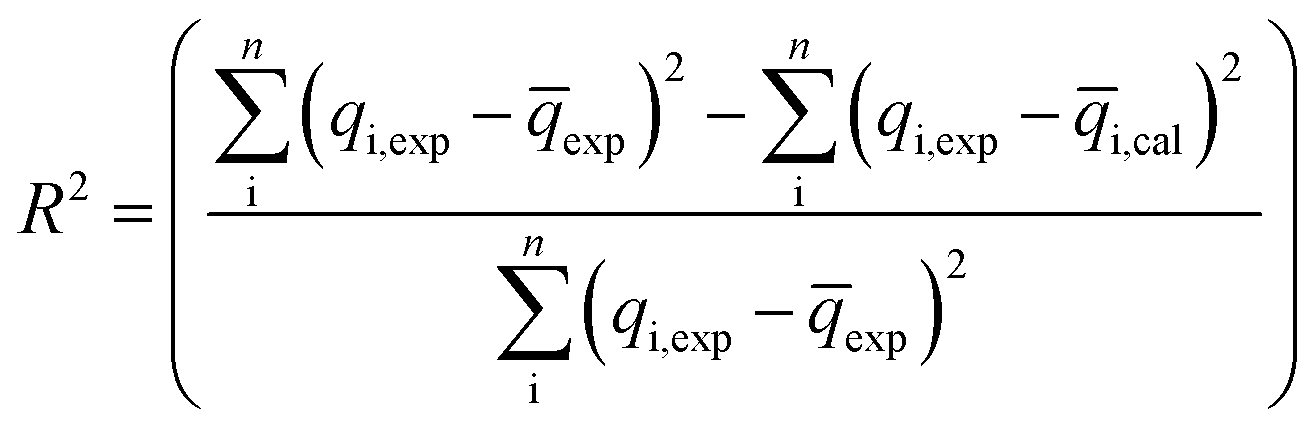 | (9) |
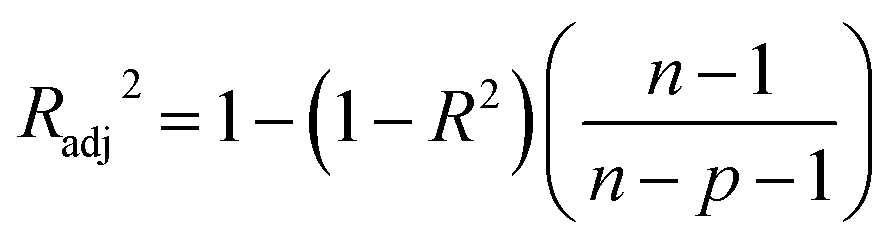 | (10) |
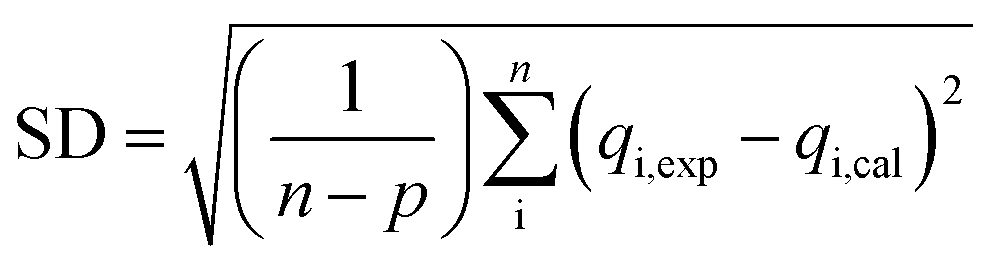 | (11) |
![[q with combining macron]](https://www.rsc.org/images/entities/i_char_0071_0304.gif) exp is the average of experimental q values; n represents the number of experiments; and p represents the number of parameters in the model.
exp is the average of experimental q values; n represents the number of experiments; and p represents the number of parameters in the model.
In Table 1, kinetic parameters obtained from model fitting are presented. High Radj2 and low SD reveals good adjustment between experimental data and theoretical models. In this case, when comparing qe,cal, qe,exp, Radj2 and SD values between the different kinetic models, it can been seen that the pseudo first order model fits better to experimental data for both nanocomposite membranes studied, meaning that the rate-controlling step might be a physical process.5,25
 |
However, kinetic models cannot be specifically used to determine an adsorption mechanism for the system under study. The prediction of the rate-limiting step is an important factor to be considered in the adsorption process. According to Rengaraj et al.43 and Kalavathy et al.,44 for the solid–liquid adsorption process, the solute transfer is characterized by external transfer and/or intra-particle diffusion, and the relative mechanisms of adsorption include three steps: (i) solute diffusion from bulk solution to the adsorbent exterior surface (external diffusion); (ii) diffusion of the adsorbate within the pores of the adsorbent (intra-particle diffusion); (iii) adsorption of the adsorbate onto active sites on the adsorbent. The last step is assumed to be fast, so it is negligible. To reach out a mechanism for As(V) adsorption, the intra-particle diffusion model can be investigated as proposed by Weber and Morris.45 According to this theory, the adsorbate uptake, qt, varies almost proportionately with the square root of contact time, t1/2 rather than t, according to eqn (12):
| qt = Kit1/2 + C | (12) |
In this work, adsorption curves fitted with Weber and Morris model (see ESI 2†) do not follow a linear behavior suggesting that the intraparticle diffusion is not the dominant process determining the kinetics of the adsorption process.29 Experimental data exhibit three linear zones suggesting that the overall adsorption process may indeed be controlled either by one or more steps, or a combination of steps. It is assumed that the first linear portion (zone 1) represents the diffusion process of mass transfer onto the adsorbent surface. The second linear portion (zone 2) is the gradual adsorption stage ascribed to intra-particle diffusion.46 The third linear portion (zone 3) can be regarded as the diffusion through smaller pores, followed by the establishment of equilibrium.47 Extended studies are being carried out to establish a possible mechanism for As(V) adsorption onto these nanocomposite membranes.
4. Conclusions
In this work, nanocomposite functionalized membranes were synthesized, characterized and tested for aqueous As(V) adsorption. Amino functionalized silica nanoparticles were cross-linked to functionalized polyacrylonitrile nanofibers, resulting in a homogenous nanocomposite fibrous membrane capable of removing As(V), fast and efficiently. Experiments showed low efficiency for arsenic adsorption using mPAN/MCM-NH2 and mPAN/MCM-PEI. However, with the addition of small amounts of Fe3+, adsorption reached almost 98% within the first 30 min. The equilibrium adsorption capacity for mPAN/MCM-NH2-Fe3+ and mPAN/MCM-PEI-Fe3+ were 4.61 mg g−1 and 5.89 mg g−1, respectively, both following a pseudo-first order kinetic model for adsorption process. Adsorption equilibrium was obtained within 30 min with mPAN/MCM-NH2-Fe3+ and 15 min with mPAN/MCM-PEI-Fe3+, the fast adsorption rate suggested that NH2 groups were readily available and easily accessible. Thus, nanocomposite membranes synthesized in this work are easy to apply and effective materials for the adsorption As(V).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by Argentina National Scientific and Technical Research Council (CONICET) and Nanoremovas project: H2020-MSCA-RISE-2014 [grant number 645024], “Advanced multifunctional nanostructured materials applied to remove arsenic in Argentinian groundwater”.References
- B. K. Mandal and K. T. Suzuki, Talanta, 2002, 58, 201–235 CrossRef CAS PubMed.
- WHO, World Health Organization Guidelines, 2011 Search PubMed.
- B. Emo, C. Eberlin, E. G. Kalaf, J. Laktas and S. Sell, J. Environ. Chem. Eng., 2017, 5, 232–239 CrossRef.
- F. Cubadda, B. P. Jackson, K. L. Cottingham, Y. O. Van Horne and M. Kurzius-Spencer, Sci. Total Environ., 2017, 579, 1228–1239 CrossRef CAS PubMed.
- C. G. Lee, P. Alvarez, A. Nam, S.-J. Park, T. Do, U.-S. Choi and S.-H. Lee, J. Hazard. Mater., 2017, 325, 223–229 CrossRef CAS PubMed.
- USEPA, National Primary Drinking Water Regulations, United States Environmental Protection Agency, 2001 Search PubMed.
- M. I. Litter, Rev. Soc. Argent. Endocrinol. Ginecol. Reprod., 2010, 17, 5–10 Search PubMed.
- P. L. Smedley and D. G. Kinnirugh, Arsenic in goundwater and the environment, Essentials of Medical Geology, 2013, ch. 12, pp. 279–310 Search PubMed.
- M. Puntoriero, A. V Volpedo and A. Fernandez Cirelli, Front. Environ. Sci., 2014, 2, 1–5 Search PubMed.
- H. B. Nicolli, J. Bundschuh, J. W. Garcia, C. M. Falcon and J. S. Jean, Water Res., 2010, 44, 5589–5604 CrossRef CAS PubMed.
- B. K. Benitez and K. T. Suzuki, Hidroarsenisismo Crónico Regional Endémico Módulo: Abatimiento del Arsénico, Ministerio de Salud la Nación, Buenos Aires, Argentina, 1st edn, 2012 Search PubMed.
- A. E. Bardach, A. Ciapponi, N. Soto, M. R. Chaparro, M. Calderon, A. Briatore, N. Cadoppi, R. Tassara and M. I. Litter, Sci. Total Environ., 2015, 538, 802–816 CrossRef CAS PubMed.
- A. Galli, O. Acosta, E. Planes, G. Pérez and M. Valiente, [D1.2] Nanoremovas Progress Report-M18, GA 645024. Groundwater status in target area. Comparison vs. regional situation, UAB, Universidad Autónoma de Barcelona (España)-INTI, Instituto Nacional de Tecnología Industrial, Argentina, 2016 Search PubMed.
- S. Kuppusamy, T. Palanisami, M. Megharaj, K. Venkateswarlu and R. Naidu, Rev. Environ. Contam. Toxicol., 2016, 236, 117–192 CrossRef CAS PubMed.
- M. Li, C. Wang and M. J. O'Connell, Environ. Sci.: Nano, 2015, 2, 245–250 RSC.
- K. Gupta, T. Basu and U. C. Ghosh, J. Chem. Eng. Data, 2009, 54, 2222–2228 CrossRef CAS.
- K. Gupta, S. Saha and U. C. Ghosh, J. Nanopart. Res., 2008, 10, 1361–1368 CrossRef CAS.
- Y. Zhang, X. M. Dou, M. Yang, H. He, C. Y. Jing and Z. Y. Wu, J. Hazard. Mater., 2010, 179, 208–214 CrossRef CAS PubMed.
- S. Zhang, H. Niu, Y. Cai, X. Zhao and Y. Shi, Chem. Eng. J., 2010, 158, 599–607 CrossRef CAS.
- Z. Veličković, G. D. Vuković, A. D. Marinković, M.-S. Moldovan, A. A. Perić-Grujić, P. S. Uskoković and M. Đ. Ristić, Chem. Eng. J., 2012, 181–182, 174–181 CrossRef.
- P. R. Aranda, I. Llorens, E. Perino, I. De Vito and J. Raba, Environmental Nanotechnology, Monitoring and Management, 2016, 5, 21–26 CrossRef.
- S. Vadahanambi, S. H. Lee, W. J. Kim and I. K. Oh, Environ. Sci. Technol., 2013, 47, 10510–10517 CAS.
- K. Wantala, S. Sthiannopkao, B. Srinameb, N. Grisdanurak and K.-W. Kim, J. Environ. Eng., 2012, 138, 119–128 CrossRef CAS.
- T. Yokoi, T. Tatsumi and H. Yoshitake, J. Colloid Interface Sci., 2004, 274, 451–457 CrossRef CAS PubMed.
- H. Alijani and Z. Shariatinia, Chemosphere, 2017, 171, 502–511 CrossRef CAS PubMed.
- Y. Wu, Y. Jin, J. Cao, P. Yilihan, Y. Wen and J. Zhou, J. Ind. Eng. Chem., 2014, 20, 2792–2800 CrossRef CAS.
- H. Yoshitake, T. Yokoi and T. Tatsumi, Chem. Mater., 2002, 14, 4603–4610 CrossRef CAS.
- A. Benhamou, J. P. Basly, M. Baudu, Z. Derriche and R. Hamacha, J. Colloid Interface Sci., 2013, 404, 135–139 CrossRef CAS PubMed.
- Y. Glocheux, A. B. Albadarin, J. Galán, E. Oyedoh, C. Mangwandi, C. Gérente, S. J. Allen and G. M. Walker, Microporous Mesoporous Mater., 2014, 198, 101–114 CrossRef CAS.
- S. El Mourabit, M. Guillot, G. Toquer, J. Cambedouzou, F. Goettmann and A. Gransjean, RSC Adv., 2012, 2, 10916–10924 RSC.
- A. D. Dwivedi, N. D. Sanandiya, J. P. Singh, S. M. Husnain, K. H. Chae, D. S. Hwang and Y.-S. Chang, ACS Sustainable Chem. Eng., 2017, 5, 518–528 CrossRef CAS.
- D. Morillo, A. Uheida, G. Pérez, M. Muhammed and M. Valiente, J. Colloid Interface Sci., 2015, 438, 227–234 CrossRef CAS PubMed.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- W.-S. Hung, C.-H. Tsou, M. De Guzman, Q.-F. An, Y.-L. Liu, Y.-M. Zhang, C.-C. Hu, K.-R. Lee and J.-Y. Lai, Chem. Mater., 2014, 26, 2983–2990 CrossRef CAS.
- F. Huang, Y. Xu, S. Liao, D. yang, Y.-L. Hsieh and Q. Wei, Materials, 2013, 6, 969–980 CrossRef CAS PubMed.
- J. M. Rosenholm, A. Penninkangas and M. Lindén, Chem. Commun., 2006, 37, 3909–3911 RSC.
- K. D. Demadis, M. Paspalaki and J. Theodorou, J. Ind. Eng. Chem., 2011, 50, 5873–5876 CrossRef CAS.
- Software Medusa Hydra, Chemical Equilibrium Diagrams Search PubMed.
- M. López-García, M. Martínez-Cabanas, T. Vilariño, P. Lodeiro, P. Rodríguez-Barro, R. Herrero and J. L. Barriada, Chem. Eng. J., 2017, 311, 117–125 CrossRef.
- A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Ed. Limusa, 1978, p. 1173 Search PubMed.
- H. Yoshitake and T. Yokoi, Chem. Mater., 2003, 15, 1713–1721 CrossRef CAS.
- M. S. Moorthy, D. J. Seo, H. J. Song, S. S. Parka and C. S. Ha, J. Mater. Chem. A, 2013, 1, 12485–12496 RSC.
- S. Rengaraj, J. W. Yeon, Y. Kim, Y. Jung, Y. K. Ha and W. H. Kim, J. Hazard. Mater., 2007, 143, 469–477 CrossRef CAS PubMed.
- M. H. Kalavathy, T. Karthikeyan, S. Rajgopal and L. R. Miranda, J. Colloid Interface Sci., 2005, 292, 354–362 CrossRef CAS PubMed.
- W. J. Weber and J. C. Morris, Proc. 1st Int. Conf. Water Pollut. Symp., 1962, vol. 2, pp. 231–266 Search PubMed.
- P. N. Diagboya, B. I. Olu-Owolabi and K. O. Adebowale, J. Environ. Manage., 2014, 146, 42–49 CrossRef CAS PubMed.
- F. M. Machado, C. P. Bergmann, E. C. Lima, B. Royer, F. E. de Souza, I. M. Jauris, T. Calveted and S. B. Fagan, Phys. Chem. Chem. Phys., 2012, 14, 11139–11153 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09866b |
| This journal is © The Royal Society of Chemistry 2019 |