
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceYttrium and europium separation by solvent extraction with undiluted thiocyanate ionic liquids†
Raju Banda,
Federica Forte ,
Bieke Onghena and
Koen Binnemans*
,
Bieke Onghena and
Koen Binnemans*
KU Leuven, Department of Chemistry, Celestijnenlaan 200F PO Box 2404, Heverlee, 3001 Leuven, Belgium. E-mail: Koen.Binnemans@kuleuven.be
First published on 7th February 2019
Abstract
An yttrium/europium oxide obtained by the processing of fluorescent lamp waste powder was separated into its individual elements by solvent extraction with two undiluted ionic liquids, trihexyl(tetradecyl)phosphonium thiocyanate, [C101][SCN], and tricaprylmethylammonium thiocyanate, [A336][SCN]. The best extraction performances were observed for [C101][SCN], by using an organic-to-aqueous volume ratio of 1/10 and four counter-current extraction stages. The loaded organic phase was afterwards subjected to scrubbing with a solution of 3 mol L−1 CaCl2 + 0.8 mol L−1 NH4SCN to remove the co-extracted europium. Yttrium was quantitatively stripped from the scrubbed organic phase by deionized water. Yttrium and europium were finally recovered as hydroxides by precipitation with ammonia and then calcined to the corresponding oxides. The conditions thus defined for an efficient yttrium/europium separation from synthetic chloride solutions were afterwards tested on a leachate obtained from the dissolution of a real mixed oxide. The purity of Y2O3 with respect to the rare-earth content was 98.2%; the purity of Eu2O3 with respect to calcium was 98.7%.
Introduction
Rare-earth elements (REEs) are a group of seventeen chemical elements, specifically the fifteen lanthanides plus yttrium (Y) and scandium (Sc). Rare earths are moderately abundant in the Earth's crust and they occur in many minerals, but the major resources are just four, including bastnäsite, monazite, xenotime and ion-adsorption clays.1 Due to their different abundance in the Earth's crust, an unwanted co-production of other REEs usually occurs together with the ones needed for the major applications. This is the so-called balance problem.2 The ideal situation is a perfect match between demand and supply, which would result in the lowest market price. To mitigate the balance problem, different solutions can be considered, such as resources diversification, recycling, urban/landfill mining, substitution, reduced use and new high-volume applications.2–4Phosphors used in fluorescent lamps contain a significant amount of the heavy REEs europium (Eu), terbium (Tb) and yttrium in comparison to primary ores and their recycling would help to keep the balance of those elements.2 A fluorescent lamp consists of a glass tube filled with an inert gas and mercury vapor at low pressure and tungsten or stainless steel cathodes which are assembled at the end of the lamps. The internal surface of the glass tube is coated with a layer of phosphor powder to obtain the desired color rendering index. The powder consists of a blend of a red phosphor (Y2O3:Eu3+, YOX), a green phosphor (LaPO4:Ce3+,Tb3+, LAP; (Gd,Mg)B5O12:Ce3+,Tb3+, CBT; (Ce,Tb)MgAl11O19, CAT) and a blue phosphor (BaMgAl10O17:Eu2+, BAM).5,6 In addition to the tri-band phosphors, the powder also contains the halophosphate phosphor (Sr,Ca)10(PO4),(Cl,F)2:Sb3+,Mn2+ (HALO), which emits broad-band cold white light. The HALO phosphor represents the largest mass fraction in the lamp phosphor waste (50 wt%), but it does not contain any REE. The red phosphor YOX accounts for about 20 wt% and it has the highest intrinsic value, since it contains high concentrations of the critical heavy REEs Y and Eu.7 Many approaches have been proposed for the recovery of REEs from lamp phosphor waste.5,6,8–10 For example, in the HydroWEEE process, the fluorescent powder is treated by dissolution in sulfuric acid, after which the obtained leachate is further treated by precipitation with oxalic acid giving a mixed Y/Eu oxalate.11–14 Our research group developed a novel separation process to recover a mixed Y/Eu oxide by selective dissolution of the red phosphor YOX with the functionalized ionic liquid [Hbet][Tf2N], followed by precipitation–stripping with oxalic acid and calcination at 950 °C to regenerate the YOX.7 In a recent paper, we described the recovery of terbium and other REEs from the residue obtained after dissolution of HALO and YOX.15 It must be stated, however, that the market demand for phosphors has moved to LED lamps.4 For this reason the mutual separation of yttrium and europium from recycled YOX is required for their use in applications other than lamp phosphors. Different approaches have been developed to separate europium and yttrium,16–18 but the most common method is solvent extraction.19–21 It must be stressed that the separation processes are often not efficient due to the low separation factors between adjacent REEs (1.5 to 2 on average), so that many extraction stages are required in order to obtain the desired purity.22
Gaudernack and coworkers filed a patent on the solvent extraction of yttrium from ammonium nitrate solutions by using the thiocyanate and nitrate form of quaternary ammonium extractants, followed by scrubbing with ammonium nitrate solution and stripping with water or dilute acid solutions.23 Other solvent extraction studies were performed with 2-ethylhexyl phosphonic acid (PC-88A) for the separation of Y, Eu and Tb from spent fluorescent lamp leach solutions.24 Yttrium and europium were recovered with >98% and 100% purity, respectively, by using process simulation of a counter-current mixer-settler cascade followed by scrubbing and precipitation with oxalic acid. PC-88A was also used in the work reported by De Carolis et al. for the separation of yttrium and europium from spent fluorescent lamp leach solutions.25 Tunsu et al. reported the results on the selective extraction of yttrium and europium with Cyanex 572 from nitrate leach solutions.19 The stripping of the REEs from the loaded organic phase was carried out with 3 mol L−1 HCl, followed by precipitation with oxalic acid and calcination at 800 °C to obtain the corresponding yttrium and europium oxides with 99.82% and 91.6% purity, respectively. A process for the extraction and separation of yttrium and europium from thiocyanate leachates was developed which makes use of acid leaching, solvent extraction with trimethylbenzyl ammonium chloride, stripping, separation of europium from yttrium by selective dissolution in ethanol and thermal reduction to the metallic form.26 Thakur reported on the Y/Eu separation by D2EHPA and PC-88A from dilute HNO3/HCl/H2SO4 solutions.27 Rhodia (presently Solvay) used molecular extractants for the processing of spent fluorescent lamp leach solution to separate yttrium and europium and other REEs by solvent extraction.28 Vu et al. reported the selective recovery of europium and yttrium using various acid lixiviants, followed by solvent extraction.29 The obtained results showed that HNO3 had the best leaching efficiency and PRIMENE-JM provided a higher selectivity than D2EHPA for the extraction of europium over yttrium.
In the present paper, the thiocyanate ionic liquids trihexyl(tetradecyl)phosphonium thiocyanate, [C101][SCN], and tricaprylmethylammonium thiocyanate [A336][SCN] were tested for the extraction and separation of yttrium(III) and europium(III) from chloride feed solutions. The structure is reported in Fig. 1. These ionic liquids in undiluted form have been proven to be useful for the extraction of REEs and transition metals.30–34 The process was first optimized using synthetic solutions and afterwards applied on a leachate obtained by the dissolution of a real mixed oxide.
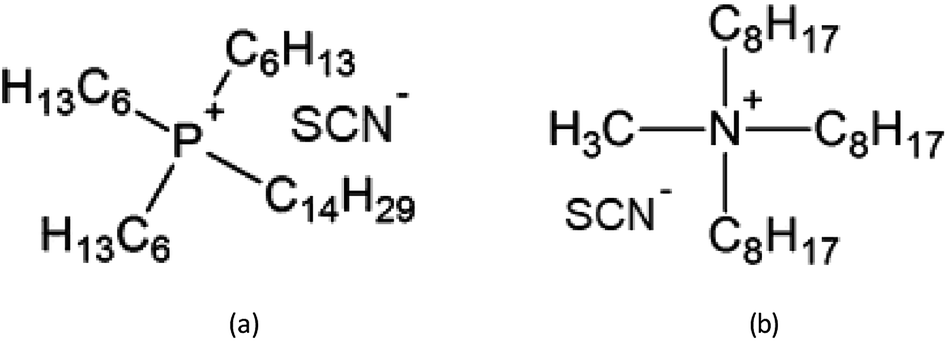 | ||
| Fig. 1 Structures of the ionic liquids used in this study: (a) [C101][SCN]; (b) [A336][SCN] (main component). | ||
Experimental
Chemicals
Ammonium thiocyanate (99%), calcium chloride dihydrate (>99.5%), and ammonium chloride (>99.8%) were purchased from Chem Lab (Zedelgem, Belgium). Ammonia solution (25%) was obtained from VWR Chemicals (Leuven, Belgium). Yttrium and europium chlorides (>99%) were purchased from Sigma Aldrich (Diegem, Belgium) and Alfa Aesar (Karlsruhe, Germany), respectively. Trihexyl(tetradecyl)phosphonium chloride (Cyphos® IL 101, [C101][Cl]) (97.7%) was purchased from Cytec Industries Inc. (Ontario, Canada). Tricaprylmethyl ammonium chloride (Aliquat 336, [A336][Cl]), a mixed quaternary ammonium salt containing mainly trioctylmethylammonium and tridecylmethylammonium chloride (88.2–90.6%) was purchased from Sigma-Aldrich (Diegem, Belgium). Oxalic acid (≥99.9%) and the standard solutions of Ga and Sm (1000 μg mL−1 in 3–5 wt% nitric acid) were obtained from Merck (Overijse, Belgium). Ethanol (PA absolute 99.8%) and Triton X-100 were purchased from Acros Organics (Geel, Belgium). The silicone–isopropanol solution was purchased from SERVA Electrophoresis GmbH (Heidelberg, Germany). Water was always of ultrapure quality, deionized to a resistivity of 18.2 MΩ cm with a Sartorius Arium® Pro ultrapure water system. All chemicals were used as received, without any further purification. The thiocyanate forms of the ionic liquids, [C101][SCN] and [A336][SCN], were synthetized according to a literature procedure.31 The chloride concentration in [C101][SCN] and [A336][SCN] was determined by both the AgNO3 test and TXRF analysis and it resulted to be negligible.Analytical techniques
The concentration of metals in the aqueous and organic phase was determined by total-reflection X-ray fluorescence spectrometry (TXRF) with a Bruker S2 Picofox TXRF spectrometer equipped with a molybdenum source.35,36 A small amount of ionic liquid (50 mg) was placed into propylene microtubes with an appropriate internal standard with the X-ray fluorescence energy as close as possible to that of the element to be determined, in order to reduce the effects caused by absorption of secondary X-rays, but minimising at the same time peaks overlap. Samples were further diluted with ethanol to 1 mL. After shaking the samples on a vibrating plate (IKA MS 3 basic), a small droplet (1.5 μL) was put on a quartz glass carrier, previously treated with a silicone–isopropanol solution to avoid spreading of the droplet. The quartz carriers were then dried for 30 min at 60 °C prior to analysis. Measurements in the aqueous phase were performed in the same way, but a 5 vol% Triton solution in water was used for diluting the sample. Each sample was measured for 500 s. Analyses were performed in duplicate and the error associated with the measurements was ±5%. A Mettler-Toledo S220 Seven Compact pH/Ion meter was used for pH measurements. The purity of the obtained oxide was measured via Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) using a Perkin Elmer Optima 8300 spectrometer equipped with an axial/radial dual plasma view, a GemTip Cross-Flow II nebulizer and a Scott double pass with inert Ryton spray chamber. The density of the two ionic liquids was measured with an Anton Paar DMA 4500M density meter and the viscosity with an Anton Paar LOVIS 2000 ME rolling-ball viscometer. Fourier Transform Infrared (FTIR) spectra of the initial ionic liquid and of the regenerated one were measured on a Bruker Vertex 70 spectrometer with a Platinum ATR module.Solvent extraction tests
Synthetic yttrium and europium solutions were prepared by dissolving hydrated YCl3 and EuCl3 in deionized water; the chloride concentration was set at 8 mol L−1 by addition of CaCl2. The general extraction and stripping experiments were carried out by using an organic-to-aqueous volume ratio (O/A) equal to 1. More specifically, 1 mL of the aqueous and organic phases were contacted at room temperature (T = 30 °C) for 30 min using a Nemus Life Thermo shaker (Model TMS-200). After each extraction and stripping experiment, samples were centrifuged using a Heraeus Megafuge 1.0 centrifuge for 3 minutes at 5000 rpm in order to separate the two phases. The distribution ratio (D) was calculated as the ratio between the metal concentration in the organic phase (Corg) to the metal concentration in the aqueous phase (Caq) at equilibrium:
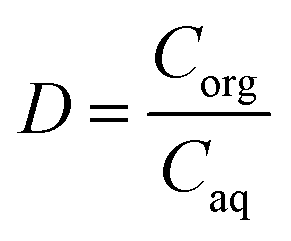 | (1) |
From the D value, the percentage extraction (%E) was calculated:
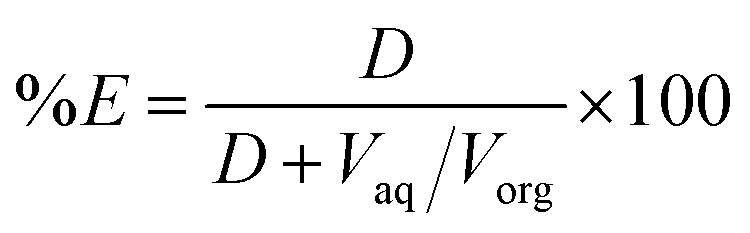 | (2) |
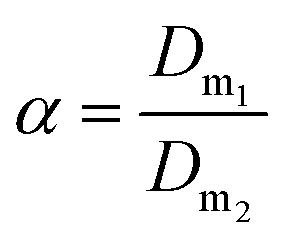 | (3) |
Precipitation and calcination tests
Precipitation of rare earths was performed by addition of an ammonia solution (25%). The precipitate was separated from the aqueous phase by filtration, placed in an oven for 24–48 h at 60–70 °C and finally calcined at 400–450 °C for 5 h to obtain the corresponding oxides Y2O3 and Eu2O3.Results and discussion
Solvent extraction tests with [C101][SCN] and [A336][SCN]
The viscosity and density of the water-saturated ionic liquids are reported in Table 1. Preliminary solvent extraction tests were carried out from synthetic solutions with yttrium and europium concentrations similar to the ones obtained by dissolving a mixed oxide from the HydroWEEE process (Y(III) = 3415 mg L−1, Eu(III) = 215 mg L−1).11–14 In order to determine the contact time necessary for the extraction system to reach the equilibrium, a set of experiments was carried with both [C101][SCN] and [A336][SCN] by varying the contact time from 5 to 30 minutes at O/A = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and T = 30 °C (Fig. S1†). It was found that equilibrium for [C101][SCN] was obtained within 15 minutes. For [A336][SCN], a slight increase in the extraction percentage was observed between 15 and 30 minutes. The percentage extraction values did not increase by increasing the contact time further in both systems. Based on these results, 30 minutes was selected as the optimal contact time. Further extraction tests were carried out as a function of the initial pH (Fig. S2†), which was varied from 1.3 to 3.8 by addition of dilute HCl or NaOH ([Cl−] = 8 mol L−1, t = 30 min, O/A = 1:1). The equilibrium pH was measured as well and the obtained values did not change by varying the initial pH, showing that there is no involvement of protons in the extraction process. The effect of temperature (30–60 °C) was also studied ([Cl−] = 8 mol L−1, t = 30 min, O/A = 1:1). It was found that extraction of Y(III) and Eu(III) slightly increased with increasing temperature in both systems, indicating that the extraction process is endothermic (Fig. S3†).
:1 and T = 30 °C (Fig. S1†). It was found that equilibrium for [C101][SCN] was obtained within 15 minutes. For [A336][SCN], a slight increase in the extraction percentage was observed between 15 and 30 minutes. The percentage extraction values did not increase by increasing the contact time further in both systems. Based on these results, 30 minutes was selected as the optimal contact time. Further extraction tests were carried out as a function of the initial pH (Fig. S2†), which was varied from 1.3 to 3.8 by addition of dilute HCl or NaOH ([Cl−] = 8 mol L−1, t = 30 min, O/A = 1:1). The equilibrium pH was measured as well and the obtained values did not change by varying the initial pH, showing that there is no involvement of protons in the extraction process. The effect of temperature (30–60 °C) was also studied ([Cl−] = 8 mol L−1, t = 30 min, O/A = 1:1). It was found that extraction of Y(III) and Eu(III) slightly increased with increasing temperature in both systems, indicating that the extraction process is endothermic (Fig. S3†).
| [C101][SCN] | [A336][SCN] | |
|---|---|---|
| Viscosity, mPa s | 224.5 | 202.1 |
| Density, g cm−3 | 0.898 | 0.892 |
Since [A336][SCN] and [C101][SCN] are both basic extractants, the anions from the aqueous phase are involved in the extraction mechanism to allow the formation of anionic complexes in the ionic liquid phase. Hence, salting-out agents play a significant role in the extraction mechanism. As shown in Fig. 2, the percentage extraction of Y(III) and Eu(III) increased by increasing the chloride concentration in the aqueous phase for both ionic liquids, indicating that chloride ions have an influence on the extraction mechanism. When [C101][SCN] was used, Y(III) was preferentially extracted over Eu(III). Y(III) extraction started at 2 mol L−1 CaCl2 and reached a maximum value (about 50%) at 3 mol L−1 CaCl2. At higher chloride concentrations, the percentage extraction did not increase significantly, but Eu(III) co-extraction occurred. Remarkably, the selectivity was reversed for extraction by [A336][SCN] (Fig. 3). This behaviour was also observed by Larsson et al.31 They reported a similar distribution ratio for Y(III) in both [A336][SCN] and [C101][SCN], while the distribution ratio of Eu(III) was significantly lower in [C101][SCN] than in [A336][SCN]. In other words, in [A336][SCN] the distribution ratio of Eu(III) was slightly higher than Y(III), implying a preference of extraction of Eu(III) over Y(III), which was also observed in this work. In [C101][SCN], the distribution ratio of Eu(III) was much lower than that of Y(III), implying a strong preference for Y(III) over Eu(III). The reason for the increase in the percentage extraction by increasing CaCl2 concentration is a combination of the adjustment of the water activity levels by the addition of a salting-out agent with high ionic strength, resulting in a weaker hydration of the lanthanide ions, and the fact that chloride anions are involved in the extraction equilibrium and have to be co-extracted in order to enable metal extraction. Therefore higher concentrations push the equilibrium to the right.31 The extraction mechanism for extraction of the trivalent rare-earth ions by the ionic liquids [C101][SCN] and [A336][SCN] can be represented by the following equation:
| M(aq)3+ + 3Cl(aq)− + x[Q][SCN](org) ⇆ [Q+]x−3[M(SCN)x3−x](org) + 3[Q]Cl(org) | (4) |
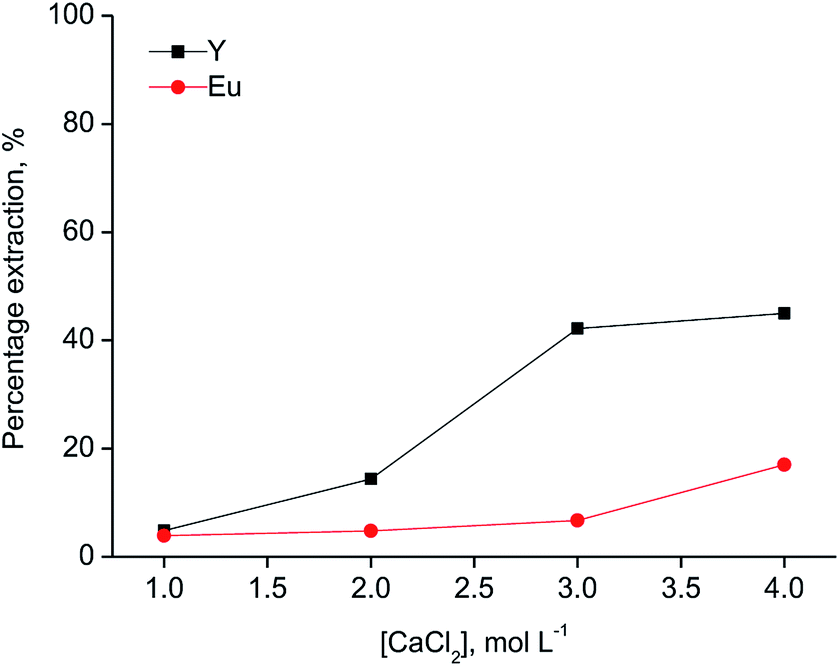 | ||
| Fig. 2 Extraction of Y(III) and Eu(III) with the ionic liquid [C101][SCN] as a function of the CaCl2 concentration (aqueous phase: [Y(III)] = 3145 mg L−1, [Eu(III)] = 215 mg L−1; O/A = 1:1, T = 30 °C). | ||
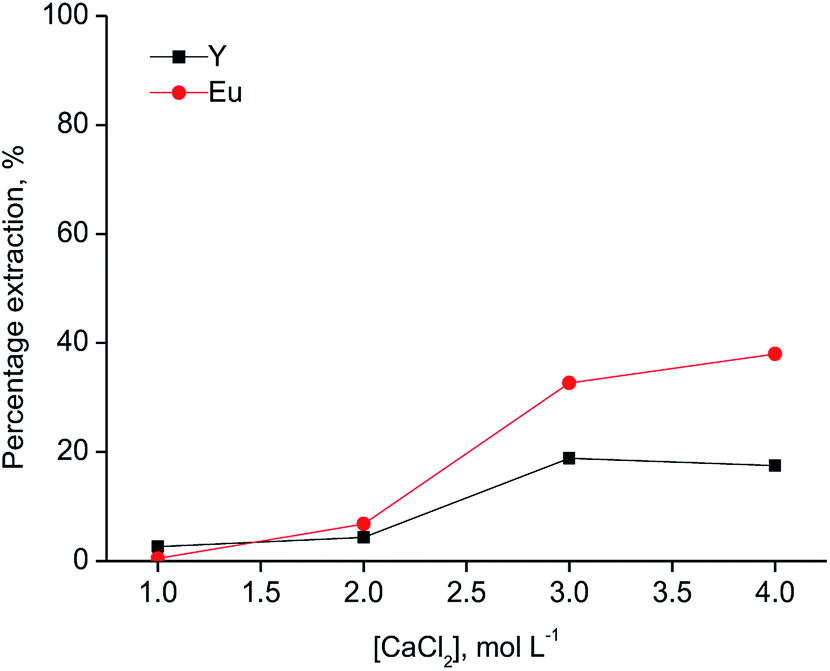 | ||
| Fig. 3 Extraction of Y(III) and Eu(III) with the ionic liquid [A336][SCN] as a function of the CaCl2 concentration (aqueous phase: [Y(III)] = 3145 mg L−1, [Eu(III)] = 215 mg L−1; O/A = 1:1, T = 30 °C). | ||
To increase the selective extraction of Y(III) over Eu(III) with [C101][SCN], further experiments were performed by addition of thiocyanate anions to the aqueous phase, with concentrations varying from 0.1 to 0.4 mol L−1 at different chloride concentration values (2–6 mol L−1). It was found that Y(III) extraction increased by increasing the thiocyanate concentration. At 1 mol L−1 CaCl2, the extraction efficiency was not significantly influenced by the thiocyanate concentration while the effect was more pronounced at 2 and 3 mol L−1 CaCl2 (Fig. 4). Quantitative extraction of Y(III) was achieved at a thiocyanate concentration of 0.2 mol L−1, with about 35% co-extraction of Eu(III). At higher thiocyanate concentrations, co-extraction of Eu(III) increases, reaching 75% at a thiocyanate concentration of 0.4 mol L−1. The increase in the percentage extraction is due to the involvement of thiocyanate ions in the extraction process.31,38 From these results, it can be stated that both chloride and thiocyanate ions are required to obtain quantitative and selective extraction of yttrium over europium. The best separation was achieved at 3 mol L−1 CaCl2 and 0.2 mol L−1 SCN, so that these conditions were selected as the optimal ones.
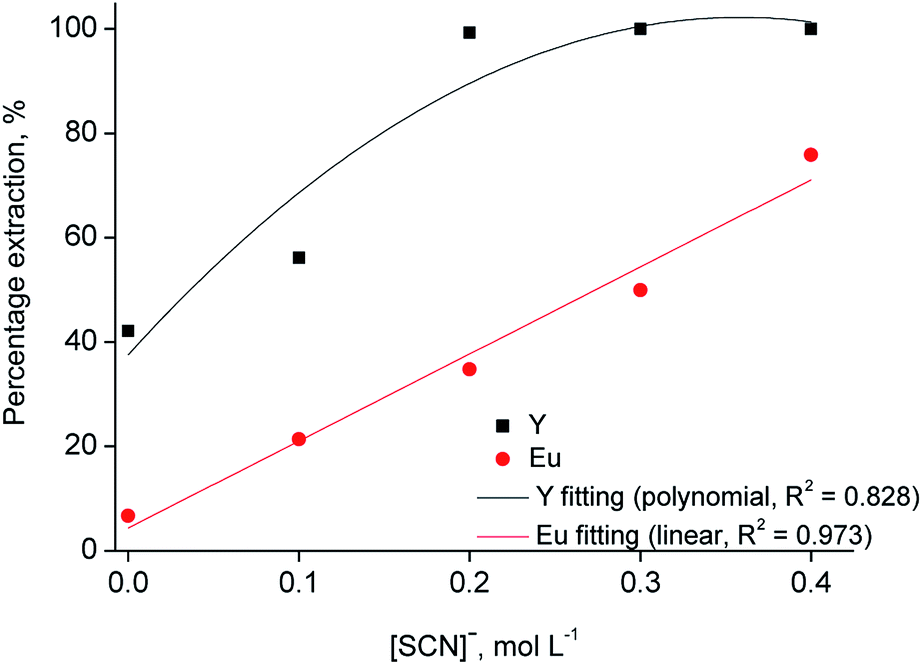 | ||
| Fig. 4 Extraction of Y(III) and Eu(III) with the ionic liquid [C101][SCN] as a function of the thiocyanate concentration (aqueous phase: [Y(III)] = 3145 mg L−1, [Eu(III)] = 215 mg L−1, [CaCl2] = 3 mol L−1; O/A = 1:1, T = 30 °C). | ||
Further tests, not reported here, were carried out by varying the thiocyanate concentration from 0.1 to 0.4 mol L−1 at different chloride concentration values (2–6 mol L−1) to get selective extraction of Eu(III) over Y(III) with [A336][SCN]. The highest separation factor (α = 17.6) was observed at 3 mol L−1 CaCl2 and 0.4 mol L−1 SCN−.
By the addition of SCN− to the aqueous phase, the extraction mechanism most likely does no longer correspond to eqn (4). Since SCN− has a higher hydrophobicity than Cl−, together with a higher affinity for coordination to rare earth ions, SCN− is most likely co-extracted to the ionic liquid phase, rather than Cl−. The expected modified extraction mechanism is presented in eqn (5):
| M3+ + 3SCN− + (x − 3)NR4SCN ⇆ NR4+(x−3)(M(SCN)x)3−x | (5) |
The selectivity for either Eu(III) or Y(III) was rather low in both ionic liquids and depends on the choice of ionic liquid. In the case of [C101][SCN], Y(III) was extracted, together with Eu(III), but the percentage extraction of Eu(III) was in all investigated experimental conditions lower than that of Y(III). For the extraction with [A336][SCN], Eu(III) was efficiently extracted, along with Y(III), and the percentage extraction of Eu(III) was always higher than that of Y(III). Therefore, the quaternary phosphonium thiocyanate ionic liquid [C101][SCN] is the most suitable for Y(III)/Eu(III) separation, due to the higher extraction efficiency and the higher separation factor values.
The theoretical number of counter-current stages required for quantitative extraction of Y(III) over Eu(III) by [C101][SCN] was predicted by constructing the McCabe–Thiele diagram. The extraction isotherm was determined by varying the organic-to-aqueous volume ratio from 1:30 to 1:1 and using a feed solution containing 0.2 mol L−1 SCN and 3 mol L−1 CaCl2. It was found that at least three counter-current stages are required for quantitative extraction of Y(III) at an O/A ratio equal to 1:1.5 (Fig. 5). The achieved loading of Y(III) into the ionic liquid phase was about 4.5 g L−1. A higher loading of Y(III) (about 29 g L−1) can be obtained from aqueous feed solutions containing 0.8 mol L−1 SCN− + 3 mol L−1 CaCl2 at O/A = 1:10 (Fig. 6). Three to four stages were found to be necessary for quantitative Y(III) extraction in a real counter-current setup.
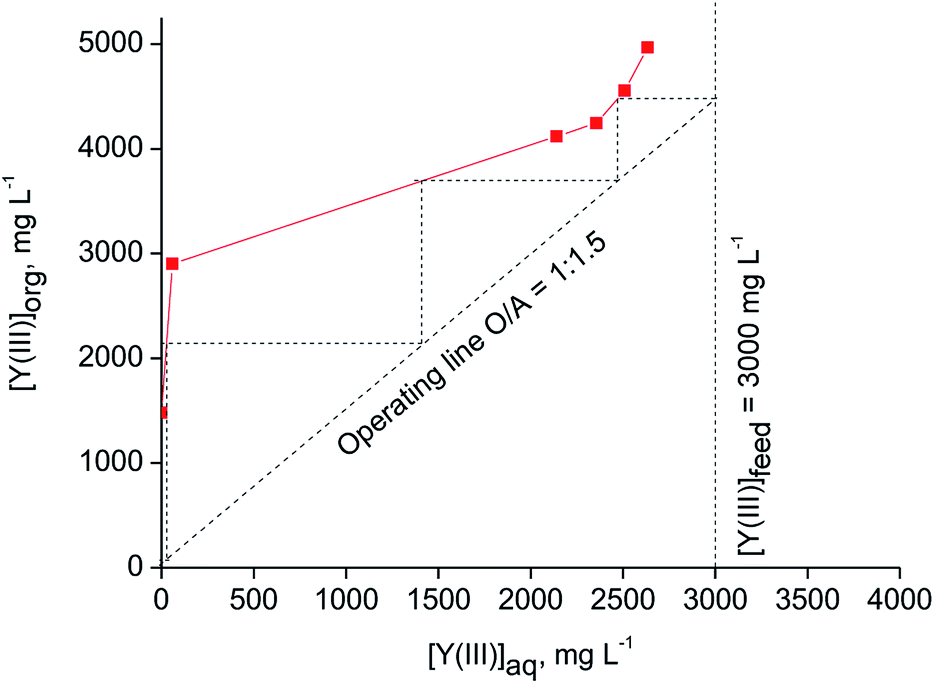 | ||
| Fig. 5 McCabe–Thiele diagram for extraction of Y(III) with [C101][SCN] (aqueous phase: [Y(III)] = 3000 mg L−1, [Eu(III)] = 215 mg L−1, [CaCl2] = 3 mol L−1, [NH4SCN] = 0.2 mol L−1; T = 30 °C). | ||
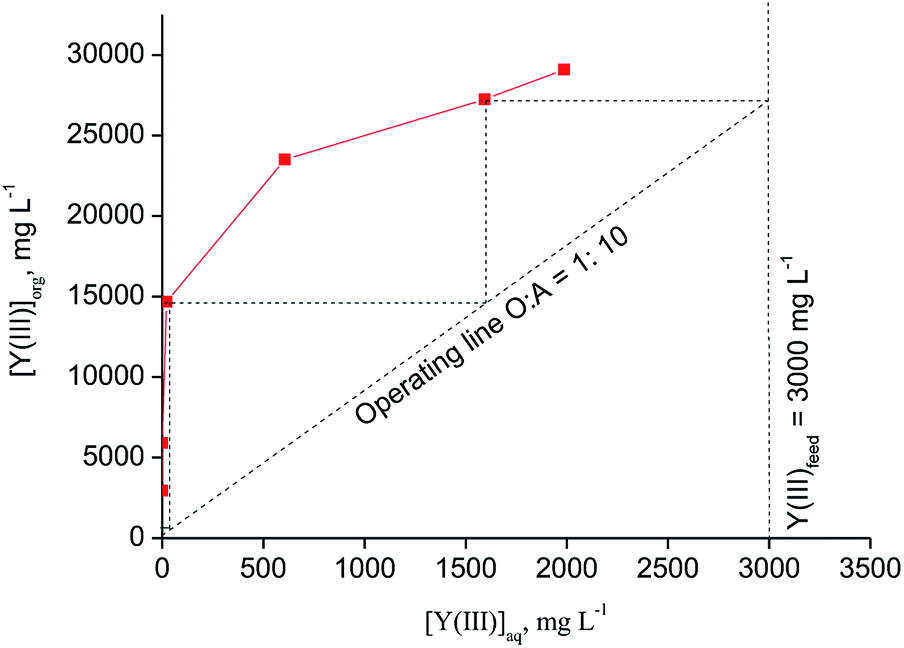 | ||
| Fig. 6 McCabe–Thiele diagram for extraction of Y(III) with [C101][SCN] (aqueous phase: [Y(III)] = 3000 mg L−1, [Eu(III)] = 215 mg L−1, [CaCl2] = 3 mol L−1, [NH4SCN] = 0.8 mol L−1; T = 30 °C). | ||
Scrubbing of [C101][SCN]
A sufficient amount of loaded ionic liquid phase [C101][SCN] was generated (O/A = 1:10, four counter-current stages, T = 30 °C, t = 30 min, 0.8 mol L−1 SCN and 3 mol L−1 CaCl2; loaded organic phase: [Y(III)] = 31709 ppm, [Eu(III)] = 338 ppm, [Ca(II)] = 989 ppm) and used for scrubbing and stripping tests. Besides Eu(III) and Y(III), also the scrubbing of Ca(II) was considered, since the mixed oxide material tested later in this work (vide infra) contains a high concentration of Ca(II). Scrubbing tests were carried out by contacting the loaded [C101][SCN] phase with various solutions (Table 2). Hot water, NH4Cl, NH4SCN, CaCl2 and NH4Cl + NH4SCN solutions did not show any selectivity towards the scrubbing of Eu(III) and Ca(II) over Y(III), while better results were achieved with the feed itself (i.e. the synthetic yttrium and europium solution): 80.7% Eu(III) was selectively scrubbed by contacting the [C101][SCN] ionic liquid phase with the feed at O/A = 1:10. Based on these one-stage scrubbing tests, it is expected that Eu(III) can be quantitatively scrubbed by applying multi-stage scrubbing. A similar scrubbing efficiency could be obtained with the mixture 3 mol L−1 CaCl2 + 0.8 mol L−1 NH4SCN at O/A = 1:1 in several cross-current simulations, but the loss of yttrium would be about 5–6%.
| Reagent | Scrubbing, % | ||
|---|---|---|---|
| Eu | Ca | Y | |
| Hot H2O | 66.2 | 70.8 | 45.2 |
| 2 mol L−1 NH4Cl | 51.8 | 53.6 | 30.5 |
| 2 mol L−1 NH4NO3 | 22.0 | 45.7 | 12.8 |
| 0.1 mol L−1 NH4SCN | 62.5 | 76.9 | 41.0 |
| 0.1 mol L−1 ascorbic acid | 64.5 | 85.8 | 44.6 |
| 6 mol L−1 NH4NO3 | 14.2 | 68.9 | 7.49 |
| 2 mol L−1 NH4Cl + 0.1 mol L−1 NH4SCN | 59.6 | 34.5 | 67.2 |
| 2 mol L−1 NH4NO3 + 0.1 mol L−1 NH4SCN | 28.7 | 17.5 | 51.7 |
| 2 mol L−1 CaCl2 | 37.8 | — | 31 |
| 3 mol L−1 CaCl2 | 21.7 | — | 15.7 |
| 3 mol L−1 CaCl2 + 0.8 mol L−1 SCN | 15 | — | 1 |
| Synthetic Y and Eu solution, O/A = 1:1 |
3.1 | — | — |
| Synthetic Y and Eu solution, O/A = 1:2 |
15.9 | — | — |
| Synthetic Y and Eu solution, O/A = 1:10 |
80.7 | — | — |
Yttrium recovery from the loaded [C101][SCN] phase
The scrubbed ionic liquid was contacted with an equal volume of various stripping agents at room temperature to recover yttrium (Table 3). About 42.4% stripping of Y(III) could be achieved in a single contact with deionized water. This percentage could be increased up to 70.1% by employing two cross-current stages. With 0.3 mol L−1 CaCl2, a similar stripping efficiency was observed. Diluted HCl and H2SO4 showed an efficient stripping of Y(III) which resulted to be quantitative at 20 vol% and 10 vol%, respectively. A white precipitate of metal salt was formed at high H2SO4 concentration (7% and 10%), which might be due to the formation of Y2(SO4)3. Similar stripping percentages were found by precipitation with oxalic acid solutions: over 99.9% of Y(III) could be stripped with 1 mol L−1 oxalic acid. Although equally good stripping results were achieved with HCl, H2SO4, oxalic acid and water, the latter is selected as the preferable stripping agent. If HCl, H2SO4 and oxalic acid would be used, an extra washing step would be necessary to allow recycling of the solvent in a continuous process. When using water, this extra washing step is avoided.| Stripping agent | Stripping, % |
|---|---|
| H2O | 42.4 |
| HCl 1% | 41.7 |
| HCl 5% | 47.9 |
| HCl 7% | 53.7 |
| HCl 10% | 59.6 |
| HCl 15% | 78.9 |
| HCl 20% | 100 |
| CaCl2 0.3 mol L−1 – stage 1 | 36.9 |
| CaCl2 0.3 mol L−1 – stage 2 | 70.4 |
| H2O – stage 2 | 70.1 |
| HCl 1% – stage 2 | 74.3 |
| HCl 5% – stage 2 | 82.9 |
| H2SO4 1% | 46.8 |
| H2SO4 5% | 52.6 |
| H2SO4 7% | 86.1 |
| H2SO4 10% | 99.8 |
| H2C2O4 0.1 mol L−1 | — |
| H2C2O4 0.5 mol L−1 | 66 |
| H2C2O4 1 mol L−1 | 99.9 |
The strip solution containing yttrium was subjected to precipitation with ammonia solution (25 vol%), followed by calcination at T = 400–450 °C for 5 h. The purity of the obtained Y2O3, as determined though ICP-OES analysis, was >99% with respect to Eu(III) and Ca(II). The stripped organic phase was recycled and tested for the extraction of Y(III) under similar conditions. The effluent streams after yttrium recovery could be treated to remove trace amounts of HSCN by activated carbon in order to meet the current environmental regulations.23
Precipitation of europium from the raffinate
To recover europium from the raffinate solutions, preliminary experiments were conducted by addition of various equivalent ratios of oxalic acid from 1 to 8 with respect to europium. The results showed no selective precipitation of Eu(III) over Ca(II). Europium recovery from the raffinate was therefore performed by precipitation in alkaline conditions by the addition of ammonia. At pH 8.6, 89% Eu(III) was precipitated as Eu(OH)3, while quantitative precipitation was achieved at pH 9.6, with a high selectivity over Ca(II). Co-precipitation of Ca(II) occurred at pH 10, so 9.6 was selected as the optimal pH value.Application of the process to the mixed REE oxide from HydroWEEE
The whole process described so far was also tested on a real mixed oxide obtained from the HydroWEEE process.11–14 The composition of the oxide was determined by dissolving the solid in 6 mol L−1 HCl. The yttrium and europium content was found to be 68.1 wt% and 5.1 wt%, respectively. Gadolinium (3.5%), cerium (0.9%) and terbium (0.6%) impurities were found to be present as well. A leaching step was then performed with 6 mol L−1 HCl by employing a liquid-to-solid ratio of 20 mL g−1. Full dissolution of the mixed oxide was achieved in 24 h at a temperature of 50 °C. The leachate was diluted ten times with deionized water and then CaCl2 and NH4SCN were added in order to adjust chloride and thiocyanate concentration to the desired values. After pH adjustment (pH = 4.2) with NaOH, the leachate ([Y(III)] = 2886 mg L−1), ([Eu(III)] = 165 mg L−1) was contacted with the undiluted ionic liquid at O/A = 1:10 in four counter-current steps. An yttrium extraction efficiency of 94% was achieved, with a few percentages of gadolinium, terbium and europium co-extracted. These results are in agreement with the ones reported from synthetic solutions, where three to four counter-current stages were necessary for quantitative Y(III) extraction. The loaded ionic liquid phase was further contacted with a mixture of 3 mol L−1 CaCl2 and 0.8 mol L−1 NH4SCN or with the feed (yttrium and europium solution coming from the leaching of the mixed oxide) to remove trace amount of co-extracted rare earths in five cross-current simulations. Yttrium was finally stripped with deionized water at O/A ratio 1:5 in two cross-current stages. Yttrium and europium were recovered from the solutions as their hydroxides by addition of an ammonia solution, followed by calcination at 400–450 °C. The purities of the yttrium and europium oxides were over 98.2% and 98.7%, respectively. Regarding the overall recovery efficiencies, they were 94% for Y(III) and 98% for Eu(III).
In the work of Nakamura et al. (2007), the purity of the obtained oxides was >98% for Y2O3 and 100% for Eu2O3, however only 53% of the Eu(III) was recovered.24 Tunsu et al. (2016) obtained Y(III) and Eu(III) oxides with purity equal to 99.82% and 91.6%, respectively; the major impurity in Eu2O3 was found to be gadolinium.19 Both processes make use of classical organic solvents for the solvent extraction step, such as PC-88A and Cyanex 572.
The regenerated ionic liquid [C101][SCN] was tested for the extraction of Y(III) under similar experimental conditions and it was found that the changes in the extraction efficiency are negligible. The FTIR of the initial ionic liquid phase and of the regenerated one have been recorded as well (Fig. S4†).
A flow sheet for the process is shown in Fig. 7. This process makes use of the undiluted ionic liquid [C101][SCN] for the separation of yttrium and europium from chloride solutions obtained by the dissolution of a mixed rare-earth oxide. Its main advantages are the use of only a few process steps, the possibility to regenerate the ionic liquid, thus reducing reagent consumption, and the relatively high purity of the obtained oxides.
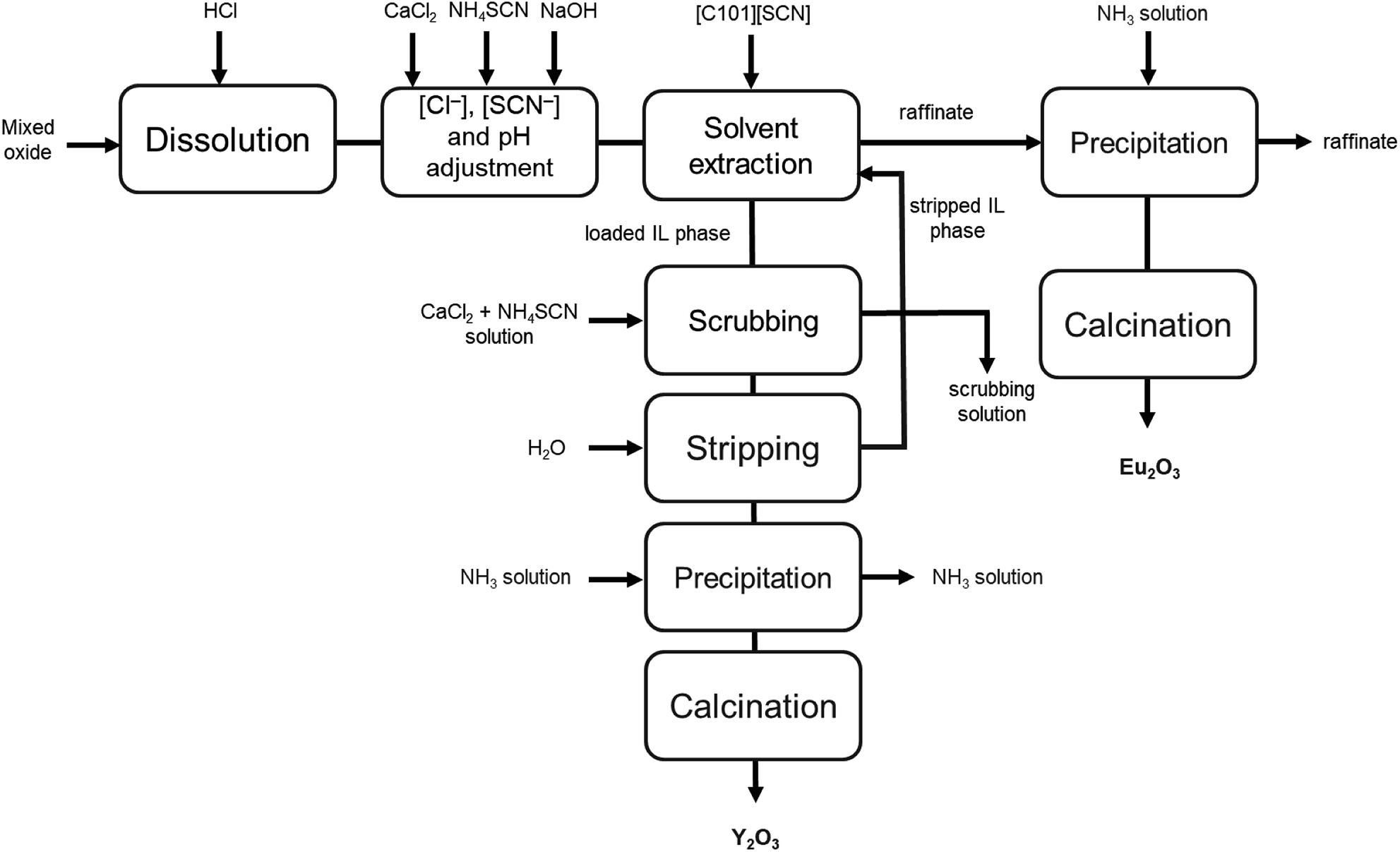 | ||
| Fig. 7 Process flow sheet for the separation of yttrium and europium from a mixed oxide recovered from lamp phosphor waste. | ||
Conclusions
In this work the separation of Y(III) and Eu(III) by undiluted quaternary ammonium and phosphonium thiocyanate ionic liquids was investigated. The process was first applied on synthetic solutions and then the conditions defined for an efficient yttrium/europium separation were tested on a real leachate obtained from the dissolution of a mixed yttrium/europium oxide recovered from lamp phosphor waste. It was found that extraction with the quaternary phosphonium ionic liquid [C101][SCN] (O/A = 1:10, four counter-current stages) was more effective than extraction with the quaternary ammonium ionic liquid [A336][SCN] under similar experimental conditions. The co-extracted Eu(III) could be scrubbed with either 3 mol L−1 CaCl2 + 0.8 mol L−1 NH4SCN or feed solution at O/A = 1:1 and 1:10, respectively, in several cross-current simulations. Y(III) could be quantitatively stripped by deionized water at O/A = 1:5 in two cross-current simulations. The raffinate and the strip solutions were further treated for the recovery of Y(III) and Eu(III) by precipitation with ammonia solution, followed by calcinations at 400–450 °C. The purity of the obtained Y(III) and Eu(III) oxides were more than 98.2% and 98.7%, respectively. The present work can be considered as a novel separation technique which makes use of non-volatile and non-flammable organic solvents and with a limited number of process steps.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation programme under Grant Agreement No 680629 (REMAGHIC: New Recovery Processes to produce Rare Earth-Magnesium Alloys of High Performance and Low Cost) (project website: http://www.remaghic-project.eu). The authors acknowledge Relight srl (Rho, Italy) for providing the mixed oxide.Notes and references
- C. K. Gupta and N. Krishnamurthy, Extractive Metallurgy of Rare Earths, CRC Press, Elsevier, 2005 Search PubMed.
- K. Binnemans and P. T. Jones, J. Sustain. Metall., 2015, 1, 22–38 CrossRef.
- K. Binnemans, P. T. Jones, K. Van Acker, B. Blanpain, B. Mishra and D. Apelian, JOM, 2013, 65, 846–848 CrossRef.
- K. Binnemans, P. T. Jones, T. Müller and L. Yurramendi, J. Sustain. Metall., 2018, 4, 126–146 CrossRef.
- K. Binnemans and P. T. Jones, J. Rare Earths, 2014, 32, 195–200 CrossRef CAS.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner. Prod., 2013, 51, 1–22 CrossRef CAS.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- T. Quanyin, L. Jinhui and Z. Xianlai, Crit. Rev. Environ. Sci. Technol., 2015, 45, 749–776 CrossRef.
- Y. Wu, X. Yin, Q. Zhang, W. Wang and X. Mu, Resour., Conserv. Recycl., 2014, 88, 21–31 CrossRef.
- C. Tunsu, M. Petranikova, M. Gergoric, C. Ekberg and T. Retegan, Hydrometallurgy, 2015, 156, 239–258 CrossRef CAS.
- V. Innocenzi, I. De Michelis, B. Kopacek and F. Vegliò, Waste Manage., 2014, 34, 1237–1250 CrossRef CAS PubMed.
- I. De Michelis, F. Ferella, E. F. Varelli and F. Vegliò, Waste Manage., 2011, 31, 2559–2568 CrossRef CAS PubMed.
- V. Innocenzi, I. De Michelis, F. Ferella and F. Vegliò, Int. J. Miner. Process., 2017, 168, 87–94 CrossRef CAS.
- V. Innocenzi, N. M. Ippolito, I. De Michelis, F. Medici and F. Vegliò, J. Environ. Manage., 2016, 184, 552–559 CrossRef CAS PubMed.
- L. Gijsemans, F. Forte, B. Onghena and K. Binnemans, RSC Adv., 2018, 8, 26349–26355 RSC.
- B. Van den Bogaert, D. Havaux, K. Binnemans and T. Van Gerven, Green Chem., 2015, 17, 2180–2187 RSC.
- C. A. de Morais and V. S. T. Ciminelli, Hydrometallurgy, 2001, 60, 247–253 CrossRef.
- A. H. Hu, C. H. Kuo, L. H. Huang and C. C. Su, Waste Manage., 2017, 60, 765–774 CrossRef CAS PubMed.
- C. Tunsu, J. B. Lapp, C. Ekberg and T. Retegan, Hydrometallurgy, 2016, 166, 98–106 CrossRef CAS.
- C. Tunsu, C. Ekberg, M. Foreman and T. Retegan, Solvent Extr. Ion Exch., 2014, 32, 650–668 CrossRef CAS.
- F. Yang, F. Kubota, Y. Baba, N. Kamiya and M. Goto, J. Hazard. Mater., 2013, 254–255, 79–88 CrossRef CAS PubMed.
- N. K. Batchu, T. Vander Hoogerstraete, D. Banerjee and K. Binnemans, RSC Adv., 2017, 7, 45351–45362 RSC.
- B. Gaudernack, G. Hannestad and I. Hundere, US Pat., 3 821 352, 1974.
- T. Nakamura, S. Nishihama and K. Yoshizuka, Solvent Extr. Res. Dev., 2007, 14, 105–113 CAS.
- R. De Carolis, D. Fontana, M. Pietrantonio, S. Pucciarmati and G. N. Torelli, Environ. Eng. Manage. J., 2015, 14, 1603–1609 CrossRef CAS.
- M. A. Rabah, Waste Manage., 2008, 28, 318–325 CrossRef CAS PubMed.
- N. V. Thakur, US Pat., 8 821 817B2, 2014.
- J. J. Braconnier and A. Rollat, EU Pat., WO 2010118967A1, 2010.
- H. N. Vu, T. D. Pham, J. Formaneck and P. Dvorak, J. Pol. Miner. Eng. Soc., 2017, 23–28 Search PubMed.
- A. Rout and K. Binnemans, Phys. Chem. Chem. Phys., 2016, 18, 16039–16045 RSC.
- K. Larsson and K. Binnemans, Hydrometallurgy, 2015, 156, 206–214 CrossRef CAS.
- S. Sobekova Foltova, T. Vander Hoogerstraete, D. Banerjee and K. Binnemans, Sep. Purif. Technol., 2019, 210, 209–218 CrossRef CAS.
- B. Onghena, S. Valgaeren, T. Vander Hoogerstraete and K. Binnemans, RSC Adv., 2017, 7, 35992–35999 RSC.
- S. Riaño, M. Petranikova, B. Onghena, T. Vander Hoogerstraete, D. Banerjee, M. StJ. Foreman, C. Ekberg and K. Binnemans, RSC Adv., 2017, 7, 32100–32113 RSC.
- S. Riaño, M. Regadío, K. Binnemans and T. Vander Hoogerstraete, Spectrochim. Acta, Part B, 2016, 124, 109–115 CrossRef.
- M. Regadío, S. Riaño, K. Binnemans and T. Vander Hoogerstraete, Anal. Chem., 2017, 89, 4595–4603 CrossRef PubMed.
- L. J. Lozano and D. Juan, Solvent Extr. Ion Exch., 2001, 19, 659–676 CrossRef CAS.
- K. Binnemans and K. Larsson, Pat., WO2015106324A1, 2015.
Footnote |
| † Electronic supplementary information (ESI) available: Preliminary solvent extraction tests as a function of time, pH and temperature; FTIR of the initial ionic liquid and of the regenerated one. See DOI: 10.1039/c8ra09797f |
| This journal is © The Royal Society of Chemistry 2019 |