
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceASK1-p38 cascaded signal mediates pulmonary microvascular endothelial barrier injury induced by the return of PHSML in rats
Muhammad Fawad ,
Muhammad Abbas,
Limin Zhang,
Yuping Zhang and
Yaxiong Guo*
,
Muhammad Abbas,
Limin Zhang,
Yuping Zhang and
Yaxiong Guo*
Institute of Microcirculation, Hebei North University, Zhangjiakou, Hebei, China. E-mail: yxguo2022@163.com; Tel: +86-0313-4029150
First published on 8th February 2019
Abstract
The return of post-hemorrhagic shock mesenteric lymph (PHSML) induces pulmonary vascular endothelial barrier dysfunction, which results in acute lung injury. Activation of the apoptosis signal-regulated kinase 1 (ASK1) and p38 mitogen-activated protein kinase (p38 MAPK) pathway has been shown to trigger inflammatory responses. However, whether the ASK1-p38 MAPK pathway is involved in the PHSML-induced pulmonary endothelial barrier dysfunction remains unclear. In the present study, permeability changes of pulmonary capillaries were found in vivo, and activation of the ASK1-p38 MAPK pathway was determined in vitro. PMVEC barrier dysfunction was determined by measuring TEER. Furthermore, junctional and cytoskeletal protein expressions were analyzed. The results showed that hemorrhagic shock led to a marked increase in the permeability of pulmonary capillaries in vivo, which was markedly alleviated by PHSML drainage. In cultured pulmonary microvascular endothelial cells (PMVECs), PHSML reduced the endothelial barrier function accompanied by upregulated p-ASK1 and p-p38 MAPK protein expression, impaired the cytoskeletal protein structure, and down-regulated junction protein expression. These adverse effects were eliminated by applying either Trx1 (ASK1 inhibitor) or SB203580 (p38 MAPK inhibitor). These results indicated that the ASK1-p38 MAPK pathway mediates PHSML-induced pulmonary vascular endothelial barrier dysfunction during hemorrhagic shock.
Introduction
Acute lung injury (ALI), challenged by lipopolysaccharide (LPS) through oxidative, inflammatory, and apoptotic effects, is a leading cause of death in severe hemorrhagic shock.1–3 Previous studies have shown that hemorrhagic shock-induced gut ischemia leads to gut barrier dysfunction, with the anti-inflammatory cytokines and bacterial toxins of gut flora directly carried into lymphatic vessels and the lungs to further cause ALI deterioration.4,5 Furthermore, intravenous infusion of post-hemorrhagic shock mesenteric lymph (PHSML) directly increases the endothelial cell layer permeability of pulmonary capillaries, which results in vascular endothelial barrier dysfunction.6 These results suggest that the backflow of mesenteric lymph derived from ischemic gut is a primary cause of ALI induced by hemorrhagic shock.7The barrier arrangement of pulmonary microvascular endothelial cells (PMVECs) is not identical to the capillary endothelial cells of other organs.8,9 The cytoskeleton that supports the PMVEC monolayer barrier mainly consists of three primary elements, namely, actin microfilaments, intermediate filaments, and microtubules.10 The endothelial barrier receives signals from extracellular matrix (ECM) fibrous proteins linked to junction proteins between the endothelium.11 Endothelial barrier integrity is stabilized by several structural proteins (inside and outside the membrane), including ZO-1, VE-cadherin, claudin-1, and F-actin.12–14 Therefore, altering the cytoskeleton elements or tight junction protein would lead to changes in pulmonary microvascular endothelial barrier function.
Many harmful factors produced by hemorrhagic shock can impair the cytoskeleton structure and reduce the barrier properties.15 The mitogen-activated protein kinase (MAPK) family plays a vital role in barrier dysfunction through the apoptosis pathway.16 There are three classical types of MAPK pathway activation cascade, namely, p38 MAPK, extracellular-signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK).17 Apoptosis signal-regulated kinase 1 (ASK1) activation plays an important role in the apoptosis pathway, and can activate the p38 MAPK pathway, which induces local production of cytokines to aggravate the inflammatory cascade, and disorganize the cytoskeleton of distinctive cells.18,19 Although activation of the ASK1-p38 MAPK pathway is involved in the cytoskeleton-mediated endothelial barrier function, it remains unclear whether this pathway also mediates PHSML-induced pulmonary microvascular endothelial barrier dysfunction following hemorrhagic shock. Therefore, the present study aimed to determine the impairing effect of PHSML on the pulmonary microvascular endothelial barrier, and confirm the mediating role of the ASK1-p38 MAPK pathway in PHSML-induced pulmonary endothelial barrier dysfunction.
Materials and methods
Pulmonary capillary permeability
A hemorrhagic shock model was established and PHSML was drained as described previously.20 The supernatant of PHSML was centrifuged and stored at −80 °C for subsequent cell experiments. A 10 mm2 window was then opened in the right thoracic wall of mechanically ventilated Wistar rats.21 To measure the permeability of pulmonary capillaries in vivo, FITC-labeled albumin (50 mg kg−1) was injected into the femoral vein.22 Using a digital camera system (dynamic in vivo imaging of microvasculature and perfusion by fluorescence microscopy, Gene & I, LTD), images were scanned and recorded, followed by analysis using Image-Pro Plus 6.0 software. Five fields were selected for evaluation in each rat. The Ao/Ai ratio (fluorescent intensity outside or inside of the alveolar capillary) was calculated as an indicator of FITC-albumin leakage (compared with the baseline). This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication no. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee of Hebei North University (Zhangjiakou, China).Morphological observations
Rat PMVECs (PriCells, China) were cultured at 37 °C in a humidified atmosphere of 5% CO2–95% air (v/v) in Dulbecco's modified Eagle's medium (DMEM, GIBCO, USA) supplemented with 10% (v/v) fetal bovine serum (FBS, GIBCO, USA), 1% antibiotic (penicillin, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1; streptomycin, 10 μg mL−1; GIBCO), and Microvascular Growth Supplement (MVGS, GIBCO, USA). The PMVECs grew to contact-inhibited monolayers with a typical cobblestone morphology and were then passaged by trypsinization (0.25% trypsin/0.02% EDTA) and resuspended in fresh culture medium. The cells (80% confluence) were incubated in FBS-free DMEM medium for 24 h before treatment. Subsequent treatment with FBS-free low-glucose DMEM medium, PHSML, lipopolysaccharide (LPS, 10 μg mL−1), PHSML + TRX1 (ASK1 inhibitor, 10 ng mL−1) or PHSML + SB203580 (p38 MAPK inhibitor, 10 μmol L−1) was performed for 6 h. Images of PMVECs were obtained using a Leica inverted phase-contrast microscope and recorded at magnifications of 50× to 200×, to observe the role of the ASK1-p38 MAPK pathway in PHSML-induced PMVEC injury.
000 U mL−1; streptomycin, 10 μg mL−1; GIBCO), and Microvascular Growth Supplement (MVGS, GIBCO, USA). The PMVECs grew to contact-inhibited monolayers with a typical cobblestone morphology and were then passaged by trypsinization (0.25% trypsin/0.02% EDTA) and resuspended in fresh culture medium. The cells (80% confluence) were incubated in FBS-free DMEM medium for 24 h before treatment. Subsequent treatment with FBS-free low-glucose DMEM medium, PHSML, lipopolysaccharide (LPS, 10 μg mL−1), PHSML + TRX1 (ASK1 inhibitor, 10 ng mL−1) or PHSML + SB203580 (p38 MAPK inhibitor, 10 μmol L−1) was performed for 6 h. Images of PMVECs were obtained using a Leica inverted phase-contrast microscope and recorded at magnifications of 50× to 200×, to observe the role of the ASK1-p38 MAPK pathway in PHSML-induced PMVEC injury.
Monolayer permeability
To assess the role of the ASK1-p38 pathway in the PHSML-induced increase in the PMVEC monolayer permeability, PMVECs were seeded at 1 × 105 cells per cm2 on Costar Transwell inserts (0.4 μm pores, surface area 0.3 cm2) using DMEM medium with different treatments. Overnight incubation ensured temperature and pH equilibration, and prevented transient electrical fluctuations, which might occur after media changes or repetitive measurements. Suspension cells (100 μL) were added into the apical compartment, and FBS-free low glucose DMEM medium (600 μL) was added into the basolateral compartment. When the cells were fully fused to form a dense monolayer, we replaced the medium with various factors, as listed above, for 6 h. Primarily, DMEM medium containing FITC-albumin (100 μL, 1 mg mL−1) was added to exchange the medium of the apical compartment. The cells were then incubated for 45 min in the dark. The trans-epithelial electrical resistance (TEER) was measured using an epithelial voltohmmeter (WPI, USA). The media were collected from the apical (2 μL) and basolateral (100 μL) compartments of the 96 well plate, with that from the apical compartments diluted with 98 μL DMEM medium (50-fold dilution), while that from the basolateral compartments was not diluted. The fluorescence intensity of each sample was measured using a Spectramax M3 fluorescence microplate reader (EX: 485 nm, EM: 525 nm). Apparent permeability coefficients (Pa) were estimated according to the following equation: Pa = ([A]/t) × (l/A) × (v/[L]), where [A] is the fluorescence absorbance of the basolateral compartment, t is time (s), A is the surface area of the chamber filter (cm2), v is the liquid volume of the basolateral compartment (mL), and [L] is the fluorescence absorbance of the apical compartment. All permeability and adsorption values were means of at least triplicate measurements using separate filters.Western blotting
Membrane and cytosolic proteins were isolated with a Membrane and Cytosol Extraction Kit (Beyotime, China). Protein concentrations were determined using a BCA Protein Assay Kit (Beyotime, China). Equal amounts of proteins were separated by 10% SDS gel electrophoresis under denaturing and nonreducing conditions, and then transferred to a PVDF membrane. The membrane was blocked with 5% non-fat milk in Tris-buffered saline and 1‰ Tween 20 (TBST) at room temperature for 1 h, and then incubated with primary antibody at 4 °C overnight. After washing in TBST, the membrane was incubated with the secondary antibody. Membranes were exposed to enhanced chemiluminescent reagents (ECL Plus Western Blotting Detection System, Beyotime, China). Images were captured and analyzed using an ImageQuant LAS 4000 instrument.Laser scanning confocal microscopy
PMVECs were seeded on sterile chamber slides at 5 × 104/mL and cultured. After 6 h, cells were fixed for 30 min in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100 for 30 min, and rinsed in PBS for 5 min. For F-actin staining, cells were incubated with FITC-conjugated phalloidin (5 μg mL−1) for 20 min at room temperature in the dark, and then washed with PBS. Phalloidin binds to F-actin. Nuclei were stained with DAPI staining solution (10 μg mL−1) for 5 min at room temperature and washed with PBS. The cells were visualized with a laser scanning confocal microscope (Nikon CISi, Japan). Transverse sections were imaged from the dorsal surface down to the cell–substratum interface at 2 μm intervals. F-actin protein expression was also detected with western blotting.Statistical analysis
Data were calculated as means ± standard deviation (SD). One-way analysis of variance (ANOVA) was used to assess significant differences between groups. P < 0.05 was considered significant. All analyses were performed using SPSS version 17.0 (SPSS, Chicago, USA).Results
Permeability changes of rat pulmonary capillaries
In this study, we used the dynamics of FITC-labeled albumin leakage to assess the permeability of pulmonary capillaries, as shown in Fig. 1. The permeability was significantly increased in the shock group. In contrast, increased permeability was markedly alleviated in the shock with drainage group.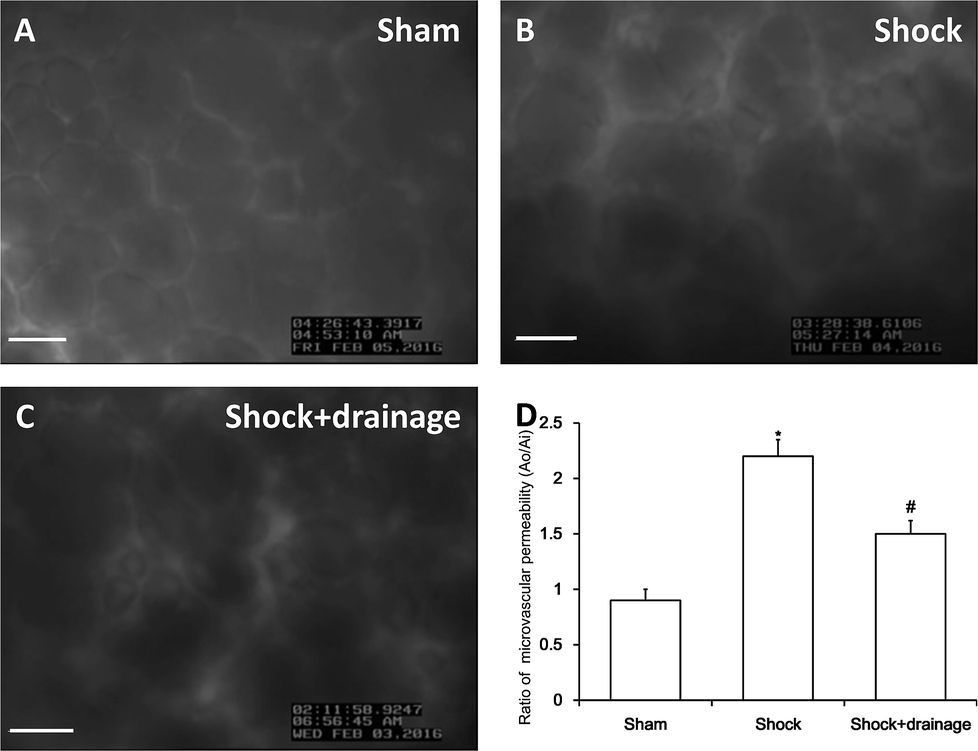 | ||
| Fig. 1 Permeability changes of pulmonary capillaries following hemorrhagic shock in rats. (A–C) Images of FITC-albumin leakage from pulmonary capillaries under fluorescence microscopy among the three groups, bar = 50 μm. (D) Ratio of FITC-albumin leakage in different groups. Results are presented as means ± SD (n = 3). *P < 0.05 vs. sham group, #P < 0.05 vs. shock group. | ||
pASK1-p38 MAPK protein activation of PMVECs
To confirm that increased permeability was linked to the ASK1-p38 MAPK pathway, we examined the p-ASK1 and p-p38 MAPK protein expression of PMVECs treated with PHSML and/or inhibitors of ASK1 and p-38 MAPK. As shown in Fig. 2, PHSML increased the activation of p-ASK1 and p-p38 MAPK proteins, which were reduced by co-administration of either TRX1 or SB203580.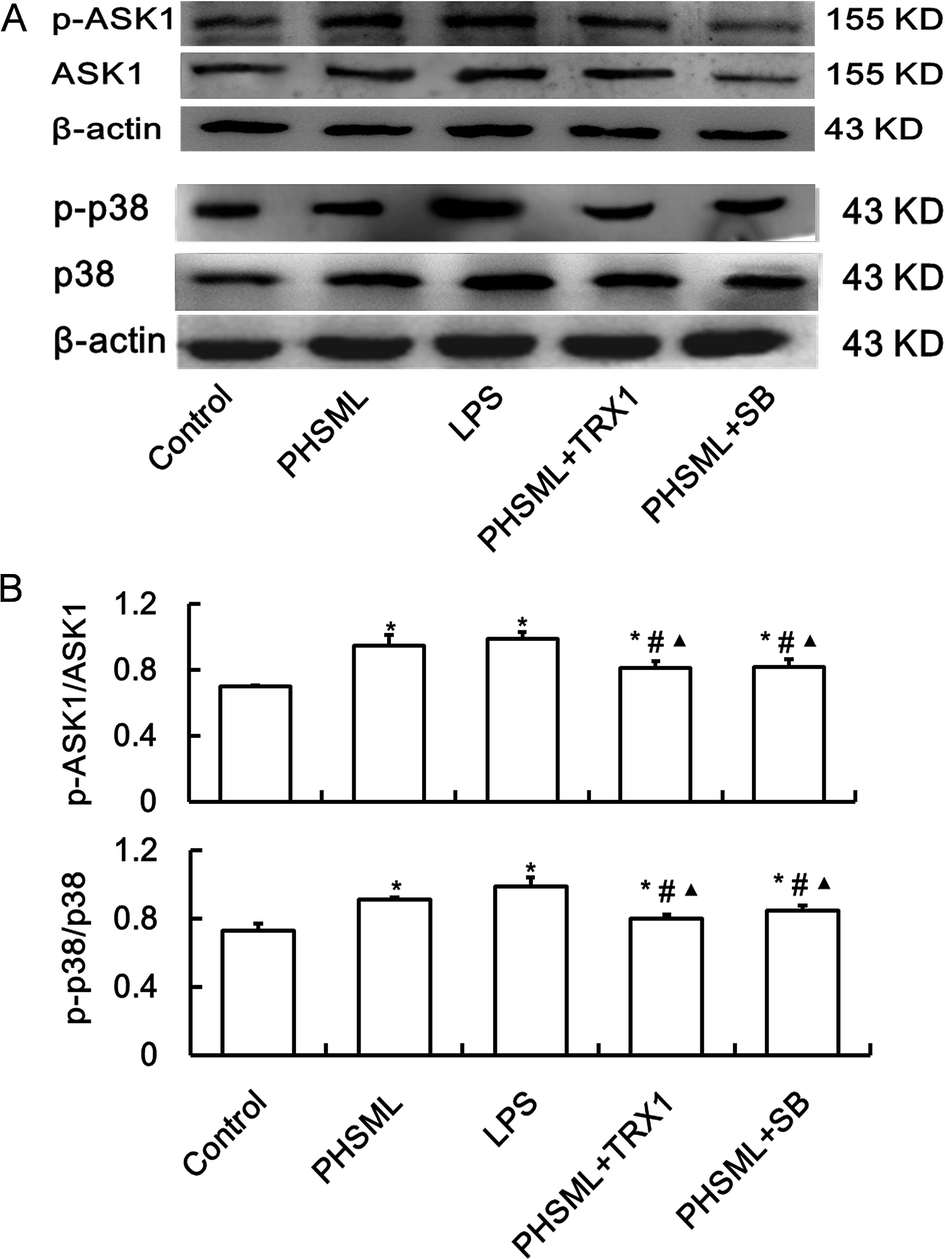 | ||
| Fig. 2 Effects of TRX1 and SB203580 on the p-ASK1 and p-p38 MAPK expressions in PMVECs treated with PHSML. (A) Images of western blotting for ASK1 and p38 MAPK. (B) Relative density analysis of concentration. Results are presented as the mean ± SD (n = 3). *P < 0.05 vs. control group, #P < 0.05 vs. PHSML group, ▲P < 0.05 vs. LPS group. | ||
Morphological changes of PMVECs
PMVECs grew vigorously to create a cobblestone wall with well-defined margins in the control group (Fig. 3A and F). However, PMVECs treated with either PHSML or LPS exhibited increased cell gaps and undifferentiated cell numbers (Fig. 3B, G, C and H). These treatments also restricted cell growth, kept cells in suspension, and altered the cytoskeleton of individual cells. Co-administration of either TRX1 or SB203580 with PHSML reversed the PHSML-induced adverse effects (Fig. 3D, I, E and J).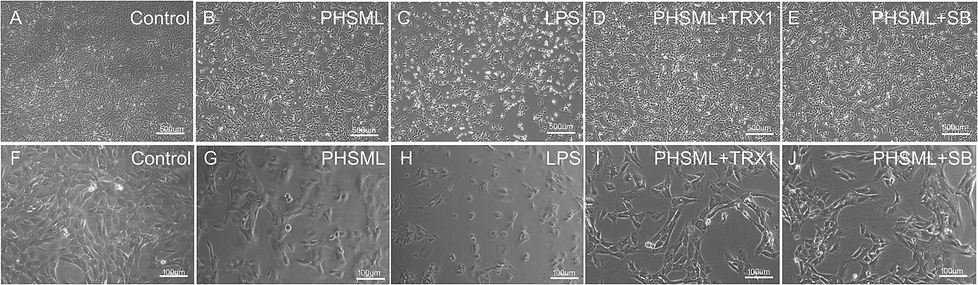 | ||
| Fig. 3 Effects of TRX1 and SB203580 on the morphology changes of PMVECs treated with PHSML. (A–J) Images of control, PHSML, lipopolysaccharide (LPS), PHSML + TRX1 (ASK1 inhibitor) or PHSML + SB203580 (p38 MAPK inhibitor) incubated for 6 h (inverted microscope, ×50 and ×200). | ||
Permeability changes of PMVECs
To examine the effects of PHSML and the ASK1-p38 MAPK pathway on PMVEC barrier dysfunction, the permeability of the PMVEC monolayer was determined by measuring TEER (Fig. 4A) and the permeability coefficient (Fig. 4B). Both PHSML and LPS significantly decreased the TEER (P < 0.05) on the PMVEC monolayer, reflecting that the endothelial barrier was compromised. The concurrent application of either TRX1 or SB203580 significantly attenuated the TEER (P < 0.05) in response to either PHSML or LPS. Similarly, by measuring the permeability coefficient, the higher permeability of the PMVEC monolayer was evident during treatment with PHSML, which was inhibited by co-administration of either TRX1 or SB203580.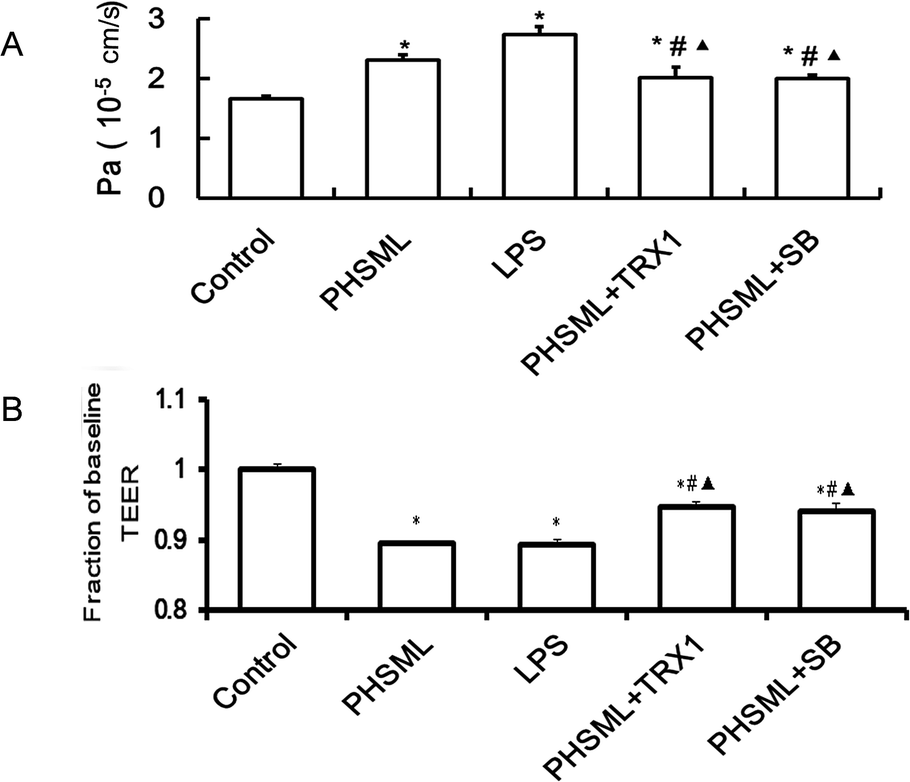 | ||
| Fig. 4 Effects of TRX1 and SB203580 on the permeability of PMVECs treated with PHSML. (A) Permeability coefficient of the PMVEC barrier. (B) TEER of the PMVEC barrier. Results are presented as means ± SD (n = 3). *P < 0.05 vs. control group, #P < 0.05 vs. PHSML group, ▲P < 0.05 vs. LPS group. | ||
Junction protein expressions of PMVECs
As PHSML and ASK1-p38 MAPK pathways play a role in the permeability of the PMVEC monolayer, we further confirmed the role of the ASK1-p38 MAPK pathway in barrier dysfunction. Changes in the junction protein expression were determined. We analyzed the expressions of proteins (VE-cadherin, ZO-1, and claudin-1) by western blotting (Fig. 5). Compared with the control group, PHSML or LPS dramatically reduced the expression of three junction proteins (P < 0.05). These reduced protein expressions were restored by treatment with either TRX1 or SB203580 (P < 0.05).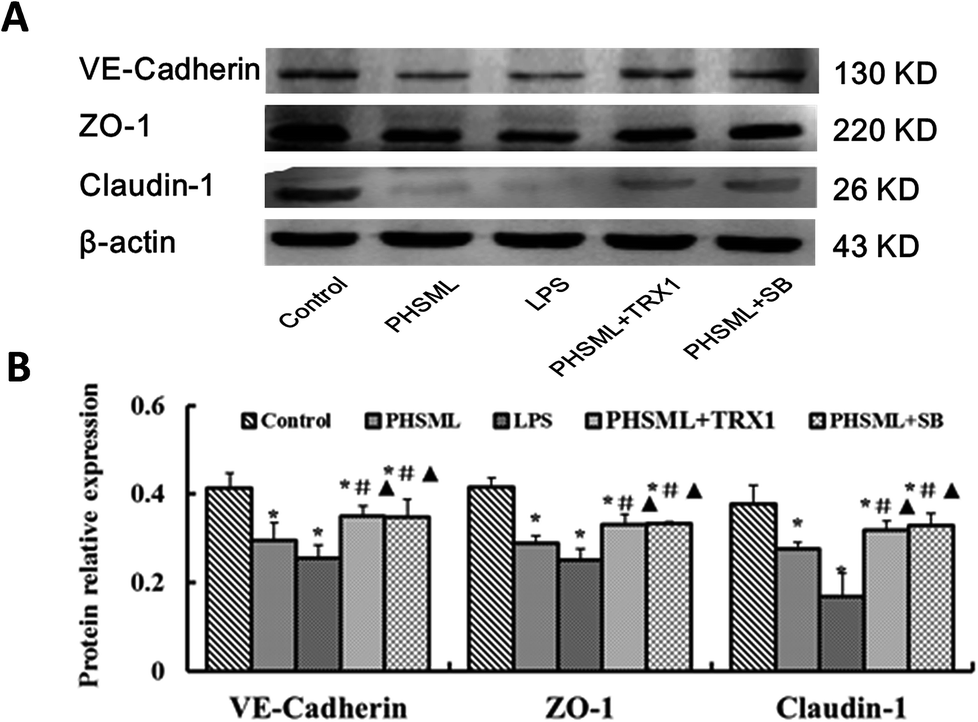 | ||
| Fig. 5 Effects of TRX1 and SB203580 on junction protein expression in PMVECs treated with PHSML. (A) Images of western blotting for VE-cadherin, ZO-1, and claudin-1. (B) Relative density analysis of concentration. Results are presented as means ± SD (n = 3). *P < 0.05 vs. control group, #P < 0.05 vs. PHSML group, ▲P < 0.05 vs. LPS group. | ||
Cytoskeleton expression of PMVECs
Cytoskeleton changes were observed using a combination of F-actin staining with fluorescein–phalloidin and nuclear staining with DAPI. F-actin is situated in the cellular cortex in the cells. This localization is observable in cells, which present a strong accumulation of actin filaments underneath the plasma membrane. The central region of the growth cone contains a F-actin meshwork, because F-actin bundles are an essential part of motility. F-actin was stained with phalloidin and analyzed using confocal microscopy. Representative confocal images of phalloidin and DAPI co-stained in PMVECs are shown as Fig. 6. Both PHSML and LPS decreased phalloidin staining and destroyed the nuclei compared with the control group, whereas treatment of either TRX1 or SB203580 with PHSML restored the nuclei and increased F-actin expression.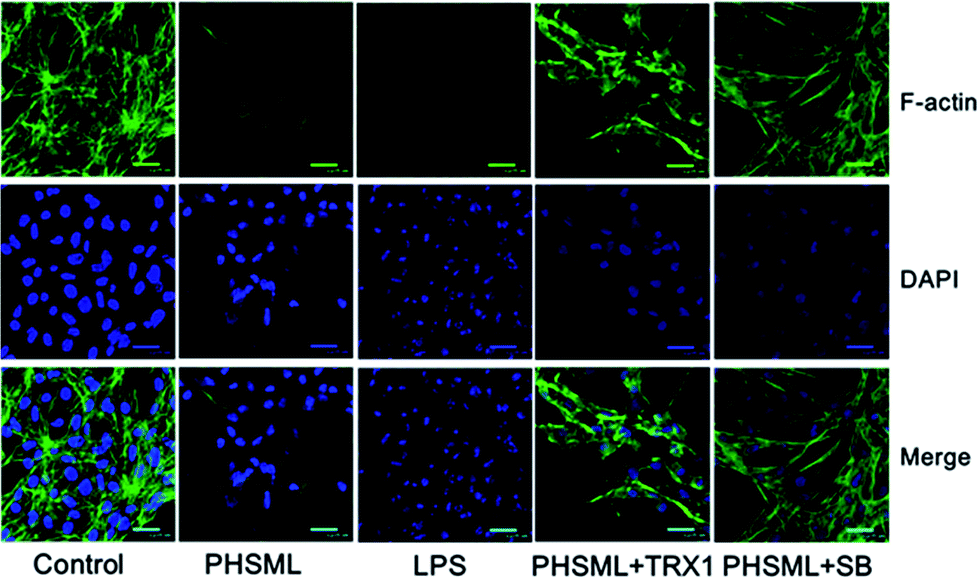 | ||
| Fig. 6 Effects of TRX1 and SB203580 on F-actin expression in PMVECs treated with PHSML. F-actin was stained with phalloidin FITC (green); nuclei were stained with DAPI (blue); cells co-expressed F-actin and DAPI in the merged image. | ||
Discussion
Pulmonary microvascular endothelial cells are irreversibly impaired after hypoxic-ischemic injury during hemorrhagic shock, which induces capillary leak syndrome or a large area of tissue edema (easily causing MOD).4,20,23 Activation of the ASK1-p38 MAPK pathway induced p38 MAPK phosphorylation and enhanced the inflammatory response and apoptosis, leading to ALI.24,25 In contrast, inhibition of shock mesenteric lymph reflux can reduce pulmonary vascular permeability and relieve ALI.7 We have previously demonstrated that the permeability of pulmonary capillaries was increased in shock rats (lung wet-to-dry weight ratio, Evan's blue dye technique).20 In the present study, we found that blocking the ASK1-p38 MAPK pathway abolished the impairing effects of PHSML on PMVEC barrier. These results indicated that ASK1-p38 MAPK pathway activation mediated the impairing effects of PHSML on the PMVEC barrier.Mesenteric lymph return can trigger a systemic inflammatory response and ALI by pro-inflammatory mediators in hemorrhagic shock.26 The MAPK pathway has been reported to play a major role in this process, especially the p38 MAPK pathway.27 Suppressing the p38 MAPK pathway reduces lung dysfunction following hemorrhagic shock, but whether hemorrhagic shock directly upregulates the expression of p38 MAPK has not been reported previously.28 Furthermore, p38 MAPK signaling has been proposed to be critical for lipopolysaccharide-induced apoptosis in PMVECs.29 ASK1 is an important kinase for apoptosis, and its activation can activate the p38 MAPK pathway.18,19 Therefore, it is necessary to establish the relationship between the ASK1-p38 MAPK pathway and PHSML in pulmonary endothelial barrier dysfunction. Our findings indicated that activation of the ASK1-p38 MAPK pathway mediated PHSML-induced pulmonary microvascular endothelial barrier dysfunction following hemorrhagic shock. Interestingly, ASK1 expression was also inhibited by p38 inhibitor, although the specific mechanism should be explored in future studies.
To mimic the environment of PMVECs during hemorrhagic shock, we used PHSML to stimulate PMVECs, and found that PHSML induced significant morphological changes, which might be related to barrier dysfunction. Blocking of the ASK1-p38 MAPK pathway could reduce these morphological changes. Our results showed that the ASK1-p38 MAPK pathway plays an important role in damage to the PMVEC morphology induced by PHSML.
PHSML has been shown to activate the ASK1-p38 MAPK pathway through inflammatory cytokines, which are responsible for several intra-cellular responses.19,30 To examine the effect of the ASK1-p38 MAPK pathway on PMVEC barrier dysfunction produced by PHSML, permeability changes were evaluated by TEER and permeability coefficient experiments. We found that PHSML could decrease TEER and increase the PMVEC monolayer permeability. Both TRX1 and SB203580 reversed the PHSML-induced changes in the TEER and PMVEC monolayer permeability. These results indicated that inhibiting ASK1-p38 MAPK significantly reduced PMVEC barrier damage and increased the permeability produced by PHSML.
Our findings were in agreement with the p38 MAPK pathway being involved in disruption of the tight junction protein induced by LPS.3 PHSML markedly reduced the expression of ZO-1, claudin-1, and VE-cadherin. Both TRX-1 and SB20358 restored PHSML-reduced protein expression. The main cytoskeleton constituent was actin filament, which was mainly linked to the junction proteins.13,14 Confocal images also showed that the F-actin contents were reduced upon stimulation of the ASK1-p38 MAPK pathway. After inhibition of either SB203580 or TRX-1, the contents were restored to near control levels. Our results supported that F-actin filaments are responsible for junction protein integrity by maintaining the barrier permeability.31 ASK1-p38 MAPK pathway activation led to actin filament derangement and disrupted the barrier properties by changing the junction protein.
Conclusions
We have demonstrated that the ASK1-p38 pathway is strongly correlated with the induction of PHSML-induced pulmonary microvascular endothelial barrier dysfunction. PHSML greatly altered the cytoskeletal and junction proteins (F-actin, VE-cadherin, claudin-1, and ZO-1). Inhibition of the ASK1-p38 pathway prevented damages of the PMVEC barrier induced by the PHSML. These results indicate that the ASK1-p38 pathway mediates PHSML-induced permeability changes of the PMVEC barrier.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81500380), the Young Top Talent Science Research Project of Colleges and Universities in Hebei Province (BJ2014004), the High-level Talent Support Project in Hebei Province (A201803058), and the Healthcare and Biomedicine Special Project of Key Research and Development Program of Hebei province (18277716D). The authors thank Dr Zigang Zhao and Dr Chunyu Niu for the conception and design of this study.References
- X. Wu, M. G. Schwacha, M. A. Dubick, A. P. Cap and D. N. Darlington, Shock, 2016, 46, 108–114 CrossRef PubMed
.
- H. Yao, Y. Sun, S. Song, Y. Qi, X. Tao, L. Xu, L. Yin, X. Han, Y. Xu, H. Li, H. Sun and J. Peng, Front. Pharmacol., 2017, 8, 120 Search PubMed
- S. Aggarwal, C. Dimitropoulou, Q. Lu, S. M. Black and S. Sharma, Front. Physiol., 2012, 3, 161 Search PubMed
- E. A. Deitch, R. Forsythe, D. Anjaria, D. H. Livingston, Q. Lu, D. Z. Xu and H. Redl, Shock, 2004, 22, 221–228 CrossRef
- G. Levy, J. E. Fishman, D. Xu, B. T. Chandler, E. Feketova, W. Dong, Y. Qin, V. Alli, L. Ulloa and E. A. Deitch, Shock, 2013, 39, 39–44 CAS
- Y. M. Zhang, S. K. Zhang and N. Q. Cui, World J. Gastroenterol., 2014, 20, 4771–4777 CrossRef PubMed
- E. A. Deitch, C. Adams, Q. Lu and D. Z. Xu, Surgery, 2001, 129, 39–47 CrossRef CAS PubMed
- M. Shao, S. T. Tang, B. Liu and H. Q. Zhu, Mol. Med. Rep., 2016, 13, 529–535 CrossRef CAS PubMed
- F. Huber, A. Boire, M. P. Lopez and G. H. Koenderink, Curr. Opin. Cell Biol., 2015, 32, 39–47 CrossRef CAS PubMed
- S. F. Rodrigues and D. N. Granger, Tissue Barriers, 2015, 3, e978720 CrossRef PubMed
- C. W. Wilson, L. H. Parker, C. J. Hall, T. Smyczek, J. Mak, A. Crow, G. Posthuma, A. De Maziere, M. Sagolla, C. Chalouni, P. Vitorino, M. Roose-Girma, S. Warming, J. Klumperman, P. S. Crosier and W. Ye, Blood, 2013, 122, 3678–3690 CrossRef CAS PubMed
- S. C. Nicklisch, J. E. Spahn, H. Zhou, C. M. Gruian and J. H. Waite, Biochemistry, 2016, 55, 2022–2030 CrossRef CAS PubMed
- C. Greene and M. Campbell, Tissue Barriers, 2016, 4, e1138017 CrossRef PubMed
- O. L. Mooren, J. Li, J. Nawas and J. A. Cooper, Mol. Biol. Cell, 2014, 25, 4115–4129 CrossRef PubMed
- S. M. Stamatovic, A. M. Johnson, R. F. Keep and A. V. Andjelkovic, Tissue Barriers, 2016, 4, e1154641 CrossRef PubMed
- J. S. Arthur and S. C. Ley, Nat. Rev. Immunol., 2013, 13, 679–692 CrossRef CAS PubMed
- C. Jiang, Y. Yuan, F. Hu, Q. Wang, K. Zhang, Y. Wang, J. Gu, X. Liu, J. Bian and Z. Liu, Biol. Trace Elem. Res., 2014, 158, 249–258 CrossRef CAS PubMed
- W. Li, B. Huang, K. Liu, J. Hou and H. Meng, J. Periodontol., 2015, 86, 917–926 CrossRef CAS PubMed
- A. Ray, N. Sehgal, S. Karunakaran, G. Rangarajan and V. Ravindranath, Free Radical Biol. Med., 2015, 87, 312–325 CrossRef CAS PubMed
- G. X. Sun, Y. X. Guo, Y. P. Zhang, L. M. Zhang, Z. G. Zhao and C. Y. Niu, J. Surg. Res., 2016, 203, 47–55 CrossRef CAS PubMed
- S. E. Gill, R. Taneja, M. Rohan, L. Wang and S. Mehta, PLoS One, 2014, 9, e88501 CrossRef PubMed
- F. Lin, Y. Y. Liu, B. Xu, K. Sun, H. Y. Wang, Q. Li, C. S. Wang, J. Y. Fan, S. W. Zhang and J. Y. Han, Shock, 2013, 39, 317–325 CrossRef CAS PubMed
- A. Gulati, Curr. Vasc. Pharmacol., 2016, 14, 187–195 CrossRef CAS PubMed
- R. K. Brobey, D. German, P. K. Sonsalla, P. Gurnani, J. Pastor, C. C. Hsieh, J. Papaconstantinou, P. P. Foster, M. Kuro-o and K. P. Rosenblatt, PLoS One, 2015, 10, e0139914 CrossRef PubMed
- J. Wu, R. Wang, Z. Ye, X. Sun, Z. Chen, F. Xia, Q. Sun and L. Liu, Biochem. Biophys. Res. Commun., 2015, 466, 155–161 CrossRef CAS PubMed
- S. Langness, T. W. Costantini, K. Morishita, B. P. Eliceiri and R. Coimbra, PLoS One, 2016, 11, e0168322 CrossRef PubMed
- J. T. Hsu, Y. C. Hsieh, W. H. Kan, J. G. Chen, M. A. Choudhry, M. G. Schwacha, K. I. Bland and I. H. Chaudry, Am. J. Physiol.: Heart Circ. Physiol., 2007, 292, H2982–H2987 CrossRef CAS PubMed
- H. Sato, K. Kasai, T. Tanaka, T. Kita and N. Tanaka, Med. Sci. Monit., 2008, 14, BR79–87 CAS
- Z. F. Liu, D. Zheng, G. C. Fan, T. Peng and L. Su, Apoptosis, 2016, 21, 896–904 CrossRef CAS PubMed
- M. Soga, A. Matsuzawa and H. Ichijo, Int. J. Cell Biol., 2012, 2012, 439587 Search PubMed
- D. Coronado-Velazquez, A. Betanzos, J. Serrano-Luna and M. Shibayama, J. Eukaryotic Microbiol., 2018, 65, 804–819 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2019 |