
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSodium tanshinone IIA silate increases melanin synthesis by activating the MAPK and PKA pathways and protects melanocytes from H2O2-induced oxidative stress†
Hui Zhong‡
 a,
Xiaohong An‡a,
Yu Li‡b,
Minxuan Caia,
Owais Ahmada,
Jing Shang*a and
Jia Zhou*a
a,
Xiaohong An‡a,
Yu Li‡b,
Minxuan Caia,
Owais Ahmada,
Jing Shang*a and
Jia Zhou*a
aSchool of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing 211198, P. R. China. E-mail: cpuzj_2016@163.com; shangjing21cn@163.com
bShanghai Key Laboratory of Crime Scene Evidence, Shanghai Research Institute of Criminal Science and Technology, Zhongshan North No. 1 Road, Shanghai 200083, China
First published on 14th June 2019
Abstract
Vitiligo is an intriguing depigmentation disorder that affects about 0.5–2% of the world population. In the past decade, first-line treatments of vitiligo have involved the use of calcineurin inhibitors and corticosteroids. Sodium tanshinone IIA sulfonate (STS) has been widely applied in the treatment of cardiovascular and cerebrovascular diseases in China. In the present study, the effect of STS on melanogenesis was confirmed in the B16F10 cells and zebrafish by direct observation. The prevention of hydrogen peroxide (H2O2)-induced oxidative stress has been proven to be beneficial to vitiligo patients, and STS that can protect the B16F10 cells against oxidative stress has been investigated in the present reversed study. Moreover, we found that pre-treatment with STS led to a concentration-dependent mitochondrial impairment and decreased cell apoptosis of the B16F10 cells in response to H2O2. In addition, we demonstrated that STS increased melanin synthesis in the B16F10 cells by activating the mitogen-activated protein kinase (MAPK) and protein kinase A (PKA) pathways. STS also increased the Cdc42 and KIF5b expression to stimulate the translocation of melanin. These results suggest that STS protects the B16F10 cells against H2O2-induced oxidative stress and exerts melanin synthesis activity in the B16F10 cells by activating the MAPK and PKA pathways; thus, it shows therapeutic potential for vitiligo.
1. Introduction
In the past decade, the number of new drugs approved by the US Food and Drug Administration (FDA) has been only around 40 per year, and no drug treatment is available for approximately 7000 rare and neglected diseases.1 Drug repurposing is a fast and effective method for the discovery and development of new drugs, and approximately 30% of new drugs approved by the FDA have been achieved via the reevaluation of existing drugs in recent years;2 moreover, drug repositioning, namely finding new uses for existing drugs, is a promising strategy for obtaining more therapeutic drugs.3,4Vitiligo is an intriguing depigmentation disorder and has significant effects on the quality of life of patients. The development of vitiligo has been linked to numerous factors such as impaired melanocyte migration, exposure to sunlight, stress, and infections.5 Histologic evidence indicates that vitiligo develops due to the progressive disappearance of epidermal melanocytes.6 Previous studies have shown that the removal of melanocytes occurs via apoptosis.7 Oxidative stress plays an important role in initiating the destruction of melanocytes, and the impairment of the redox balance can be one possible mechanism of vitiligo. The melanocytes of vitiligo are particularly vulnerable to damage or apoptosis under certain circumstances such as excessive ROS production.8,9 ROS can attack melanocytes and interfere with the normal metabolism, proliferation, and differentiation of melanocytes, causing cell apoptosis and defects.10 Excessive accumulation of ROS in the epidermis of vitiligo patients leads to the disorders of melanocytes, resulting in damage to the mitochondrial electron transport chain; this further forms a vicious cycle and leads to the destruction of melanocytes.11
The first-line treatment of vitiligo involves the use of calcineurin inhibitors and corticosteroids, such as clobetasol and tacrolimus, for topical treatment;12 skin atrophy will appear after the use of these drugs for a trial period of more than 2 months.13 Thus, a new drug for the treatment of vitiligo is urgently needed; however, the development of new drugs is restricted by the ambiguity of exact pathogenesis. Repositioning of existing drugs offers an effective way for the quick development of new drugs for many diseases.1 Sulfotanshinone sodium injections have been clinically approved for the treatment of cardiovascular diseases by the China Food and Drug Administration (CFDA) with the annual turnover of US$ 3 billion. The active ingredient of the sulfotanshinone sodium injection is sodium tanshinone IIA silate (STS), a water-soluble derivative of tanshinone IIA, which has been isolated from danshen.14 Some studies suggest that STS can work as a blood–brain barrier protective agent to enhance recovery after thrombolysis treatment.15 STS inhibited the proliferation and migration of vascular smooth muscle cells induced by high glucose through the AMPK-p53-p21 signaling and AMPK/NF-κB signaling axis.14 Moreover, STS plays a protective role against apoptosis in cardiomyocytes via JNK and PI3K signaling.16 The signaling of MAPK and JNK is also the key pathway for vitiligo. In the keratinocytes of vitiligo patients, protein kinase B (PKB)/mitogen-activated protein kinase (MAPK) signaling has been revealed, and the oxidative stress and apoptotic cell death are decreased by SIRT1 through the MAPK pathway.17,18 In both the keratinocytes and the melanocytes, the expression of DNA methyltransferases (DNMTs) was inhibited by oxidative stress via the JNK pathway and caused many genes, which was proven to play an important role in the abnormal development of vitiligo.19
Based on the abovementioned findings, the purpose of this study was to investigate the stimulating activity of STS for melanin synthesis and its protective effect on the B16F10 cells against H2O2-induced oxidative damage. Furthermore, our action mechanism investigation results revealed that MAPK signaling and PKA signaling might be the potential targets of STS. Moreover, STS increased the expression of Cdc42, KIF5b, Rab27a and Rab27a to promote the transfer of melanosomes; these findings supported the use of STS as a potential therapeutic strategy against vitiligo.
2. Materials and methods
STS was synthesized from tanshinone IIA. The data for the synthetic route are provided in the ESI (Fig. S1†). The structure of STS is shown in Fig. 1A. The purity of STS was determined by HPLC, and the identification of the structure was confirmed by NMR spectra (Bruker Advance 600 MHz) and high-resolution mass spectra (HR-MS). 1H NMR (600 MHz, DMSO-d6) δ: 7.85 (d, J = 6 Hz, 1H), 7.55 (d, J = 6 Hz, 1H), 3.09 (t, J = 12 Hz, 2H), 2.32 (s, 3H), 1.73 (d, J = 24 Hz, 2H), 1.62 (d, J = 12 Hz, 2H), 1.29 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ: 182.82, 175.77, 158.43, 154.65, 149.87, 143.31, 133.91, 127.43, 126.95, 120.37, 120.14, 117.18, 37.87, 34.83, 31.96, 31.96, 30.03, 19.21, 9.81. [MS + 1] = 375.39. The purity of STS was 96.8597%. The chromatogram and spectrogram of STS are shown in the ESI (Fig. S2–S5†). | ||
| Fig. 1 Zebrafish toxicity tests. (A) The structure of STS. (B) Zebrafishes were treated with STS at the different doses of 0.1, 1, 10, 50, and 100 μM for 24, 48, 72, 96 and 120 hpf, respectively. Body length and trunk-tail angle of the zebrafish were observed. | ||
1-Phenyl-2-thiourea (PTU) (P7629), dimethyl sulfoxide (DMSO) (D8418), L-3,4-dihydroxyphenylalanine (L-DOPA) (D1507), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (M2128), melanin (M8631), SB203580 (S8307), PD98059 (P215), SB600125 (S5567), H89 (B1427), and horseradish peroxidase-conjugated secondary antibodies were purchased from Sigma-Aldrich (MO, USA). Tyrosinase (C-19) and TRP-1 antibodies were obtained from Santa Cruz Biotechnology (CA, USA); MITF, DCT, gp100, Cdc42, KIF5b, Rab27a, and Rab17 antibodies were obtained from Abcam Technology (UK); antibodies against p-p38, p38, p-ERK1/2, ERK1/2, p-JNK, JNK, p-CREB, CREB, cytochrome c, cleaved-caspase 3, Bax, Bcl-2 and β-actin were obtained from Cell Signaling Technology (MA, USA), and enhanced BCA protein assay kit (P0012), phenylmethylsulfonyl fluoride (PMSF) (ST506) and cell lysis buffer (P0013) for Western and IP analyses were obtained from Beyotime Institute of Biotechnology.
2.1 Zebrafish and treatment
The experimental protocols for using zebrafish were approved by the Institutional Animal Care and Use Committee at the Model Animal Research Center, China Pharmaceutical University. All animal experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.Adult zebrafishes were purchased from China Zebrafish Resource Center, and the feeding conditions were as follows: 28.5 °C, with a 14/10 h light/dark cycle. The zebrafishes were fed with brine shrimps three times a day, and embryos were obtained every morning after being induced by turning on the light. The embryos were pre-treated with PTU at 6 hpf when the total concentration of PTU reached 0.2 mM; then at 35 hpf, the solution of PTU was washed with the fresh egg water followed by treatment with STS at the different doses of 0.1, 1, 10, 50, and 100 μM for further 25 h.20
2.2 Cell culture
The B16F10 cells (CRL-6475) were obtained from the CAS (Chinese Academy of Sciences). The cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS) (GIBICO), 100 U ml−1 penicillin and 100 mg ml−1 streptomycin (GIBCO) under a humidified atmosphere with 5% CO2 at 37 °C. The FBS used herein was incubated in a 56 °C water bath for 30 min to destroy serum complements. Significantly fast proliferation can affect melanin production. When the cells were treated with STS for melanogenesis, 2.5% FBS was used to slow down the proliferation.2.3 Determination of cell viability by the MTT assay
MTT (5 mg ml−1) was added to the wells, and the plates were incubated for 4 h at 37 °C. The MTT reaction was stopped by adding DMSO (150 μl per well), followed by stirring for 10 min. The optical absorbance at 490 nm of each well was measured using a multi-well plate reader. The B16F10 cells were incubated with STS at the concentrations of 0.01, 0.1, 1, 10, 20, 40, 80, and 100 μM for 48 h.2.4 Embryo cytotoxicity assays
The embryos were dipped in the egg water and processed with STS at 6 hpf. The mortality of the embryos was determined after exposure to STS at 24 hpf (the end of the segmentation stage). The hatching rate and survival rate were also appraised at specified time points (48, 72, 96, and 120 hpf), and malformations, including tissue ulceration and pericardial edema, were observed among the larvae for both the control and STS-treated groups.2.5 Tyrosinase activity and melanin content assay
Tyrosinase activity, as the dopa oxidase activity herein, was measured by the rate of L-DOPA oxidation as reported in the literature.21 The B16F10 cells were treated with STS for 48 h, washed with ice-cold PBS, and lysed by incubation in cell lysis buffer [1 mM PMSF] at 4 °C for 20 min; then, the lysates were centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min to obtain the supernatant for the activity assay and centrifugation for the melanin content assay. Protein concentrations were determined by the BCA kit with bovine serum albumin (BSA) as a standard. Herein, 100 μl of supernatant containing the same 30 μg total proteins was added to each well in a 96-well plate and then mixed with 100 μl 0.1% L-DOPA in 0.1 MPBS (pH 6.8) (m/v). After incubation at 37 °C for 0.5 h, dopachrome was monitored by measuring the absorbance at 475 nm.
000 rpm for 10 min to obtain the supernatant for the activity assay and centrifugation for the melanin content assay. Protein concentrations were determined by the BCA kit with bovine serum albumin (BSA) as a standard. Herein, 100 μl of supernatant containing the same 30 μg total proteins was added to each well in a 96-well plate and then mixed with 100 μl 0.1% L-DOPA in 0.1 MPBS (pH 6.8) (m/v). After incubation at 37 °C for 0.5 h, dopachrome was monitored by measuring the absorbance at 475 nm.
Total melanin in the cell pellet was dissolved in 100 μl of 1 N NaOH/10% DMSO for 1 h at 80 °C and the solubilized melanin was measured at 405 nm. Melanin content was calculated as a percent of the control.
2.6 Fontana-Masson staining for melanin
For the detection of the melanin pigment, the B16F10 cells were plated on 13 mm glass coverslips in a 24-well plate. Then, 48 hours after the STS administration, cells were fixed with formalin. After this, they were stained according to the Masson-Fontana ammoniacal silver staining method.22 Briefly, the cells were incubated in an ammoniacal silver solution overnight. After fixation in an acid-fixing solution, they were stained with Kernechtrot. Images were obtained using a CCD camera (Nikon Ni-U).2.7 Reactive oxygen species measurement
The embryos were dipped in the egg water and processed with PTU at 6 hpf. PTU was washed at 5 dpf, and the zebrafishes were treated with STS at different doses for 48 h and then with H2O2 (500 μM) for 4 h. Then, DCFH-DA (10 μM) was added to the zebrafish for 30 min. Images were obtained using a CCD camera (Nikon Ni-U).23The B16F10 cells were grown in 24-well plates and treated with STS at different concentrations for 48 h and then with H2O2 (100 μM) for 4 h. ROS generation was measured using 2′,7′-dichlorodihydrofluorescein (D6883, Sigma), which was oxidized to fluorescent dichlorodihydrofluorescein in the presence of ROS. Cells were incubated with 10 μM 2′,7′-dichlorodihydrofluorescein for 30 min; after this, the reaction was quenched via the addition of the cell lysis buffer provided with the kit.
2.8 Mitochondrial membrane potential assay
The B16F10 cells were evaluated by JC-1 [5,5,6,6-tetrachloro-1,1,3,3-tetraethylbenzimidazolylcarbocyanine iodide] staining. Briefly, the B16F10 cells were incubated on coverslips in 24-well plates. The cells were treated with STS at different concentrations for 48 h and then with H2O2 (500 μM) for 4 h. Then, the cells were incubated with an equal volume of the JC-1 staining solution (5 μg ml−1) at 37 °C for 20 min and rinsed twice with dye buffer (1×). Mitochondrial membrane potentials have been monitored by the determination of the relative amounts of dual emissions: the red emission of the dye represents a potential-dependent aggregation in the mitochondria that reflects ΔΨm, and the green fluorescence represents the monomeric form of JC-1 appearing in the cytosol after mitochondrial membrane depolarization. The ratio of the aggregated JC-1 to the monomeric JC-1 represented ΔΨm of the B16F10 cells. The detailed analysis procedure was described in the manufacturer's protocol (Beyotime Institute of Biotechnology, China).2.9 Apoptosis analysis
The B16F10 cells were treated with STS at different concentrations for 48 hours and washed with PBS; then, they were treated with H2O2 (100 μM) for 4 hours. The cells were washed twice with cold PBS, fixed with paraformaldehyde and stained with the fluorescent dye Hoechst 33258 (1 μg ml−1, Hoechst Staining Kit, Beyotime) for 15 min in the dark. The cells were imaged via fluorescence microscopy.Cell apoptosis was also investigated by FACS (Becton Dickinson, San Jose, CA, USA) according to the instruction provided in the Annexin V/PI kit (BD Biosciences). Briefly, after treatment, the floating and trypsinized adherent cells were obtained and suspended in a binding buffer (1×); after this, an aliquot of 100 μl of cell suspension was incubated with 5 μl of Annexin V-FITC and 5 μl of PI for 15 min in the dark, and then, 400 μl binding buffer (1×) was added to each sample. The stained cells were analyzed directly by flow cytometry using the Cell Quest program (Becton Dickinson, Franklin, NJ).
2.10 Western blot analysis
The protein suspension was obtained according to the abovementioned method. Western blot was performed as described previously.21 The primary antibodies used were MITF (ab20663), TYR (C-19) (SC7833), TRP-1 (SC25543), DCT (ab74073), p-CREB (CST 9198), CREB (CST 9197), cAMP (ab76238), p-p38 (CST4511), p38 (CST8690), p-ERK1/2 (CST4370), ERK1/2 (CST4695), p-JNK (CST4668), JNK (CST9252), gp100 (ab137078), Rab27a (ab55667), Rab17 (ab118998), Cdc42 (ab187643), KIF5b (ab5629), cytochrome c (CST 11940), cleaved-caspase 3 (CST 9661), Bax (CST 2772), Bcl-2 (CST2870), and β-actin (CST3700). Proteins were visualized using an enhanced chemiluminescence detection system. Densitometric analysis was again performed using the Tanon 5200 Multi to scan the signals. The Western blot assay results reported herein are representative of at least 3 experiments.2.11 Statistical analyses
All data are expressed as mean ± SD. Statistical analysis was performed via one-way ANOVA followed by Tukey's post hoc test for multiple comparison tests. Significant differences were accepted when P < 0.05.3. Results
3.1 Zebrafish toxicity tests
The toxicity of STS in zebrafish was observed. The mortality and the developmental progression, including the body length and trunk-tail angle, were measured. As shown in Fig. 1B, taking the survival, body length and trunk-tail angle of the zebrafish as indices, no significant difference was observed after STS-treatments (0.1, 1, 10, 50, and 100 μM) for the control group at 24, 48, 72, 96 and 120 hpf. Malformations were determined and imaged, and pericardial edema and tissue ulceration were not observed.3.2 STS increases melanin synthesis and tyrosinase activity in zebrafish
Zebrafish has been approved as an efficient whole-animal model for the screening of melanogenic inhibitors or stimulators.20 The change in the melanin content of zebrafish is one of the most visual indices to evaluate the activity of STS. The embryos were preprocessed with PTU (0.2 mM) at 6 hpf. The PTU-1 group was treated with PTU for the whole course (from 6 hpf to 60 hpf). The removal of pretreated PTU (via treatment from 6 hpf to 35 hpf and then replacement with water) repaired the pigmentation of the zebrafish (named the PTU-2 group), and the STS treatments promoted this recolor process in a dose-dependent manner (Fig. 2A). Moreover, tyrosinase is a rate-limiting enzyme in melanin synthesis; thus, tyrosinase activity has been confirmed to measure the ability of melanogenesis.24 Tyrosinase activity of zebrafish treated with STS was measured and compared with the case of the blank group; it was found that STS increased the tyrosinase activity in a dose-dependent manner (Fig. 2B). The melanin content in the zebrafish was quantified using the homogenate, and the melanin content of zebrafish was increased in a dose-dependent manner by treatment with STS at the concentrations of 10 μM, 50 μM, and 100 μM (Fig. 2C). | ||
| Fig. 2 STS increases melanin synthesis and tyrosinase activity in zebrafish. (A) The embryos were obtained and treated with STS at different doses (1, 10, 50, and 100 μM) from 6 hpf to 35 hpf. (B) The homogenate of each group of zebrafishes was used to examine the tyrosinase activity and melanin content. (C) Zebrafish were treated with PTU (0.2 mM) from 6 hpf to 35 hpf; then, PTU was removed using water with or without different doses of STS for another 25 hours (from 35 hpf to 60 hpf). STS increased the melanin recovery. Data were analyzed by one-way analysis of variance (ANOVA) followed by post hoc Tukey test. *P < 0.05, **P < 0.01, compared with control. | ||
3.3 STS regulates melanin synthesis and the related pathway in the B16F10 cells
The viability of the B16F10 cells treated by STS was measured, and the results are shown in the ESI (Fig. S6†); the viability of the B16F10 cells remained almost unchanged as compared to that in the case of the control group at the concentrations of 0.01, 0.1, 1, and 10 μM. Moreover, the melanin content and tyrosinase activity (Fig. 3A) were increased in a dose-dependent manner after STS treatment. Our data demonstrated that the expression levels of MITF, tyrosinase, TRP-1 and DCT were significantly enhanced 48 h after the administration of STS to the B16F10 cells (Fig. 3B). To further clarify the mechanism underlying the effect of STS on melanogenesis, the mitogen-activated protein kinase (MAPK) intracellular signal transduction cascade was examined. As shown in Fig. 3C, the phosphorylation of p38, JNK and ERK1/2 was enhanced after treatments with STS at different concentrations, and 10 μM of STS exerted the most significant effect. Moreover, 10 μM of STS induced p38 and JNK phosphorylation from 5 min onwards that reached the peak at 30 min, whereas ERK1/2 phosphorylation reached the peak earlier at 5 min. The PKA signaling pathway was also examined. The activation of CREB and cAMP was detected by Western blot analysis. STS showed a concentration-dependent promoting effect on the PKA pathway. As shown in Fig. 3D, a delayed increase in the p-CREB levels was observed following 10 μM of STS treatment at 6 h and 12 h. The cells were treated with STS at various concentrations (0.1, 1, and 10 μM) for 48 h, and the cAMP level increased in a concentration-dependent manner (Fig. 3E). Using the combination of STS and inhibitors, herein, SB203580, PD98059, SP600125 and H89, we demonstrated the effect of STS mainly on the p38 and JNK MAPK signaling pathways and secondarily on the ERK1/2 and PKA signaling pathways (Fig. 3F).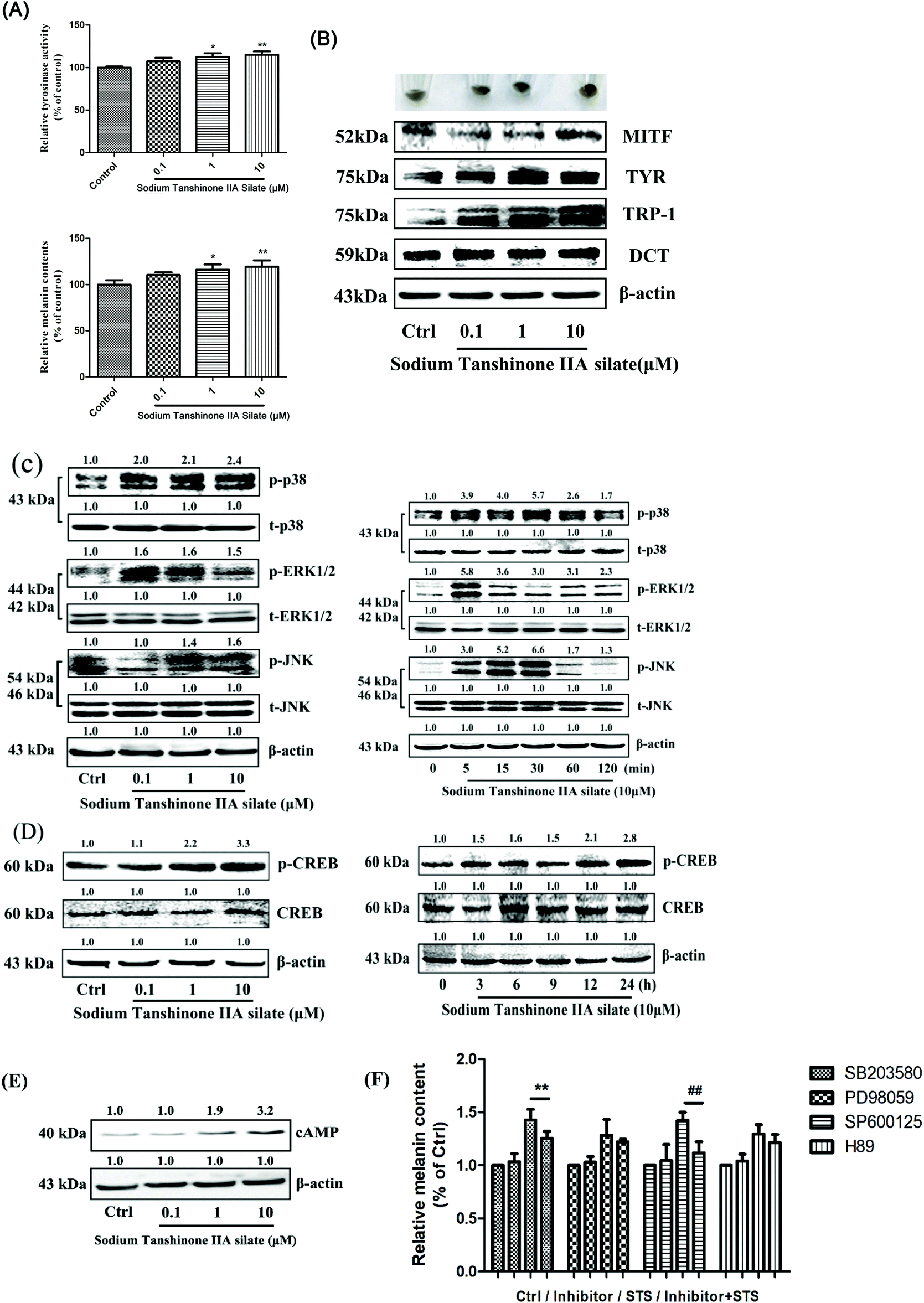 | ||
| Fig. 3 STS regulates melanin synthesis and the related pathway in B16F10 cells. (A) After incubation of cells with STS at various concentrations (0.1, 1, and 10 μM) for 48 h, tyrosinase activity and the melanin content were examined. (B) The expression levels of MITF, tyrosinase, TRP-1 and DCT were examined after STS treatments for 48 h. (C) The mitogen-activated protein kinase (MAPK) intracellular signal transduction cascade was examined. Cells were treated with STS at various concentrations (0.1, 1, and 10 μM) for 48 h, or with STS (10 μM) for the indicated time period (0, 5, 15, 30, 60, and 120 min), and the phosphorylations of the p38 MAPK, ERK, and JNK were measured. (D) Cells were treated with STS at various concentrations (0.1, 1, and 10 μM) for 48 h, or with STS (10 μM) for the indicated time period (0, 3, 6, 9, 12, and 24 h); the phosphorylations of the CREB were measured. (E) Cells were treated with STS at various concentrations (0.1, 1, and 10 μM) for 48 h, and the cAMP level was examined. (F) The inhibitors of MAPK (p38 inhibitor: SB203580; ERK1/2 inhibitor: PD98059; JNK inhibitor: PD600125; PKA inhibitor: H89) and PKA pathway were applied, and the melanin content was measured. Results were normalized against β-actin expression. Results shown are means ± SD (n = 3). Data were analyzed by one-way analysis of variance (ANOVA) followed by the post hoc Tukey test. **P < 0.01, (STS + SB203580) compared with STS; ##P < 0.01, (STS + SP600125) compared with STS. | ||
3.4 STS regulates melanocytic filopodia formation to increase melanosome trafficking
The B16F10 cells were treated with STS (0.1, 1, and 10 μM) for 48 hours. Then, Fontana-Masson staining was applied, and as shown in Fig. 4A, STS increased melanogenesis in the B16F10 cells. Via the images, we found that STS promoted melanin transfer from the area near the nucleus to the membrane-surrounding area, particularly to the dendrite tips. We hypothesized that STS not only promoted melanin synthesis but also stimulated the translocation of melanin. Filopodia are known to play a role in melanosome transfer. Cdc42 stimulates filopodia formation25 and mediates melanosome transfer in human melanocytes.26 Kinesin superfamily proteins (KIFs) function as motors: for example, KIF5b contributes to the outward transport of melanosomes.27 Rabs participate in melanosome maturation and trafficking, and Rab27a is the most notable example.28 Rab17 may regulate the final steps of melanosome release from the cell.29 Herein, we demonstrated that STS increased the Cdc42 and KIF5b expression to stimulate filopodia formation. Furthermore, the expressions of both Rab27a and Rab17 were promoted; this benefited melanosome transfer (Fig. 4B).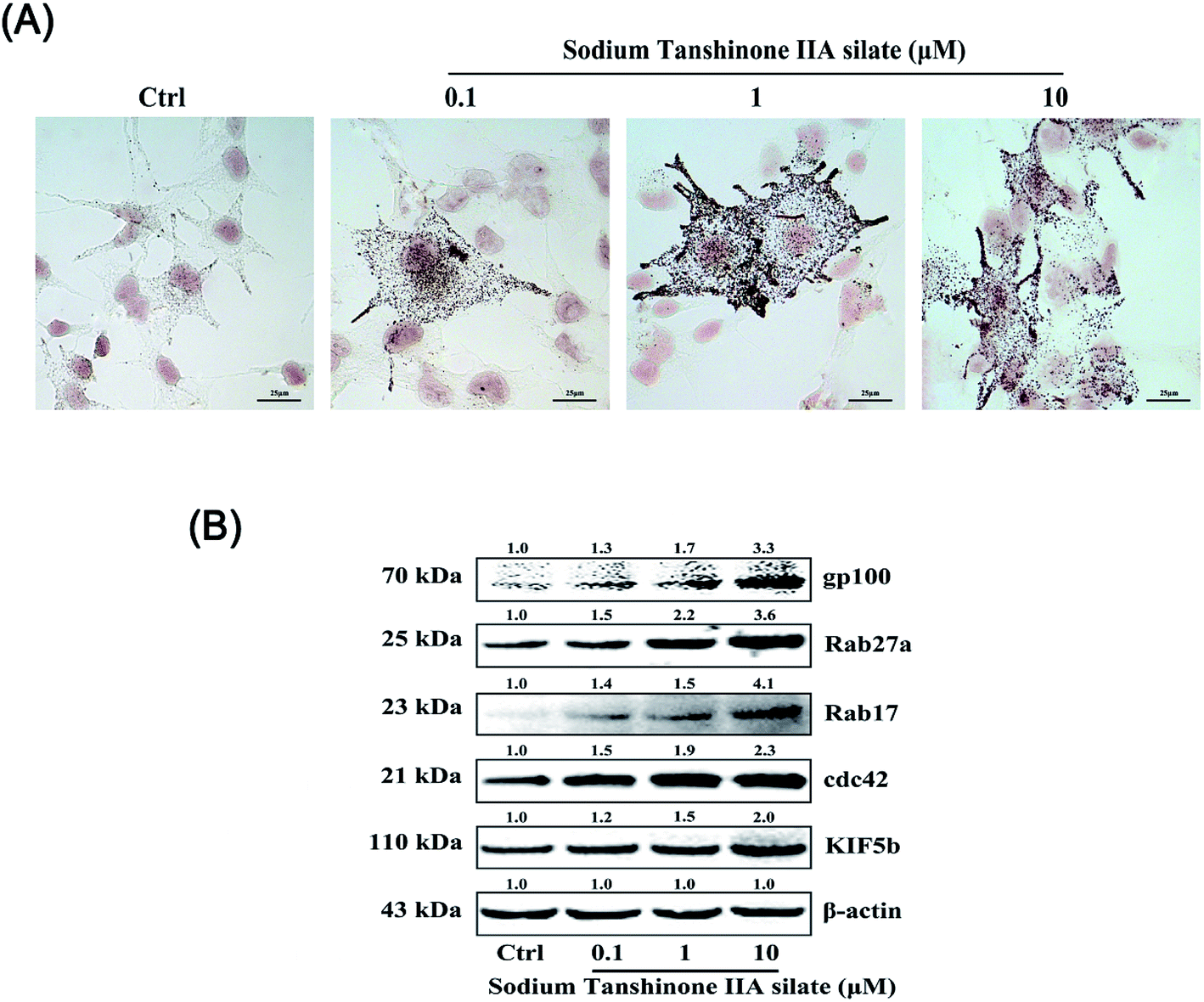 | ||
| Fig. 4 STS regulates melanocytic filopodia formation to increase melanosome trafficking. (A) The B16F10 cells were stained with Masson-Fontana ammoniacal silver stain. The B16F10 cells were plated on 13 mm glass coverslips in a six-well plate and then treated with STS at different concentrations for 48 h. (B) The expression levels of gp100, Rab27a, Rab17, Cdc42, and KIF5b were examined after STS treatments for 48 h. Results were normalized against the β-actin expression. | ||
3.5 STS-meditated ROS elimination contributes to melanogenesis
Since zebrafish remain optically transparent after PTU treatment, they have been used to observe ROS in vivo, and several fluorescent probes have been commercialized.30Images are shown in Fig. 5A, and the difference between the green fluorescence of the control group and H2O2-model group was remarkable. Compared to the case of the model group, the ROS content was significantly decreased by the STS treatment. To illustrate the antioxidant activity of STS, the superoxide-specific fluorescent probe dihydroethidium (DHE) was used in the B16F10 cells. The images of the DHE-stained B16F10 cells are shown in Fig. 5B, and the fluorescence intensity of the model group (H2O2-treated) is higher than that of the control group. STS concentration-dependently decreased the superoxide anion as detected by the lower DHE fluorescence as compared to that in the case of the model group.
 | ||
| Fig. 5 STS-meditated ROS elimination contributes to melanogenesis. (A) Zebrafishes (7 dpf) were treated with STS at different doses for 24 h and then with H2O2 (500 μM) for 4 h. DCFH-DA (10 μM) was added to zebrafish for 30 min. Images were obtained. (B) The B16F10 cells were grown in 24-well plates and treated with STS at different concentrations for 24 h and then with H2O2 (100 μM) for 4 h. Cells were incubated with 10 μM 2′,7′-dichlorodihydrofluorescein for 30 min. Scale bars, 100 μm. | ||
3.6 STS reverses mitochondrial impairment of the B16F10 cells
Mitochondrial impairment in the lesion area of vitiligo is determined by the production of intracellular ROS.11 To further discover the role of STS in antioxidation, the mitochondrial impairment of B16F10 cells was measured by 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1). JC-1 is a fluorescent probe used to determine the mitochondrial membrane potential, and JC-1 imaging also shows the comparison of mitochondrial heterogeneity under different treatment conditions.31 As shown in Fig. 6, the mitochondrion of the model group emits green fluorescence, which means that the mitochondrion is depolarized, and the mitochondrial membrane potential (MMP) is low. CCCP is a positive drug that is used to decrease the MMP. After CCCP treatment, the MMP became low, and the fluorescence was green.32 Compared to that of the control group, the MMP of the B16F10 cells was remarkably decreased after H2O2 treatment. Thus, STS could reverse the mitochondrial impairment of the B16F10 cells induced by H2O2 in a concentration-dependent manner. | ||
| Fig. 6 STS reverses mitochondrial impairment of the B16F10 cells. The B16F10 cells were treated with STS at different concentrations (0.1, 1, and 10 μM) for 48 hours and then with H2O2 (100 μM) for 4 hours. CCCP is the positive drug used to decrease the MMP. Mitochondrial membrane potential was measured using the JC-1 dye. Representative photomicrographs were obtained by the Nikon Ni-U fluorescence microscope. Scale bars, 50 μm. | ||
3.7 STS inhibits the H2O2-induced apoptosis of B16F10 cells
The apoptotic death of melanocytes has been discovered in the skin of vitiligo, and “the autocytotoxic theory” has been proposed for it and accepted.33 Oxidative stress and the production of ROS are the possible causes of the apoptosis of melanocytes. The apoptosis of H2O2-treated B16F10 cells was measured by Hoechst 33258 staining and flow cytometry analysis. H2O2 treatment remarkably induced apoptotic features when compared with the case of the blank group. STS pre-treatment for 48 hours dose-dependently inhibited the apoptosis (Fig. 7A). The flow cytometry assay showed that the apoptotic cells increased significantly after H2O2 treatment for 4 hours from 34.54% to 48.06% as compared to the case of the control group. STS pre-treatment could inhibit the H2O2-induced apoptosis (Fig. 7B). Moreover, STS could repair cell damage induced by excessive oxidative stress. In various cells, phospho-Akt works as an anti-apoptotic factor by phosphorylating Bad to prevent its binding to Bcl-2.34 Our data demonstrated that STS reduced the expressions of Bax, cleaved-caspase 3 and cytochrome c, at the same time increased Bcl-2 (Fig. 7C), hence, the Bcl-2/Bax ratio was significantly increased (Fig. 7D). | ||
| Fig. 7 STS inhibits H2O2-induced apoptosis of the B16F10 cells. (A) Morphology of the cells exposed to STS at different concentrations for 48 h and imaged using a fluorescence microscope. The white arrows indicate apoptotic cells. Scale bars, 50 μm. (B) After treatments with STS for 48 h, apoptosis was evaluated using Annexin-V-FITC/PI staining detected by flow cytometry assay. (C) Western blot of the cytosol fraction of melanocytes demonstrated an increase in the Bax, cleaved-caspase 3 and cytochrome c protein levels and a decrease in the Bcl-2 levels. Results were normalized against the β-actin expression. (D) The band densities of the interest proteins were measured by the Tanon 5200 Multi analysis software program, and then, the Bcl-2/Bax ratios were obtained. Results shown are means ± SD and are representative of 3 independent experiments. Data were analyzed by one-way analysis of variance (ANOVA) followed by the post hoc Tukey test. ***P < 0.001, compared with control. | ||
4. Discussion
Vitiligo is a common autoimmune disease of the skin that has a major impact on patients' quality of life. Current treatments for vitiligo are time-consuming and expensive and have low efficacy.35 For drug exploitation, pathogenesis and further comprehensive study on signal transduction are thought to be vital points. Numerous factors, including impaired melanocyte migration, exposure to sunlight, stress, trauma, infections, and neural abnormalities, have been implicated in the development of vitiligo.5 This disease becomes worse when accompanied by systemic inflammation and oxidative stress. Oxidative stress can disrupt the homeostasis of melanocytes.10,19,36 Apoptosis has been reported as the mechanism for the removal of melanocytes. Confirmation of the mechanisms of apoptosis induction and identification of ways to block apoptosis are thought to be one possible way to prevent vitiligo progression.37In numerous studies, the signature of IFN-γ as a driver of vitiligo autoimmunity has been confirmed.38–41 Tofacitinib is a JAK 1/3 inhibitor for the treatment of rheumatoid arthritis. A patient with facial and acral vitiligo acquired repigmentation after approximately 5 months of therapy with oral tofacitinib citrate.42 Moreover, a recent case study indicates that the topical Janus kinase inhibitor ruxolitinib 1.5% cream may offer a new therapeutic option for vitiligo treatment.43 Simvastatin is widely used in the treatment of dyslipidemia and cardiovascular diseases. Interestingly, a patient with both vitiligo and hypercholesterolemia has been reported to gain rapid repigmentation of skin after receiving high-dose simvastatin.44
STS is a water-soluble derivative of tanshinone IIA, which is a main bioactive component isolated from the roots of the Chinese herb Salvia miltiorrhiza Bunge (danshen). STS has been widely used for the treatments of cardiovascular and cerebrovascular diseases in China.15,45 It has been reported that STS protects cardiomyocytes against oxidative stress-mediated apoptosis by inhibiting JNK activation.46 STS inhibits the formation of reactive oxygen radicals in rat heart mitochondria.47 In the present study, the effect of STS on melanogenesis in vivo and in vitro was investigated to clarify its underlying molecular mechanism. Our results indicate no toxicity of STS at different doses on the development of zebrafish embryos as pericardial edema and tissue ulceration, which are key indices of malformations, have not been observed (Fig. 1). Compared with the case of the control group, after treatment from 6 hpf to 35 hpf, STS significantly increased melanin synthesis in zebrafish in a dose-dependent manner (Fig. 2A). PTU (0.2 mM) is used as a tyrosinase inhibitor that does not interfere with the developmental process of zebrafish (1). In this study, PTU was added from 6 hpf to 60 hpf (PTU-1 group), or from 6 hpf to 35 hpf and washed with brine water with or without STS (PTU-2 group). As shown in Fig. 2C, compared with the case of the control group, melanogenesis of the PTU-1 group was significantly inhibited. STS facilitated the re-pigmentation process in a dose-dependent manner (compared with the case of the PTU-2 group). The melanin content and tyrosinase activity, as quantitative indicators, were also measured, which reconfirmed the pro-melanogenesis effect of STS on zebrafish (Fig. 2B). STS increases melanin synthesis in the B16F10 cells, activating the MAPK and PKA pathways (Fig. 3). Skin pigmentation depends not only on the production of melanin by melanocytes but also importantly on the transfer of the uniformly distributed melanin-containing vesicles, the melanosomes, to adjacent keratinocytes and their distribution towards the surface of the skin;48 as shown in Fig. 4A, STS promoted melanin synthesis and transfer from the area near the nucleus to the membrane-surrounding area, particularly to the dendrite tips. The expressions of the representative Rabs, Rab27a and the behind Rab17, which regulate melanosome maturation and trafficking, have been increased by the STS treatment. In addition, we demonstrated that STS increased the Cdc42 and KIF5b expression to stimulate filopodia formation (Fig. 4B). Vitiligo is a dermatological disease characterized by the loss of melanocytes from the epidermis. Several theories have been reported to explain the pathogenesis, and “autoimmune theories” are most accepted.33 Experimental and clinical evidence suggest that oxidative stress closely interacts with the immune system and plays an important role in the pathogenesis of the depigmentation process.49 The presence of reactive oxygen species (ROS) in active vitiligo skin has been adequately reported. Oxidative stress is one of the inducing factors causing vitiligo. In vivo and in vitro experimental results reveal that STS-meditated ROS elimination contributes to melanogenesis (Fig. 5). Mitochondrial dysfunction is emerging as a mechanism underlying various inflammatory and autoimmune diseases. The repair of injured melanocytes and the renewal of melanocytes are keys for the treatment of vitiligo. STS could concentration-dependently reverse the mitochondrial impairment of the B16F10 cells induced by H2O2 (Fig. 6). STS reduces the expressions of Bax, cleaved-caspase 3 and cytochrome c and simultaneously increases the expression of Bcl-2 to repair the cell damage induced by excessive oxidative stress (Fig. 7).
The procedures for detecting the ability of STS to reduce ROS-mediated damage induced by H2O2 were followed from literature, and cells or zebrafish exposed to STS for 24/48 h prior to damage induction were the most accepted models.50,51
5. Conclusion
Since cost and long-term safety are major concerns in the treatment of vitiligo, topical formulations may be a better option in this regard. STS may provide a breakthrough in the treatment of vitiligo in the future. Its use may be repurposed to provide an option for patients with vitiligo, and it can also be reformulated as a topical treatment option.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81503176), the Fundamental Research Funds for the Central Universities (2632019ZD03), Shanghai Scientific and Technological Innovation Project (17DZ1205400), and Science and Technology Development Fund of Shanghai Municipal Public Security Bureau (2018002).References
- W. Sun, P. E. Sanderson and W. Zheng, Drug discovery today, 2016, 21, 1189–1195 CrossRef CAS PubMed.
- G. Jin and S. T. Wong, Drug discovery today, 2014, 19, 637–644 CrossRef PubMed.
- S. M. Strittmatter, Nat. Med., 2014, 20, 590–591 CrossRef CAS.
- R. Pryor and F. Cabreiro, Biochem. J., 2015, 471, 307–322 CrossRef CAS PubMed.
- J. M. Richmond, M. L. Frisoli and J. E. Harris, Curr. Opin. Immunol., 2013, 25, 676–682 CrossRef CAS PubMed.
- V. R. Attili and S. K. Attili, Int. J. Dermatol., 2008, 47, 663–669 CrossRef.
- R. Kumar and D. Parsad, Indian J. Dermatol. Venereol. Leprol., 2012, 78, 19–23 CrossRef PubMed.
- M. L. Dell'Anna, M. Ottaviani, B. Bellei, V. Albanesi, A. Cossarizza, L. Rossi and M. Picardo, J. Cell. Physiol., 2010, 223(1), 187–193 Search PubMed.
- Z. Qiao, X. Wang, L. Xiang and C. Zhang, Oxid. Med. Cell. Longevity, 2016, 2016, 3401570 Search PubMed.
- L. Denat, A. L. Kadekaro, L. Marrot, S. A. Leachman and Z. A. Abdel-Malek, J. Invest. Dermatol., 2014, 134, 1512–1518 CrossRef CAS PubMed.
- M. L. Dell'Anna, V. Maresca, S. Briganti, E. Camera, M. Falchi and M. Picardo, J. Invest. Dermatol., 2001, 117, 908–913 CrossRef PubMed.
- A. Taieb, A. Alomar, M. Böhm, M. L. Dell'anna, A. De Pase, V. Eleftheriadou, K. Ezzedine, Y. Gauthier, D. J. Gawkrodger, T. Jouary, G. Leone, S. Moretti, L. Nieuweboer-Krobotova, M. J. Olsson, D. Parsad, T. Passeron, A. Tanew, W. van der Veen, N. van Geel, M. Whitton, A. Wolkerstorfer and M. Picardo, Vitiligo European Task Force (VETF); European Academy of Dermatology and Venereology (EADV); Union Européenne des Médecins Spécialistes (UEMS), Br. J. Dermatol., 2013, 168, 5–19 CrossRef CAS PubMed.
- D. J. Gawkrodger, A. D. Ormerod, L. Shaw, I. Mauri-Sole, M. E. Whitton, M. J. Watts, A. V. Anstey, J. Ingham and K. Young, Therapy Guidelines and Audit Subcommittee, British Association of Dermatologists, Clinical Standards Department, Royal College of Physicians of London, Cochrane Skin Group, Vitiligo Society, Br. J. Dermatol., 2008, 159, 1051–1076 CrossRef CAS PubMed.
- W. Y. Wu, H. Yan, X. B. Wang, Y. Z. Gui, F. Gao, X. L. Tang, Y. L. Qin, M. Su, T. Chen and Y. P. Wang, PLoS One, 2014, 9, e94957 CrossRef.
- B. Ji, F. Zhou, L. Han, J. Yang, H. Fan, S. Li, J. Li, X. Zhang, X. Wang, X. Chen and Y. Xu, Transl. Stroke Res., 2017, 8, 334–340 CrossRef CAS PubMed.
- W. Y. Wu, W. Y. Wang, Y. L. Ma, H. Yan, X. B. Wang, Y. L. Qin, M. Su, T. Chen and Y. P. Wang, Br. J. Pharmacol., 2013, 169, 1058–1071 CrossRef CAS PubMed.
- M. Becatti, C. Fiorillo, V. Barygina, C. Cecchi, T. Lotti, F. Prignano, A. Silvestro, P. Nassi and N. Taddei, J. Cell. Mol. Med., 2014, 18, 514–529 CrossRef CAS PubMed.
- S. A. Birlea, G. E. Costin, D. R. Roop and D. A. Norris, Med. Res. Rev., 2017, 37, 907–935 CrossRef PubMed.
- Q. Shi, W. Zhang, S. Guo, Z. Jian, S. Li, K. Li, R. Ge, W. Dai, G. Wang, T. Gao and C. Li, Cell Death Differ., 2016, 23, 496–508 CrossRef CAS PubMed.
- T. Y. Choi, J. H. Kim, D. H. Ko, C. H. Kim, J. S. Hwang, S. Ahn, S. Y. Kim, C. D. Kim, J. H. Lee and T. J. Yoon, Pigm. Cell Res., 2007, 20, 120–127 CrossRef CAS PubMed.
- J. Zhou, J. Shang, J. Song and F. Ping, Int. J. Biochem. Cell Biol., 2013, 45, 308–316 CrossRef CAS.
- S. Noguchi, M. Kumazaki, Y. Yasui, T. Mori, N. Yamada and Y. Akao, J. Invest. Dermatol., 2014, 134, 461–469 CrossRef CAS PubMed.
- M. Kitada, D. Koya, T. Sugimoto, M. Isono, S. Araki, A. Kashiwagi and M. Haneda, Diabetes, 2003, 52, 2603–2614 CrossRef CAS PubMed.
- M. K. Kim, C. Y. Bang, M. Y. Kim, J. H. Lee, H. Ro, M. S. Choi, D. I. Kim, Y. P. Jang and S. Y. Choung, BMC Complementary Altern. Med., 2016, 16, 223 CrossRef PubMed.
- S. A. Hobson, P. A. Vanderplank, R. J. Pope, N. C. Kerr and D. Wynick, J. Neurochem., 2013, 127, 199–208 CrossRef CAS PubMed.
- G. A. Scott, M. Arioka and S. E. Jacobs, J. Invest. Dermatol., 2007, 127, 668–675 CrossRef CAS.
- M. Hara, M. Yaar, H. R. Byers, D. Goukassian, R. E. Fine, J. Gonsalves and B. A. Gilchrest, J. Invest. Dermatol., 2000, 114, 438–443 CrossRef CAS PubMed.
- A. N. Hume, L. M. Collinson, A. Rapak, A. Q. Gomes, C. R. Hopkins and M. C. Seabra, J. Cell Biol., 2001, 152, 795–808 CrossRef CAS PubMed.
- K. A. Beaumont, N. A. Hamilton, M. T. Moores, D. L. Brown, N. Ohbayashi, O. Cairncross, A. L. Cook, A. G. Smith, R. Misaki, M. Fukuda, T. Taguchi, R. A. Sturm and J. L. Stow, Traffic, 2011, 12, 627–643 CrossRef CAS PubMed.
- L. Fang and Y. I. Miller, Free Radical Biol. Med., 2012, 53, 1411–1420 CrossRef CAS PubMed.
- V. C. Keil, F. Funke, A. Zeug, D. Schild and M. Müller, Pflügers Arch., 2011, 462, 693–708 CrossRef CAS PubMed.
- H. Xie, J. Guan, L. A. Borrelli, J. Xu, A. Serrano-Pozo and B. J. Bacskai, J. Neurosci., 2013, 33, 17042–17051 CrossRef CAS PubMed.
- N. C. Laddha, M. Dwivedi, M. S. Mansuri, A. R. Gani, M. Ansarullah, A. V. Ramachandran, S. Dalai and R. Begum, Exp. Dermatol., 2013, 22, 245–250 CrossRef CAS.
- Y. Hosotani, S. Kashiwamura, A. Kimura-Shimmyo, A. Sekiyama, H. Ueda, T. Ikeda, O. Mimura and H. Okamura, J. Dermatol., 2008, 35, 514–524 CrossRef CAS.
- P. Agarwal, M. Rashighi, K. I. Essien, J. M. Richmond, L. Randall, H. Pazoki-Toroudi, C. A. Hunter and J. E. Harris, J. Invest. Dermatol., 2015, 135, 1080–1088 CrossRef CAS PubMed.
- N. G. Gavalas, S. Akhtar, D. J. Gawkrodger, P. F. Watson, A. P. Weetman and E. H. Kemp, Biochem. Biophys. Res. Commun., 2006, 345, 1586–1591 CrossRef CAS PubMed.
- C. L. Huang, J. J. Nordlund and R. Boissy, Am. J. Clin. Dermatol., 2002, 3, 301–308 CrossRef.
- J. E. Harris, T. H. Harris, W. Weninger, E. J. Wherry, C. A. Hunter and L. A. Turka, J. Invest. Dermatol., 2012, 132, 1869–1876 CrossRef CAS PubMed.
- L. Yang, S. Yang, J. Lei, W. Hu, R. Chen, F. Lin and A. E. Xu, J. Dermatol., 2018, 45, 31–38 CrossRef CAS PubMed.
- R. Speeckaert, M. Speeckaert, S. De Schepper and N. van Geel, Autoimmun. Rev., 2017, 16, 937–945 CrossRef CAS PubMed.
- M. Rashighi and J. E. Harris, Ann. Transl. Med., 2015, 3, 343 Search PubMed.
- B. G. Craiglow and B. A. King, JAMA Dermatol., 2015, 151, 1110–1112 CrossRef.
- B. Rothstein, D. Joshipura, A. Saraiya, R. Abdat, H. Ashkar, Y. Turkowski, V. Sheth, V. Huang, S. C. Au, C. Kachuk, N. Dumont, A. B. Gottlieb and D. Rosmarin, J. Am. Acad. Dermatol., 2017, 76, 1054–1060 CrossRef CAS PubMed.
- M. Noël, C. Gagné, J. Bergeron, J. Jobin and P. Poirier, Lipids Health Dis., 2004, 3, 7 CrossRef PubMed.
- Q. Shang, H. Wang, S. Li and H. Xu, J. Evidence-Based Complementary Altern. Med., 2013, 756519 Search PubMed.
- R. Yang, A. Liu, X. Ma, L. Li, D. Su and J. Liu, J. Cardiovasc. Pharmacol., 2008, 51, 396–401 CrossRef CAS PubMed.
- G. Zhou, W. Jiang, Y. Zhao, G. Ma, W. Xin, J. Yin and B. Zhao, Biochem. Pharmacol., 2003, 65, 51–57 CrossRef CAS PubMed.
- V. J. Hearing, Pigm. Cell Res., 2007, 20, 334–335 Search PubMed.
- R. Colucci, F. Dragoni and S. Moretti, Oxid. Med. Cell. Longevity, 2015, 631927 Search PubMed.
- Z. Jian, L. Tang, X. Yi, B. Liu, Q. Zhang, G. Zhu, G. Wang, T. Gao and C. Li, J. Cell. Mol. Med., 2016, 20, 1307–1318 CrossRef CAS.
- Y. Chang, S. Li, W. Guo, Y. Yang, W. Zhang, Q. Zhang, Y. He, X. Yi, T. Cui, Y. An, P. Song, Z. Jian, L. Liu, K. Li, G. Wang, T. Gao, L. Wang and C. Li, J. Invest. Dermatol., 2017, 137, 1286–1296 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09786k |
| ‡ These three authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |