
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of novel elongator protein 2 inhibitors by compound library screening using surface plasmon resonance†
Chang-Peng
Xu‡
a,
Yong
Qi‡
a,
Zhuang
Cui
b,
Ya-Jun
Yang
c,
Jian
Wang
d,
Yan-Jun
Hu
b,
Bin
Yu
b,
Fa-Zheng
Wang
e,
Qing-Po
Yang
e and
Hong-Tao
Sun
 *a
*a
aDepartment of Orthopaedics, Guangdong Second Provincial General Hospital, Guangzhou, Guangdong, P. R. China. E-mail: htsun_bw@163.com
bDepartment of Orthopaedics and Traumatology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, P. R. China
cDepartment of Pharmacology, Guangdong Medical College, Zhanjiang, Guangdong, P. R. China
dDepartment of Orthopaedics, The Inner Mongolia People's Hospital, Hohhot, Inner Mongolia, P. R. China
eDepartment of Orthopaedics, The First People's Hospital of Kashgar Prefecture, Kashgar, Xinjiang, P. R. China
First published on 14th January 2019
Abstract
Tumour necrosis factor-α (TNF-α) is a pleiotropic cytokine that becomes elevated in chronic inflammatory states, including slowing down osteogenic differentiation, which leads to bone dysplasia in long-term inflammatory microenvironments. The elongator complex plays a role in gene regulation and association with various cellular activities, including the downstream signal transduction of TNF-α in osteogenic cells. To find an inhibitor of Elongator Protein 2 (Elp2), we performed a compound library screen and verified the pharmaceutical effects of candidate compounds on the mouse myoblast cell (C2C12) and mouse osteoblastic cells (MC3T3-E1). The commercial FDA-approved drug (FD) library and the bioactive compound (BC) library were used as candidate libraries. After a label-free, high-throughput affinity measurement with surface plasmon resonance (SPRi), seven kinds of compounds showed binding affinity with mouse Elp2 protein. The seven candidates were then used to perform an inhibition test with TNF-α-induced C2C12 and MC3T3-E1 cell lines. One candidate compound reduced the differentiation suppression caused by TNF-α with resuscitated alkaline phosphatase (ALP) activity, mineralization intensity and expression of osteogenic differentiation marker genes. The results of our study provide a competitive candidate to mitigate the TNF-α-induced osteogenic differentia.
Introduction
The evolutionarily conserved elongator complex (EC) plays an imperative role in gene regulation, associates with various cellular activities, and has attracted cultural attention.1 The EC structure contains two parts, each with three subunits. The Elp123 (Elp1–Elp3 subunit) is considered to be the core subcomplex of EC, and Elp456 (Elp4–Elp6 subunit) is an ancillary subcomplex.2 Elongator protein 2 (Elp2), also named STAT3 interacting protein 1 (StIP1), is the second largest subunit of EC with two WD40 propeller domains. The domains function as a histone acetyltransferase component of the RNA polymerase II (Pol II) holoenzyme involved in transcriptional elongation. Maria et al. demonstrated that Elp2 contributes to the stability of the Elp123 subcomplex and integrates different signals to regulate elongator activity.3 Wang et al. also reported that the conserved loop regions of Elp2 are crucial for elongator function.4 As a result, Elp2 plays a crucial role in conferring total enzyme activity and occupies an important place in downstream regulation.The human skeleton is constantly renewing itself and maintaining bone mass through a balance between the osteogenic activity of osteoblasts and the bone resorption activity of osteoclasts.5 However, persistent inflammation, such as traumatic fractures, rheumatoid arthritis (RA) and systemic lupus erythaematosus (SLE), is usually associated with bone formation and affects this balance by reducing bone mass and significantly impacting bone regeneration.6 Our previous proteomics study revealed that Elp2 showed a vital regulating effect on TNF-α-induced inhibition of osteoblast differentiation and regulation of ligand-dependent STAT3 activation. The verification on the C2C12 cell line showed that Elp2 activated the STAT3 pathway and down-regulated BMPR2 expression to block BMP-2-induced osteogenic differentiation. Elp2 activation leads to slower bone tissue formation in the inflammatory microenvironment.7
Surface plasmon resonance (SPR) is a label-free bioanalytical technique for interrogating biomolecular interactions at a metal/dielectric interface.8 It can be used to study and quantify molecular interactions such as protein–protein and protein–nucleic acid binding affinities, as well as other interactions.9–12 One challenge facing classical SPR analysis is large-scale screening of interaction between chemical molecules and protein. As an improvement, the SPRi image (SPRi) technique was developed with improvements in throughput and applied to the ligand–protein screening field.13–16 SPRi allows for the parallel evaluation of hundreds or thousands of compounds simultaneously.17 Moreover, it provides a rapid identification of biomolecular interactions along with their kinetic parameters in real time.18 A variety of small molecules have been reported using SPRi to measure protein–ligand interactions and protein–protein inhibition.19,20
Here, we report on our efforts to unite the compound library and SPRi technique to screen ligand–protein interactions, specifically to find new inhibitors of the Elp2 protein. After the screening stage, different verification strategies were applied to evaluate the pharmacodynamics of the candidate compounds in vivo. The result showed that ALP activity and mineralization of compound-treated cells were recovered during suppression of TNF-α. The mRNA content and expression levels of osteogenic differentiation marker genes were distinctly up-regulated compared to the TNF-α treated group. The result confirmed the antagonistic role of the candidates in TNF-α-induced osteoblast differentiation inhibition and revealed a possibility for further optimization and developed into a potent clinical inhibitor of Elp2.
Results
Chip preparation and SPRi affinity analysis
The two compound libraries, FD and BC, were prepared and printed on photocross-linker biosensor chips with a density of 1800 dot per chip (50 points per line, 36 lines). Thus, the FD and BC libraries were separately printed onto two and three chips; three duplicated arrays were printed as replications. With the help of photocross-linker biosensor chips and ultraviolet radiation, the molecules in each sample dot were immobilized on the chip surface. Optical inspection confirmed that the samples were fixed on the chip surface (data not shown), and the scalogram images of chip surface were captured by the bScreen LB 991 Label-free Microarray System (BERTHOLD TECHNOLOGIES, Germany) before affinity analysis in real time. During the test, five gradient concentrations (200 nM, 400 nM, 800 nM, 1600 nM and 3200 nM) of Elp2 protein diluted in PBS flowed through the surface. The programme was described in the methods section and the raw sensor gram was collected in real time. The binding of each compound to the Elp2 protein during each cycle was represented by the response unit (RU) of surface resonance. The raw data was calculated and 10−4 M was set as a cut-off for the equilibrium dissociation constant (KD). According to the affinity measurement, seven compounds in the two libraries showed obvious affinity for Elp2 protein. Among those, six compounds showed a strong affinity (KD < 10−6 M for Elp2 protein and one compound showed lower binding capacity with a high koff. A higher koff indicated a high dissociate rate and would presumably be difficult to utilize in vivo. The association rate constants (kon), dissociation rate constants (koff) and KD of positive samples and controls are shown in Table 1. Their binding curves during the test are shown in Fig. 1.| No. | Protein name | Compound name | CAS # | k on (1/Ms) | k off (1/s) | K D (M) |
|---|---|---|---|---|---|---|
| 1 | ELP2 | Epinephrine bitartrate | 51-42-3 | 7.83 × 102 | 5.29 × 10−4 | 6.76 × 10−7 |
| 2 | ELP2 | Isoprenaline hydrochloride | 51-30-9 | 9.87 × 102 | 4.54 × 10−3 | 4.60 × 10−6 |
| 3 | ELP2 | Gluconolactone | 90-80-2 | 2.64 × 103 | 8.19 × 10−2 | 3.10 × 10−5 |
| 4 | ELP2 | Temozolomide | 85622-93-1 | 1.82 × 102 | 4.62 × 10−4 | 2.53 × 10−6 |
| 5 | ELP2 | PLX-4720 | 918505-84-7 | 1.32 × 102 | 2.22 × 10−5 | 1.68 × 10−7 |
| 6 | ELP2 | RK-33 | 1070773-09-9 | 5.38 × 101 | 2.59 × 10−5 | 4.81 × 10−7 |
| 7 | ELP2 | 5-Hydroxymethyl tolterodine | 207679-81-0 | 1.13 × 104 | 1.66 × 10−2 | 1.47 × 10−6 |
| Blank | ELP2 | DMSO (solvent) | 67-68-5 | 1.71 × 100 | 7.40 × 10−1 | 4.32 × 10−1 |
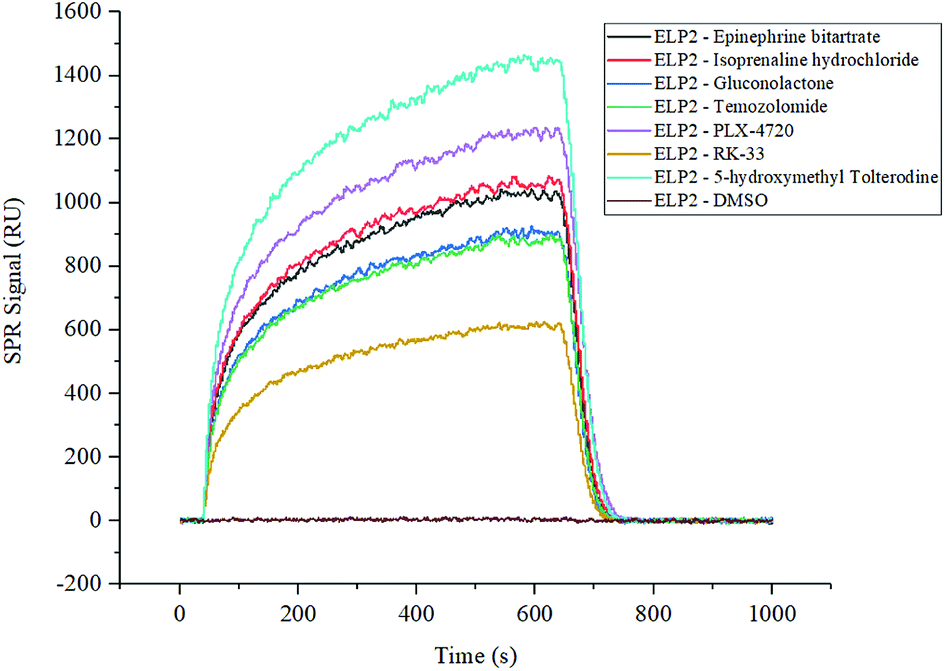 | ||
| Fig. 1 SPRi binding signal curve of positive compounds and controls to Elp2 protein. The DMSO solution of each compound from the FD and BC libraries was printed on the biosensor chips. The binding signals were filtered with a cut-off of KD < 10−3. The positive interaction and negative control are shown in this figure individually. | ||
Cell model establishment and culture
Regarding biological processes and drug function, an inflammatory differentiation model (ID model, C2C12 + BMP-2 model, C2C12 cells treated with 100 ng mL−1 BMP-2 to induce osteoblast differentiation) and a self-differentiation model (SD model, MC3T3-E1 model, MC3T3-E1 cells with no additive) were used to investigate the ability of candidate compounds to inhibit osteoblast differentiation. TNF-α (5 ng mL−1) was present as the antagonist of cell osteogenic differentiation. The ID model and SD model cells were cultured in 24-well plates and divided into four groups as follows: positive control (treated with no external conditions inhibiting differentiation), negative control (treated with TNF-α to ease differentiation), test group 1 (treated with TNF-α to ease differentiation and candidate to suppress the effect of TNF-α), test group 2 (treated with candidate only without TNF-α).Alkaline phosphatase stain and activity measurement
Cells in each group were cultured for 7 days in a suitable environment and alkaline phosphatase staining was performed using the Alkaline Phosphatase Assay Kit (Beyotime Biotechnology, China) according to the manufacturer's instruction. The stain observation field and relative ALP activity were compared. The results for the positive control in the ID and SD models demonstrated strong ALP activity, indicating cell differentiation. The negative controls with the addition of TNF-α remarkably reduced their ALP activity and were light in colour during ALP staining. Compound PLX-4720 showed similar ALP activity to the positive control. The effects of other candidate compounds were not as strong as PLX-4720 and only slightly eased TNF-α inhibition (data not shown). ALP staining and activity are shown in Fig. 2.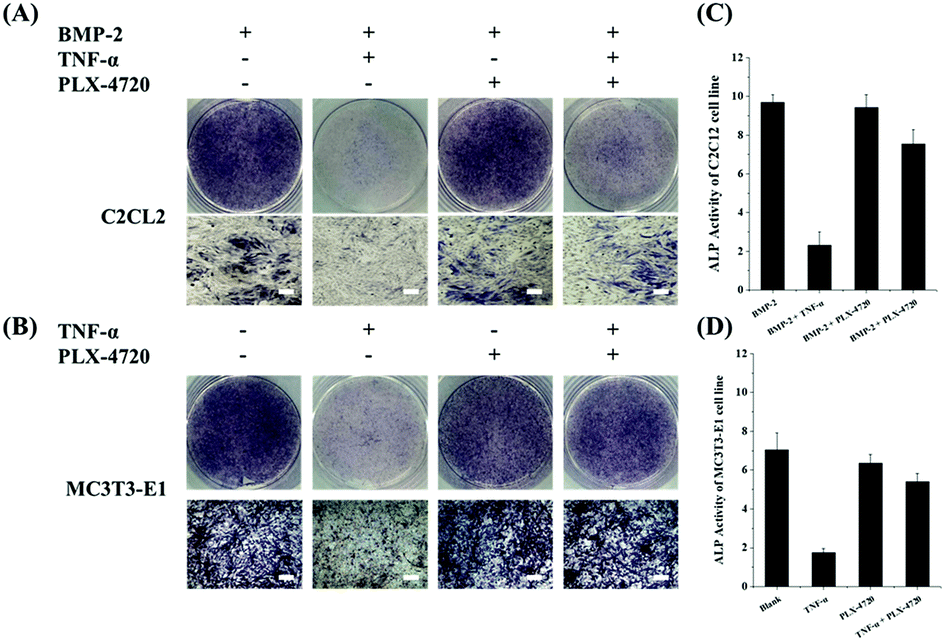 | ||
| Fig. 2 The ALP stain and activity measurements on C2C12 and MC3T3-E1 cells lines. AB shows ALP staining of C2C12 (A) and MC3T3-E1 (B) cells on day 10 with combination treatments of BMP-2/TNF-α/50 μM and PLX-4720 (bar = 50 μm). The ALP activity showed a positive correlation with the area of the dyed region (blue). The ALP activity of the C2C12 (C) and MC3T3-E1 (D) groups were measured and compared; an asterisk indicates significant differences with p < 0.05. With the presence of 2# or 5# compounds, the TNF-α suppression of ALP activity was partially reversed. Compounds 2# and 5# did not suppress TNF-α activity. | ||
Determination of effective concentration
The effective concentration (EC50) was calculated based on ALP activity. The gradient of compound concentrations ranged from 10 nM to 800 μM in the presence of osteoblast differentiation medium (with TNF-α). The EC50 of PLX-4720 was 10.35 μM in C2C12 cells in the ID model, while the EC50 in MC3T3-E1 cells in the SD model was 16.73 μM. The dose–effect curve is shown in Fig. 3.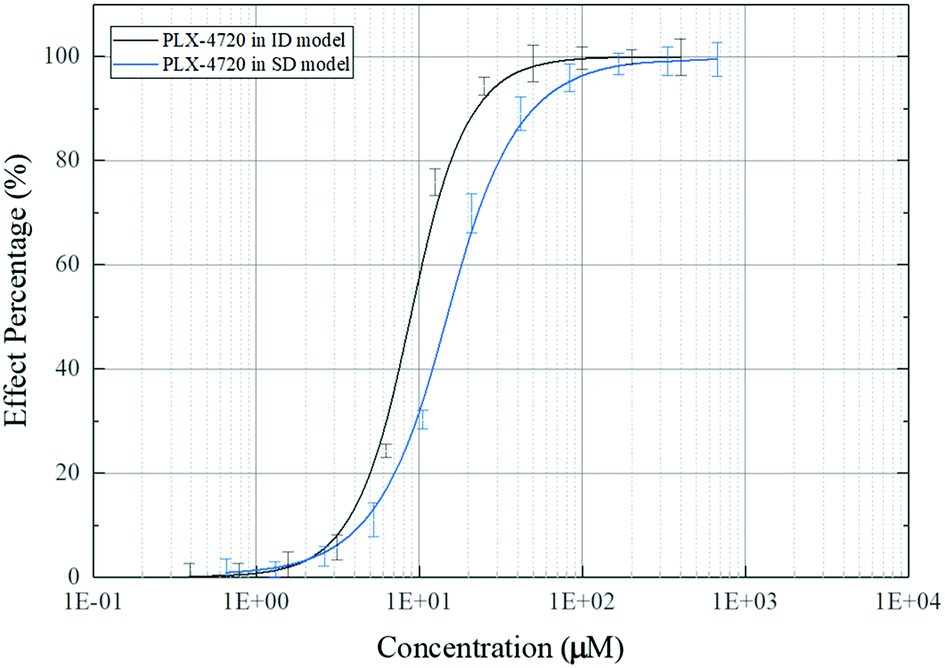 | ||
| Fig. 3 Dose–effect curve of PLX-4720 based on ALP activity measurement. | ||
Mineralization assessment
To investigate the osteoblasts mineralization activity derived by different candidate, the SD and ID model cells were stained with Alizarin Red S at day 35 of culture. The number of stained spots was used to estimate the mineralization activity for each group. As shown in Fig. 4, extracellular mineralization was visualized by Alizarin Red S staining after osteogenic induction. The positive control showed a high quantity of spots, indicating calcium deposits caused by osteogenic differentiation. The negative control showed fewer spots due to inhibition of TNF-α. The PLX-4720 groups showed more stained spots compared with the negative control (p < 0.05). These results suggested that the cell mineralization activity changes with the addition of PLX-4720, indicating that the osteoblast differentiation altered by TNF-α was reversed.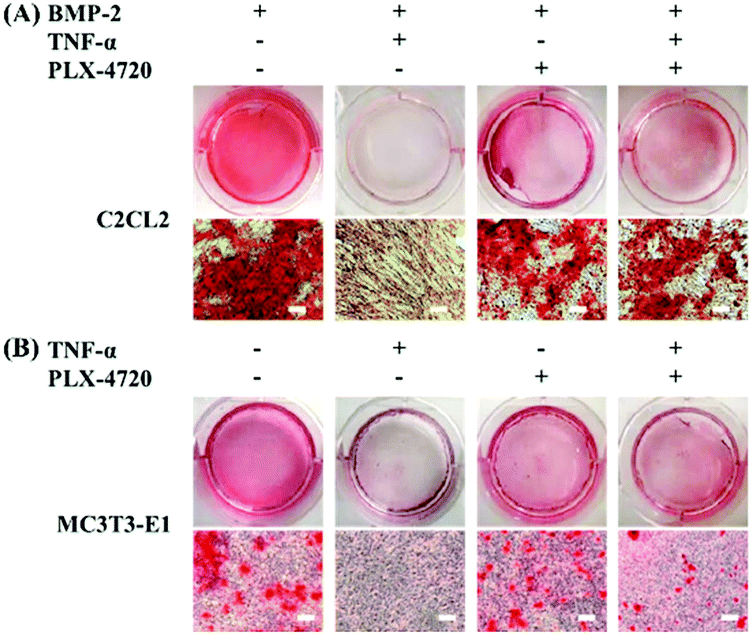 | ||
| Fig. 4 Alizarin Red S staining of differentiated osteogenic cells to determine mineralization. The intensity of mineralization in C2C12 (A) and MC3T3-E1 (B) cells with Alizarin Red S staining on day 35 (bar = 50 μm). The mineralization intensity showed a positive. | ||
Expression levels of osteoblast differentiation related genes
Further Q-PCR and western blot assays were performed in triplicate on the ID and SD model cell samples from day 7 to distinguish the altered expression of the osteogenic differentiation marker genes including COL-I, ALP, OCN, BMP-2, RUNX-2 and OSX. Comparisons of the ΔΔCt showed a significant increase in expression on day 7 in the positive control group versus the negative control group. The expression levels of COL-I, ALP, OCN, BMP-2, RUNX-2 and OSX were elevated in the PLX-4720 group with a cellular morphology indicating differentiation. This result indicated that the mRNA levels of osteogenic differentiation marker genes increased with ALP activity and mineralization in the positive control group and PLX-4720 group and remained steady in the negative control group (Fig. 5). This indicated the cells treated with PLX-4720 and TNF-α demonstrated stronger differentiation than the TNF-α only treatment group (negative control). Further investigation of the protein-level expression of osteoblast markers was performed by western blot analysis. The relative protein content is shown in Fig. 6 and was consistent with the Q-PCR results. Therefore, combined with the cell staining and ALP activity analysis, the Q-PCR and WB results suggest that the Elp2 inhibitor candidate PLX-4720 ameliorated TNF-α-induced osteoblast differentiation inhibition.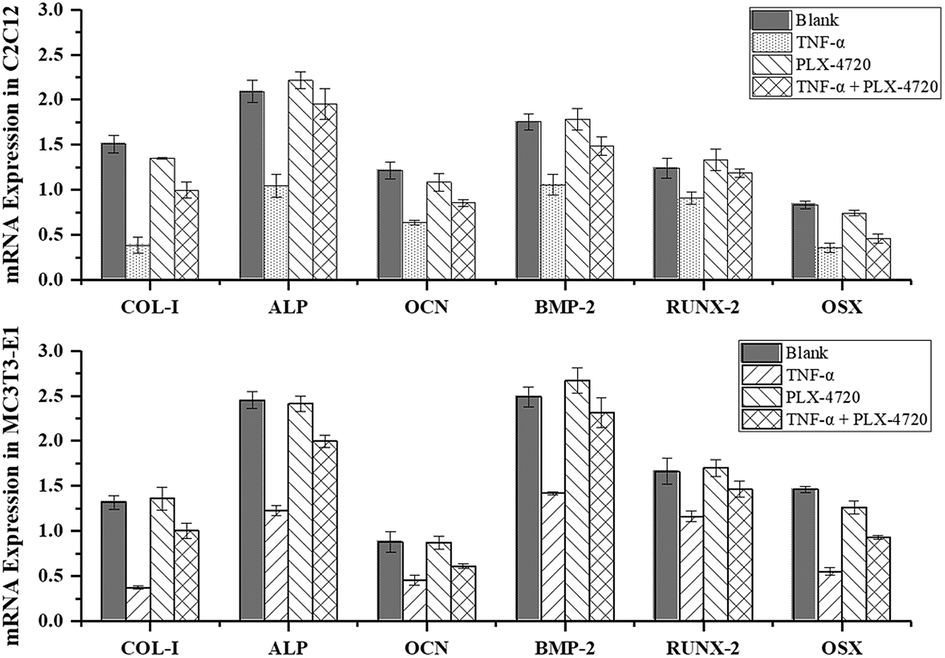 | ||
| Fig. 5 Validation of differentially expressed marker genes by real-time PCR. The mRNA expression levels of COL-I, ALP, OCN, BMP-2, RUNX-2 were measured with RT-Q-PCR. Under most circumstances, TNF-α down-regulated the mRNA levels of the six genes and it returned to a relatively higher level (p < 0.05) with the presence of PLX-4720. | ||
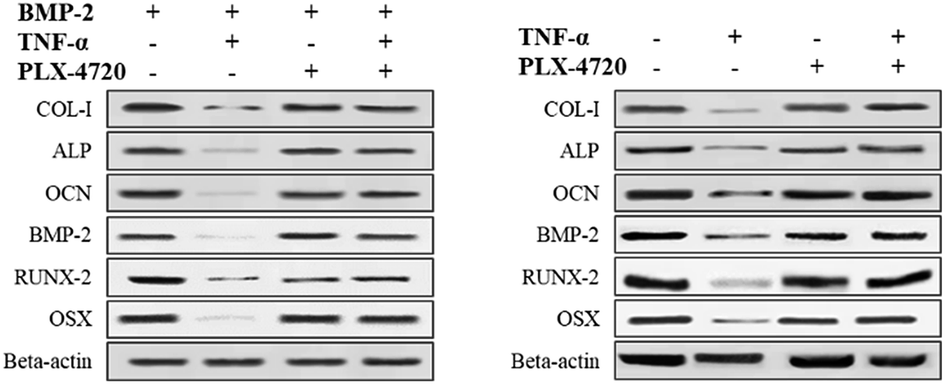 | ||
| Fig. 6 Western blot verification of marker gene expression. The protein expression levels of COL-I, ALP, OCN, BMP-2, RUNX-2 were measured with a western blot. The results showed the same trend as the mRNA levels. TNF-α down-regulated the protein expression of the six genes and it returned to a relatively high level (p < 0.05) with the presence of PLX-4720. | ||
Discussion
The elongator complex has been reported to have multiple functions in different organisms. Several loss of function studies have demonstrated the critical role of this complex in cell development by regulating various cellular activities. For example, yeast elongator complex mutants were allergic to hyperthermia and osmotic conditions and exhibit exocytosis, telomere gene silencing, DNA damage responses and defects in adapting to new growth media.21 In Arabidopsis, mutations in the elongator complex result in impaired root growth,22 and deletion of Elp3 in Drosophila melanogaster lead to death at the larval stage.23 In mice, knockdown of the transgenic Elp2 mRNA resulted in embryonic lethality due to failed neuromodulation and vascular system formation.24 In humans, some neurological diseases are related to a defect in the elongator complex.25Bone has a highly structured matrix and is still a very dynamic tissue constantly being renewed through cycles of resorption and formation. A previous study on Elp2 function7 reported that over-expression of Elp2 lead to an up-regulation of TNF-α-induced inhibition of osteoblast differentiation. Since inhibition of osteoblast differentiation impacts bone tissue regeneration and new bone tissue formation in clinical settings, Elp2 controls bone tissue dysplasia caused by excessive TNF-α due to long-term infection.
The commercial FD library is a unique collection of 1591 FDA approved drugs for high-throughput screening (HTS) and high content screening (HCS). The bioactivity and safety of each compound were confirmed by clinical trials in past reports. The BC library is a collection of 3051 bioactive chemical compounds that have been used in many research preclinical trials, which can also easily trace back to the progress of mechanistic research. These two compound libraries can provide suitable objects to be investigated for new inhibitors.26–33 Thus, we focused on these two commercial compound libraries and performed high-throughput screening to seek new Elp2 inhibitors.
The binding affinity between the Elp2 protein and compounds was determined by the SPRi technique; 7 compounds in the libraries showed fair bind affinity to the mouse Elp2 protein. Six candidate compounds were tested in vivo to determine any potential drug effects. The candidates were tested in ID and SD model cell lines and verified by ALP activity measurement and alizarin red-S staining. Further Q-PCR and western blot analyses on osteogenic differentiation genes were performed and the results complied with cell phenotypes. Among the candidates, PLX-4720 demonstrated its inhibitory effect on TNF-α.
The candidate PLX-4720 is a 7-azaindole derivative and a potential inhibitor of B-RafV600E in precious reports. It significantly inhibits the ERK phosphorylation in melanoma cells bearing B-RafV600E but has no toxic effect in wild-type B-Raf cells.34 The specific inhibitory effect results in a cell cycle arrest and apoptosis exclusively in tumor cells carrying B-RafV600E oncogenes (such as COLO205, A375, WM2664, and COLO829), which enables advancement to human clinical treatments.35 Some previous studies have shown that PLX-4720 may have clinical benefits in anti-tumor therapy. The study of Prahallad A., et al.36 shows that PLX-4720 can also act on colon cancer cells and inhibit the rapid response of B-RafV600E to EGFR activation with no evidence of toxicity. Tsai J., et al.37 reported PLX-4720 inhibited the active B-RafV600E kinase with marked selectivity in both biochemical and cellular assays. Through orally dosed PLX-4720 in B-RafV600E dependented tumor xenograft models, it caused tumor growth delays without evidence of cytotoxicity. Also, the GI50 of PLX-4720 in the common melanoma cells bearing wild-type B-Raf genes were between 13 nM and 41 nM. Compared to our result, we believe it is necessary to study if PLX-4720 will impose an adverse influence on normal body cell. Then we further performed a set of ERK phosphorylation tests and cell proliferation tests to determine if PLX-4720 inhibit the growth of normal body cells without a B-RafV600E mutation (such as MC3T3-E1 cell and C2C12 cell). The method was according to Tsai J.'s.37 The results of the ERK phosphorylation test suggested that even a very high concentration (100 μM) of PLX-4720 did not cause a significant down-regulating effect on the ERK phosphorylation level of MC3T3-E1 cell and C2C12 cell lines (Fig. S1A†). The results of cell proliferation assay showed that the growth of MC3T3-E1 cell was inhibited at 100 μM while the growth of C2C12 cells showed insignificant inhibition. It is speculated that the reason for the phenotypic difference may be the pluripotent stem cell characteristics of MC3T3-E1 cells (Fig. S1B†). Further study will pay more attention to the dosage regimen and the side effect of PLX-4720 on stem cells to pave the way becoming a clinical drug.
SPR-based biosensors have been developed for biomolecular interaction and various SPR and chemical surface modification based biosensors have been developed for assaying.38–50 The technique for immobilizing compounds in this study is photocross-linker chemical modification. With this technique, a plurality of random- orientated sub-structure of compound was formed in one sample dot due to each carbon–hydrogen bond in immobilized molecule was equivalent in free radical substitution. It is possible that a few of these sub-structures was endowed with unwanted binding activities to Elp2 and consequently cause false positive in SPR screening. So, we went through a set of in vivo analysis in the downstream workflow to verify the results of SPR step and ensure the reliability of drug candidates.
In this study, osteoblast differentiation indicators were evaluated by ALP activity, mineralization and osteogenic differentiation marker genes. Tissue-nonspecific alkaline phosphatase (ALP) is associated with mineral tissue forming cells as well as stem cells.51 Differentiated osteoblasts express matrix proteins (e.g., α-1-collagen) and alkaline phosphatase (ALP), a protein associated with mineralization, which also functions as a biochemical marker for osteoblastic differentiation and bone-forming capacity.52 The five osteoblast marker genes, COL-I, ALP, OCN, BMP-2, RUNX-2 and OSX, were reported early and used for osteogenic differentiation in several other studies.53–56 Therefore, these models were used to study the mechanism of inhibition of osteoblast differentiation in this manuscript.
According to a previous study, Elp2 regulates the JAK-STAT cascade and affects the inflammatory response through this downstream pathway. These analyses suggest that Elp2 exerts regulatory effects on osteoblast differentiation in response to TNF-α. The C2C12 cells can switch from their normal myotube formation pathway to the osteoblast differentiation pathway after a BMP-2 trigger.57 According to the aforementioned results, to study the inhibition of candidate compounds on Elp2 function, inflammatory differentiation model cell lines received 100 ng mL−1 BMP-2 in the culture medium of the C2C12 cell line to trigger differentiation. On the other hand, the preosteoblast MC3T3-E1 cells were regarded as a self-differentiation model from pluripotent stem cells to osteocyte without trigger molecules.
Growing evidence suggests that the inflammatory environment adversely affects the recovery of bone tissue.58–61 Among the inflammatory factors, TNF-α is a pleiotropic cytokine that becomes elevated during chronic inflammation.62 Persistent inflammation and infection have obvious effects on bone regeneration, and possible strategies for circumventing inflammatory inhibition of tissue regeneration can be pursued once the signalling pathways involved have been elucidated.63 There is an urgent demand in clinical orthopaedics to find auxiliary drugs to ease the disadvantage of long-term TNF-α excess.
In summary, we have screened one competitive inhibitor PLX-4720 against mouse Elp2 protein. The in vivo verification suggested the potential use of this compound to suppress TNF-α-induced osteogenic differentiation in C2C12 and MC3T3-E1 cells. The EC50 of PLX-4720 is 10.35 μM in C2C12 cells in the ID model, while the EC50 in MC3T3-E1 cells in the SD model is 16.73 μM. The results of the ALP staining, ALP activity, Alizarin Red S staining, and differentiation-related marker gene expression analysis met the initial expectations. PLX-4720 is a potential new drug in clinical orthopaedics. However, in-depth studies need to be performed to investigate the complete drug effect mechanism, metabolic processes and possible side effects of the candidates. Moreover, this study was performed with mouse source protein to improve the convenience of animal experiments at the early stage and reduce the costs of model animals. However, PLX-4720 may have a different mechanism of action between mouse and human cells. This may be why the other 6 positive compounds from SPR screening showed no efficacy during in vivo verification. This compound may also lose treatment efficacy during clinical testing. To mitigate this risk, cell experiments were performed using human cells. In the future, further study will illuminate additional pharmacodynamics and metabolic dynamics of PLX-4720 to develop it into a clinical drug preventing the inhibition of bone differentiation due to long-term inflammation.
Experimental methods
Compound library preparation and array printing
The FDA-approved drug (FD) library and bioactive compound (BC) library were purchased from Selleck Chemicals LLC (USA) and contained 1591 compounds (Cat#. L1300, Bat#. C1591) and 3051 compounds (Cat#. L1700, Bat#. C3051), respectively. The compounds were dissolved in DMSO or ddH2O according to their solubility and stored in a dark −80 °C refrigerator. Before printing, the compounds were restored to room temperature and transiently spun with a horizontal centrifuge to concentrate the solution. Concentrations of each compound were adjusted to 100 μM using DMSO or ddH2O according to the product specification and then aliquoted at 20 μL into 384-well plates. Microarrays chips used in this experiment were 25 × 75 × 1.1 mm glass slides coated with 47.5 nM thick gold layer and photocross-linker chemical modification (Betterways Inc., China). This sensor chip is capable of immobilizing small chemical molecules (MW > 60 Da) with no chemical label linking. The pre-treatment procedure of the microarrays chips was performed according to the standard operating procedure provided by the manufacturer. During the immobilization process, the C–H bonds were attacked by azo radicals excited by ultraviolet light and connected to the chip surface through a hydrogen substitution reaction. This improvement enhanced the molecules' spatial freedom and prevented inactivation by the labelling step.For spotting, a BioDot™ AD-1520 Array Printer (BIODOT Inc., USA) printed samples and controls on the chip surface. The instrument was maintained and operated in accordance with official instructions and the pins for printing were originally supplied with the instruments. According to the printing programme, each compound solution was printed with a volume of 1.875 nL per dot on four parallel chips with a formation of a 40 × 32 array (1280 dots per chip). Twelve positive control dots (6 rapamycin dots and 6 biotin dots) and 12 negative control dots (6 DMSO dots and 6 ddH2O dots) were printed around the sample printing area. Since one chip cannot contain more than 1280 samples, the FD library was separated into two different chips and the BC library was separated in three chips. Subsequently, the solvent in the sample dots was evaporated in a dark N2 atmosphere, and the sensor chips were quickly transferred to a UV spectroirradiator for a photocross-linking reaction. A UV Spectroirradiator 1020 (Amersham Life Science, USA) was used for the photocross-linking reaction quickly after the array print. The irradiation programme was 100 μW cm−2, 2 min; pause 2 min; 100 μW cm−2, 2 min pause 2 min; 25 μW cm−2, 15 min. Two more sensor chips were prepared as replicates. One of the four parallel chips was stored in a −80 °C refrigerator as a backup for troubleshooting and the other three chips underwent SPRi analysis. Then, the flow cell chambers were assembled, and the sensor chips were inserted and tested in the SPRi instrument.
SPRi interaction and affinity analysis
To understand the specific interactions between candidate compounds and Elp2 protein, we performed an affinity measurement using surface plasmon resonance (SPRi) technology. The SPRi validation experiment was performed with the bScreen LB 991 Label-free Microarray System (Berthold Technologies, Germany). To validate detection of the chemical compound–protein interactions using the photo-cross-linker SPRi sensor chip, we selected rapamycin and biotin as system controls and arranged kinetic constant tests with FKBP12 and streptavidin immediately after the sample tests. Additionally, the solvents for chemical compounds (DMSO, ddH2O) were also tested during the Elp2 flow. At the end of the affinity test, proteins solutions (PBS, pH = 7.0) were crosswise tested with all compounds on the chip as the background noise control.During the SPRi test, the surface was first primed three times with HBS-EP running buffer (containing 10 mM HEPES, pH 7.0, 150 mM NaCl, 3 mM EDTA and 0.005% (v/v) of P20 surfactant) at rate of 2 μL s−1 for 40 s and one time with running buffer (1 × PBS with 5% DMSO) at rate of 2 μL s−1 for 40 s. The mouse Elp2 protein (Antibodies online, Germany) was diluted separately with running buffer to 200 nM, 400 nM, 800 nM, 1600 nM and 3200 nM and injected for 600 s at a flow rate of 0.5 μL s−1 at associating stage, followed by running buffer for 360 s at a flow rate of 0.5 μL s−1 at each dissociating stage. At the end of each associating–dissociating cycle, the surface was regenerated to remove any remaining bound material with a pulse of 10 mM glycine–HCl (pH 2.5) at 20 μL min−1 for 30 s.
The raw sensor grams and measurements of the binding process of ligands and proteins were recorded in real time. The response unit (RU) of surface resonance was compared to determine the different binding affinities between each sample dot. The response unit data collected on the SPRi biosensor was further processed to eliminate any artefacts such as non-specific binding and differences in buffer composition. The process and analysis of association and dissociation rate constants (ka/kon and kd/koff respectively) and the equilibrium dissociation constant (KD, kd/ka) were performed using the data analysis software of the bScreen LB 991 unlabelled microarray system according to a single-site binding model (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Langmuir binding) with mass transfer limitations for binding kinetics determination.
:1 Langmuir binding) with mass transfer limitations for binding kinetics determination.
Cell culture and experimental group
C2C12 and MC3T3-E1 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, HyClone, GE Healthcare, USA) and supplemented with 10% foetal bovine serum (FBS; HyClone, GE Healthcare, USA), as well as 1% of penicillin/streptomycin (Thermo Scientific, USA). All cell lines were cultured at 37 °C in 5% CO2 and the culture media was replaced every 2 days. One day before tests, cells in the exponential growth phase were plated in 24-well plates at a density of 50% confluence, followed by incubation for 24 h, then grouped and treated separately. For the inflammatory differentiation model (ID model, C2C12 + BMP-2 model, and C2C12 cells treated with 100 ng mL−1 BMP-2 to induce osteoblast differentiation), four groups were treated on day 0 as follows: (ID-a) with 100 ng mL−1 BMP-2; (ID-b) with 100 ng mL−1 BMP-2 and 5 ng mL−1 TNF-α; (ID-c) with 100 ng mL−1 BMP-2 and a gradient concentration of candidate compounds; (ID-d) with 100 ng mL−1 BMP-2, 5 ng mL−1 TNF-α and a gradient concentration of candidate compounds. On the other hand, four groups were treated in the self-differentiation model (SD model, MC3T3-E1 model, and MC3T3-E1 cells with no additive) and treated on day 0 as follows: (SD-a) with equal volume solvent (ddH2O); (SD-b) with 5 ng mL−1 TNF-α; (SD-c) a gradient concentration of candidate compounds; (SD-d) with 5 ng mL−1 TNF-α and a gradient concentration of candidate compounds. The gradient concentrations used for the candidate compounds ranged from 10 nM to 800 μM (10 nM, 100 nM, 500 nM, 1 μM, 5 μM, 10 μM, 50 μM, 100 μM, 200 μM, 400 μM, 800 μM, eleven titters in total).Alkaline phosphatase staining and activity measurements
The osteogenic differentiation of each group was evaluated as a function of ALP activity on day 7. ALP activity was assessed using an Alkaline Phosphatase Assay Kit (Abcam Inc., Cambridge, UK) according to the manufacturer's protocol. Optical density was measured at 405 nm using an Epoch 2 Microplate Spectrophotometer (BioTek Instruments Inc., Virginia, USA). ALP activity values were normalized with respect to the total protein content obtained from the same cell lysate and expressed as units per microgram of total proteins. Total protein content was determined using the Pierce BCA Protein Assay Kit (Thermo Scientific, USA). The absorbance of the reaction product was measured at 562 nm. The protein concentration was calculated and compared between each group.Mineralization assessment
Alizarin (1,2-dihydroxyanthraquinone), which emits a red signal under fluorescent green light, has been used for labelling mineralization in osteocytes for many decades. In both cell modules and all experimental groups, the culture medium was changed every two days until day 35. Mineralization was visualized by staining with an Optimized Alizarin Red S Stain Kit (Solarbio Life Science, Beijing, China). The procedures were in accordance with the instructions.Expression levels of osteoblast differentiation related genes
After treatment and culturing for 7 days, 2 × 105 C2C12 and MC3T3-E1 cells were harvested separately. Total RNA was isolated from cultured cells using TRIzol reagent (Thermo Scientific, USA) and was purified using the RNeasy Mini Kit (Qiagen, Germany) and treated with RNase-Free DNase (Qiagen, Germany). RNA concentrations were measured using NanoDrop™ 8000 spectrophotometry (Thermo Scientific, USA). RNA integrity for all samples was confirmed with 1% agarose gels electrophoresis. For cDNA synthesis, 0.25 μg of cDNA was transcribed from total RNA using the High Capacity cDNA Reverse Transcription Kit (Thermo Scientific, USA). Gene expression profiles for collagen alpha-1 (COL-I), alkaline phosphatase (ALP), osteocalcin (OCN), bone morphogenetic protein 2 (BMP-2), runt-related transcription factor 2 (RUNX-2) and osterix (OSX) were determined by quantitative Q-PCR with the Thermal Cycler Dice™ Real Time System III (TaKaRa, Japan). Specific PCR primers were synthesized and optimized for amplification of each cDNA. Primers are listed in Table 2. Beta-actin (β-actin) was used as an internal reference for the experiment and gene expression analysis was normalized to β-actin, using delta–delta CT (ΔΔCt).64 Another set of harvested cells was processed for western blot analysis. The cell samples were washed with PBS (pH = 7.0) and lysed in RIPA Lysis Buffer (Beyotime Biotechnology, China). Then, the lysates were subjected to SDS-PAGE and immunoblotting analysis in triplicate. Protein concentrations of the cell lysates were determined using a Pierce BCA Protein Assay Kit (Thermo Scientific, USA).| Gene symbol | Primer sequence (5′–3′) | Product size (bp) | Accession no. |
|---|---|---|---|
| COL-I | F: 5′-GACCCTAACCAAGGATGCAA-3′ | 200 | NG_007404.1 |
| R: 5′-GGAAGTTCAGGATTGCCGTA-3′ | |||
| ALP | F: 5′-CCACGTCTTCACATTTGGTG-3′ | 196 | NG_008940.1 |
| R: 5′-AGACTGCGCCTGGTAGTTGT-3′ | |||
| OCN | F: 5′-GTGCAGAGTCCAGCAAAGGT-3′ | 202 | NG_047015.1 |
| R: 5′-CGATAGGCCTCCTGAAAGC-3′ | |||
| BMP-2 | F: 5′-TCAAGCCAAACACAAACAGC-3′ | 197 | NG_023233.1 |
| R: 5′-ACGTCTGAACAATGGCATGA-3′ | |||
| RUNX-2 | F: 5′-CTCTTCCCAAAGCCAGAGTG-3′ | 206 | NG_008020.1 |
| R: 5′-CAGCGTCAACACCATCATTC-3′ | |||
| OSX | F: 5′-TAATGGGCTCCTTTCACCTG-3′ | 198 | NG_023391.2 |
| R: 5′-GAGCCATAGGGGTGTGTCAT-3′ |
Protein samples (20 μg each) were mixed with denaturing buffer, heated to 98 °C for 10 min and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Total protein was transferred to a nitrocellulose membrane (Millipore, USA) using a semi-dry transfer apparatus (Bio-Rad, USA). The membranes were blocked with 5% skimmed milk in Tris-buffered saline (TBS)-0.1% Tween-20 (TBST) for 1 h and then incubated with the following corresponding primary antibodies: anti-COL-1 antibody (mouse monoclonal, ab90395, Abcam Inc., Cambridge, UK), anti-ALP antibody (rabbit monoclonal, ab133602, Abcam Inc., Cambridge, UK), anti-OCN antibody (mouse monoclonal, ab13420, Abcam Inc., Cambridge, UK), anti-BMP-2 antibody (mouse monoclonal, ab6285, Abcam Inc., Cambridge, UK), anti-RUNX2 antibody (mouse monoclonal, ab76956, Abcam Inc., Cambridge, UK), anti-Osterix (rabbit monoclonal, ab209484, Abcam Inc., Cambridge, UK), and anti-β-actin (mouse monoclonal, ab8226, Abcam Inc., Cambridge, UK). NC films were washed with TBS-0.1% Tween-20 (TBST) and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies as appropriate (rabbit anti-mouse IgG, ab6728, Abcam; goat anti-rabbit IgG, ab6721, Abcam) in a blocking buffer. Then, NC films were washed with TBST, incubated with a DAB Horseradish Peroxidase Color Development Kit (Beyotime Biotechnology, China) for 1 min and quickly removed the reaction solution. The NC film was washed with TBST twice according to the instructions. Images were captured with an AlphaImager HP imaging system (ProteinSimple LLC., USA). The relative quantification of each sample in the western blots was normalized to β-actin.
Statistical analysis
Multiple comparisons were made using a two-way ANOVA. Single comparisons were made using two-tailed Student's t tests (P < 0.05 was considered statistically significant). Analyses were performed using the SPSS 13.0 statistical package (SPSS Inc., Chicago, USA) and Origin 2017 (OriginLab Corporation, Massachusetts, USA). Data were presented as the mean ± SEM, performed in triplicate and repeated at least one time.Conclusions
We screened two chemical libraries with a high-throughput SPR technique and found one compound related to the signal transmission of TNF-α inhibition of osteogenic differentiation based on downstream factor Elp2. The compound reduced TNF-α-induced differentiation inhibition on the C2C12 and MC3T3-E1 cell lines. These results suggest PLX-4720 is a candidate for bone dysplasia treatment during long-term inflammation.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Fundation for Youths of China (No. 81501902), Guangdong Province Natural Science Foundation (No. 2014A030310451, 2017A030313736), Science and Technology Planning Project of Guangzhou (No. 201804010226), Science Foundation of Guangdong Second Provincial General Hospital (No. YQ2016-003), National Natural Science Fundation of China (No. 81560368), Science and Technology Assistance Project of Xinjiang Province (No. 2018E02056), Natural Science Foundation of Xinjiang Province (No. 2018D01C014).References
- K. Tony, T. Hasan, A. K. M. F. Mahmud, X. Fu, X. Hao and A. S. Byström, RNA Biol., 2014, 11, 1519–1528 CrossRef PubMed.
- S. Glatt, B. Seraphin and C. W. Muller, Transcription, 2012, 3, 273–276 CrossRef PubMed.
- M. I. Dauden, J. Kosinski, O. Kolaj-Robin, A. Desfosses, A. Ori, C. Faux, N.A. Hoffmann, O. F. Onuma, K. D. Breunig, M. Beck, C. Sachse, B. Séraphin, S. Glatt and C.W. Müller, EMBO Rep., 2017, 18, 264–279 CrossRef CAS PubMed.
- W. Yongsheng, A. Chuanfu, Z. Xudong, Y. Jiqiang, Z. Yanping, S. Yijun, Y. Fahong, A. David Moraga and M. Zhonglin, Plant Cell, 2013, 25, 762–776 CrossRef PubMed.
- M. Almeida, M. R. Laurent, V. Dubois, F. Claessens, C. A. O'Brien, R. Bouillon, D. Vanderschueren and S. C. Manolagas, Physiol. Rev., 2017, 97, 135–187 CrossRef PubMed.
- S. H. Mcbridegagyi, J. A. Mckenzie, E. G. Buettmann, M. J. Gardner and M. J. Silva, Bone, 2015, 81, 533–543 CrossRef CAS PubMed.
- C. P. Xu, X. Li, Y. J. Hu, Z. Cui, L. Wang, L. Liang, Y. L. Zhou, Y. J. Yang and B. Yu, J. Proteomics, 2015, 114, 234–246 CrossRef CAS PubMed.
- V. Kanda, J. K. Kariuki, D. J. Harrison and M. T. Mcdermott, Anal. Chem., 2004, 76, 7257–7262 CrossRef CAS.
- A. K. Anders, M. J. Call, M. S. E. D. Schulze, K. D. Fowler, D. A. Schubert, N. P. Seth, E. J. Sundberg and W. W. Kai, Nat. Immunol., 2011, 12, 54 CrossRef CAS.
- C. Boozer, G. Kim, S. Cong, H. Guan and T. Londergan, Curr. Opin. Biotechnol., 2006, 17, 400–405 CrossRef CAS PubMed.
- S. Deng, P. Wang and X. Yu, Sensors, 2017, 17, 2819 CrossRef.
- M. Ritzefeld and N. Sewald, J. Amino Acids, 2012, 2012, 816032 CrossRef.
- S. Li, M. Yang, W. Zhou, T. G. Johnston, R. Wang and J. Zhu, Appl. Surf. Sci., 2015, 355, 570–576 CrossRef CAS.
- J. Zhang, V. Singh, Z. Li, X. Zhou, X. Xu, J. Xu, A. Nand, H. Wen, H. Li and J. Zhu, RSC Adv., 2016, 6, 3213–3218 RSC.
- Z. Zhao, L. Zhu, X. Bu, H. Ma, S. Yang, Y. Yang and Z. Hu, Chem. Commun., 2014, 51, 718–721 RSC.
- Y. Jia, H. Tian, H. Li, Q. Yu, L. Wang, J. Friml and Z. Ding, J. Exp. Bot., 2015, 66, 4631–4642 CrossRef CAS.
- K. T. von der Trenck, R. Konietzka, A. Biegel-Engler, J. Brodsky, A. Hädicke, A. Quadflieg, R. Stockerl and T. Stahl, Environ. Sci. Eur., 2018, 30, 19 CrossRef.
- N. H. Hiep, P. Jeho, S. Kang and K. Moonil, Sensors, 2015, 15, 10481 CrossRef PubMed.
- M. C. D. Moraes, C. L. Cardoso, C. Seidl, R. Moaddel and Q. B. Cass, Curr. Pharm. Des., 2016, 22(39), 5976–5987 CrossRef PubMed.
- E. Helmerhorst, D. J. Chandler, M. Nussio and C. D. Mamotte, Clin. Biochem. Rev., 2012, 33, 161–173 Search PubMed.
- S. Gudipati, R. Muttineni, A. U. Mankad, H. A. Pandya and Y. T. Jasrai, Bioinformation, 2018, 14, 15–20 CrossRef.
- M. Kojic and B. Wainwright, Front. Mol. Neurosci., 2016, 9, 115 Search PubMed.
- J. Walker, S. Y. Kwon, P. Badenhorst, P. East, H. McNeill and J. Q. Svejstrup, Genetics, 2011, 187, 1067–1075 CrossRef CAS.
- Y. Jia, H. Tian, H. Li, Q. Yu, L. Wang, J. Friml and Z. Ding, J. Exp. Bot., 2015, 66, 4631–4642 CrossRef CAS.
- L. Nguyen, S. Humbert, F. Saudou and A. Chariot, Trends Mol. Med., 2010, 16, 1–6 CrossRef CAS.
- S. M. Corsello, J. A. Bittker, Z. Liu, J. Gould, P. Mccarren, J. E. Hirschman, S. E. Johnston, A. Vrcic, W. Bang and M. Khan, Nat. Med., 2017, 23, 405–408 CrossRef CAS.
- S. P. Mcelroy, P. S. Jones and D. V. Barrault, Drug Discovery Today, 2016, 22, 199–203 CrossRef.
- Y. M. Wang, J. W. Lu, C. C. Lin, Y. F. Chin, T. Y. Wu, L. I. Lin, Z. Z. Lai, S. C. Kuo and Y. J. Ho, Antiviral Res., 2016, 135, 81–90 CrossRef CAS.
- T. Hayashi, M. Jean, H. Huang, S. Simpson, N. G. Santoso and J. Zhu, Antiviral Res., 2017, 146, 76 CrossRef CAS.
- S. Martens, M. Jeong, W. Tonnus, F. Feldmann, S. Hofmans, V. Goossens, N. Takahashi, J. H. Bräsen, E. W. Lee and P. V. D. Veken, Cell Death Dis., 2017, 8, e2904 CrossRef CAS.
- H. Nishioka, N. Tooi, T. Isobe, N. Nakatsuji and K. Aiba, Sci. Rep., 2016, 6, 33427 CrossRef CAS.
- B. Preuett, J. S. Leeder and S. Abdel-Rahman, J. Biomol. Screening, 2015, 20, 1171–1177 CrossRef CAS.
- M. A. de Graaff, S. Malu, I. Guardiola, A. B. Kruisselbrink, J. Y. De, W. E. Corver, H. Gelderblom, P. Hwu, T. O. Nielsen and A. J. Lazar, Transl. Oncol., 2017, 10, 546–554 CrossRef.
- K. H. T. Paraiso, X. Yun, V. W. Rebecca, E. V. Abel, C. Y. Ann, M. A. Cecilia, W. Elizabeth, I. V. Fedorenko, V. K. Sondak and A. R. A. Anderson, Cancer Res., 2011, 71, 2750–2760 CrossRef CAS.
- N. Carmelo, P. Alessandro, A. Zeus Andrea, M. Michal, N. Matthew A, G. Thomas J, G. Damien, B. Laura E, P. Carmen and P. Efisio, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10649–10654 CrossRef.
- P. Anirudh, S. Chong, H. Sidong, D. N. Federica, S. Ramon, Z. Davide, B. L. Roderick, B. Alberto and B. René, Nature, 2012, 483, 100–103 CrossRef.
- J. Tsai, J. T. Lee, W. Wang, J. Zhang, H. Cho, S. Mamo, R. Bremer, S. Gillette, J. Kong, N. K. Haass, K. Sproesser, L. Li, K. S. Smalley, D. Fong, Y. L. Zhu, A. Marimuthu, H. Nguyen, B. Lam, J. Liu, I. Cheung, J. Rice, Y. Suzuki, C. Luu, C. Settachatgul, R. Shellooe, J. Cantwell, S. H. Kim, J. Schlessinger, K. Y. Zhang, B. L. West, B. Powell, G. Habets, C. Zhang, P. N. Ibrahim, P. Hirth, D. R. Artis, M. Herlyn and G. Bollag, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3041–3046 CrossRef CAS.
- C. V. Hebel, S. Rudolph, A. Mester, J. A. Huisman, P. Kumbhar, H. Vereecken and J. V. D. Kruk, Water Resour. Res., 2014, 50, 2732–2748 CrossRef.
- P. Michael, A. Nasser, J. H. M. Austin, R. B. Cameron, R. N. Cataneo, G. Joel, K. Robert, R. A. Maxfield, M. I. Munawar and H. I. Pass, Cancer Biomarkers, 2007, 3, 95 Search PubMed.
- G. Tricoles and N. H. Farhat, Proc. IEEE, 1977, 65, 108–121 Search PubMed.
- C. D. Natale, A. Macagnano, E. Martinelli, R. Paolesse, G. D'Arcangelo, C. Roscioni, A. Finazzi-Agrò and A. D'Amico, Biosens. Bioelectron., 2003, 18, 1209–1218 CrossRef.
- L. Ma, A. Hunt and M. Soleimani, Int. J. Multiphase Flow, 2015, 72, 198–209 CrossRef CAS.
- P. J. Mazzone, W. Xiao-Feng, X. Yaomin, M. Tarek, M. C. Beukemann, N. Jie, J. W. Kemling, K. S. Suslick and S. Madhu, J. Thorac. Oncol., 2012, 7, 137–142 CrossRef.
- M. Peter J, H. Jeffrey, D. Raed, N. Jie, C. Carmen, L. Daniel and M. Tarek, Thorax, 2007, 62, 565 CrossRef.
- T. Itoh, T. Miwa, A. Tsuruta, T. Akamatsu, N. Izu, W. Shin, J. Park, T. Hida, T. Eda and Y. Setoguchi, Sensors, 2016, 16, 1891 CrossRef.
- P. Michael, R. N. Cataneo, A. R. C. Cummin, A. J. Gagliardi, G. Kevin, G. Joel, R. A. Maxfield and W. N. Rom, Chest, 2003, 123, 2115–2123 CrossRef PubMed.
- P. Michael, A. Nasser, J. H. M. Austin, R. B. Cameron, R. N. Cataneo, K. Robert, R. A. Maxfield, M. I. Munawar, H. I. Pass and R. Asif, Clin. Chim. Acta, 2008, 393, 76–84 CrossRef.
- M. Han, X. Cheng and Y. Xue, Physiol. Meas., 2016, 37, 683–697 CrossRef PubMed.
- O. Barash, N. Peled, U. Tisch, P. A. B. Jr, F. R. Hirsch and H. Haick, Nanomedicine, 2012, 8, 580–589 CrossRef CAS.
- Z. Zakaria, R. A. Rahim, M. S. B. Mansor, S. Yaacob, N. M. N. Ayub, S. Z. M. Muji, M. H. F. Rahiman and S. M. K. S. Aman, Sensors, 2012, 12, 7126–7156 CrossRef.
- Z. Yu, P. Gauthier, Q. T. Tran, I. Elayachi, F. U. Bhatti, R. Bahabri, M. Alhabib and G. T. Huang, J. Stem Cell Res. Ther., 2015, 5(7), 292 Search PubMed.
- C. Koskinen, E. Persson, P. Baldock, Å. Stenberg, I. Boström, T. Matozaki, P. A. Oldenborg and P. Lundberg, J. Biol. Chem., 2013, 288, 29333–29344 CrossRef CAS.
- H. Wang, C. Li, J. Li, Y. Zhu, Y. Jia, Y. Zhang, X. Zhang, W. Li, L. Cui and W. Li, Iran. J. Basic Med. Sci., 2017, 20, 408–414 Search PubMed.
- X. Sun, W. Su, X. Ma, H. Zhang, Z. Sun and X. Li, Regener. Biomater., 2018, 5, 93–103 CrossRef CAS.
- M. H. Siddiqi, M. Z. Siddiqi, S. Ahn, S. Kang, Y. J. Kim, N. Sathishkumar, D. U. Yang and D. C. Yang, J. Ginseng Res., 2013, 37, 261–268 CrossRef CAS.
- I. Kanazawa, T. Yamaguchi, S. Yano, M. Yamauchi, M. Yamamoto and T. Sugimoto, BMC Cell Biol., 2007, 8, 1–12 CrossRef.
- O. Nelson, C. J. Carlos and B. Enrique, BMC Cell Biol., 2009, 10, 73 CrossRef.
- D. V. Novack and G. Mbalaviele, Microbiol. Spectrum, 2017, 18(1), 112 Search PubMed.
- A. Robaszkiewicz, C. Qu, E. Wisnik, T. Ploszaj, A. Mirsaidi, F. A. Kunze, P. J. Richards, P. Cinelli, G. Mbalaviele and M. O. Hottiger, Sci. Rep., 2016, 6, 21131 CrossRef CAS.
- K. Ali, L. Catharina and U. H. Lerner, PLoS One, 2016, 11, e0156708 CrossRef.
- C. Gao, S. Peng, P. Feng and C. Shuai, Bone Res., 2017, 5, 17059 CrossRef CAS.
- V. D. Ramseyer and J. L. Garvin, Am. J. Physiol. Renal. Physiol., 2013, 304, F1231 CrossRef CAS.
- E. Tsialogiannis, I. Polyzois, T. Q. Oak, G. Pavlou, E. Tsiridis, M. Heliotis and E. Tsiridis, Expert Opin. Ther. Targets, 2009, 13, 123–137 CrossRef CAS.
- J. S. Yuan, A. Reed, F. Chen and C. N. Stewart, BMC Bioinf., 2006, 7, 85 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09640f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |