
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA proof of concept application of aptachain: ligand-induced self-assembly of a DNA aptamer†
Miguel A. D. Neves‡
 *,
Sladjana Slavkovic,
Oren Reinstein,
Aron A. Shoara and
Philip E. Johnson*
*,
Sladjana Slavkovic,
Oren Reinstein,
Aron A. Shoara and
Philip E. Johnson*
Department of Chemistry & Centre for Research on Biomolecular Interactions, York University, 4700 Keele St., Toronto, Ontario, Canada M3J 1P3. E-mail: pjohnson@yorku.ca; nevesm@smh.ca
First published on 14th January 2019
Abstract
A challenge for the use of aptamers as biosensors is how to signal the occurrence of their ligand binding event into a signal that can be exploited in a detection scheme. Here, we present the concept of “aptachain” formation, where an aptamer is split into two overlapping or staggered strands and assembles into an extended oligomer upon ligand binding. This assembly of aptamers can then be used as a way to detect ligand binding by the aptamer. As an example of this concept, we employed the cocaine-binding aptamer as a model system, used its ability to tightly bind quinine and demonstrated its capability in a gold nanoparticle-based biosensing application. We used isothermal titration calorimetry to demonstrate that, when split into two overlapping DNA strands, the aptamer remains functional. Size-exclusion chromatography showed that the quinine-bound oligos form a larger assembly of aptamer units than in the absence of ligand. Finally, we used the oligomer forming ability of the aptachain oligos in a biosensor application for quinine that brings gold nanoparticles closer together resulting in a shift in their plasmonic resonance to a longer wavelength and an observed colour shift. We propose that splitting aptamers into overlapping strands that form oligomers in the presence of a ligand, aptachain formation, will be generally applicable to aptamers and prove useful in a variety of biotechnology applications.
1. Introduction
DNA is a versatile material for building nanoscale structures since it can be used to construct large assemblies that form as a result of hybridization between strands.1–5 These assemblies can form a large array of different shapes and come together in a passive manner through base pairing interactions. An important class of functional nucleic acids are aptamers. Aptamers can be selected to bind a wide variety of ligands, often with high affinity and selectivity and are commonly employed in biosensor applications.6,7 We are interested in exploiting the ligand-binding ability of DNA aptamers to trigger the formation of DNA assemblies in a manner dependent on ligand binding.In order to demonstrate this concept of ligand-driven aptamer assembly, we employed the cocaine-binding aptamer.8 This aptamer is widely used in biosensor development and forms a three-way junction structure and has been shown to function when split into two or three separate strands.8–12 An unusual feature of the cocaine-binding aptamer is that it is able to bind quinine 50-fold tighter than cocaine13–19 and other quinine-based antimalarial compounds even tighter.20 In the work presented here, we split the cocaine-binding aptamer in such a way that the strands overlap and can elongate in a manner resembling an oligomer-forming chain (Fig. 1). Independent to this study, similar chain-forming split aptamers have been demonstrated with the theophylline-binding RNA aptamer21 and on two occasions using the ATP-binding DNA aptamer.22,23 Significantly, in this study we use size-exclusion chromatography to show that the aptamer chain formed (aptachain) is longer in the presence of ligand than in the absence of ligand and that aptachain formation can be employed in a biosensor application.
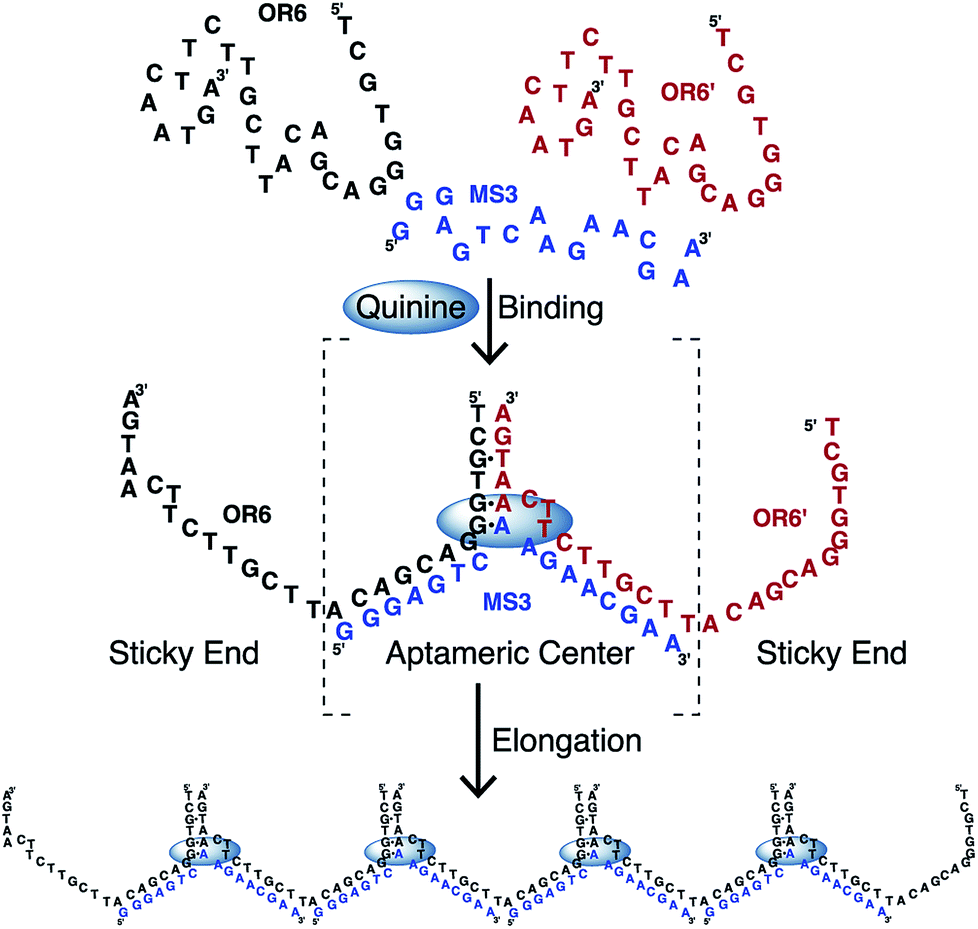 | ||
| Fig. 1 Schematic outlining the formation of aptachain. The parent cocaine-binding aptamer is split into two overlapping strands that associate in the presence of a ligand for the aptamer, such as quinine. | ||
2. Experimental
2.1 Materials
Unmodified DNA samples (MS3: 5′-GGG AGT CAA GAA CGA A-3′; OR6: 5′-TCG TGG GAC GAC ATT CGT TCT TCA ATG A-3′; AC3: 5′-GGG AGA CAA GGA AG-3′; AC4: 5′-TCG TGG GTC GAC ACT TCC TTC AAC GA-3′) were obtained from Integrated DNA Technologies (Coralville, Iowa, USA). 5′ thiol-modified OR6 (OR6–(CH2)6–S–S–(CH2)6–O–DMT) samples were obtained from the University of Calgary Core DNA Service (Calgary, Alberta, Canada). Prior to use, unmodified DNA samples were dissolved in distilled deionized water (ddH2O) with a measured resistivity ≥ 18.0 MΩ cm and then exchanged three times using a 3 kDa molecular weight cutoff concentrator with sterilized 1 M NaCl followed by at least three exchanges into ddH2O. Aptamer and ligand concentrations were determined by UV absorbance spectroscopy using the extinction coefficients supplied by the manufacturers. Unless otherwise stated, all other reagents were obtained from Sigma-Aldrich and used as received. Furthermore, all buffers were prepared using ultrapure ddH2O and the pH adjusted at room temperature.2.2 Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed using a MicroCal VP-ITC instrument in a manner similarly as described previously.24 Samples were degassed prior to use with a MicroCal ThermoVac unit. All experiments were acquired at 15 °C and corrected for the heat of dilution. Data were fit to a one-site binding model using Origin 7 software. Quinine binding experiments were performed with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of AC3:AC4 with the concentration of each strand at 20 μM using a ligand concentration of 0.312 mM in the needle. For the titration of AC3 into AC4, the AC3 was in the cell at 40 μM and the AC4 in the needle at 0.624 mM. All DNA samples were heated in a 65–70 °C water bath for 3 minutes and left to cool to room temperature preceding the ITC experiments to allow the aptamer to anneal. Binding experiments consisted of 35 successive 8 μL injections spaced every 300 s, where the first injection was 2 μL to account for diffusion from the syringe into the cell.
:1 molar ratio of AC3:AC4 with the concentration of each strand at 20 μM using a ligand concentration of 0.312 mM in the needle. For the titration of AC3 into AC4, the AC3 was in the cell at 40 μM and the AC4 in the needle at 0.624 mM. All DNA samples were heated in a 65–70 °C water bath for 3 minutes and left to cool to room temperature preceding the ITC experiments to allow the aptamer to anneal. Binding experiments consisted of 35 successive 8 μL injections spaced every 300 s, where the first injection was 2 μL to account for diffusion from the syringe into the cell.
2.3 Size-exclusion chromatography
Size-exclusion chromatography (SEC) experiments were performed using an ÄKTA FPLC system (GE Healthcare) with a HiPrep 16/60 Sephacryl S-100HR column (GE Healthcare). The column was equilibrated with 3 column volumes (CV) of running buffer (10 mM sodium phosphate, 100 mM NaCl, pH 7.4) prior to experiments. 100 μL of sample was injected onto the column and experiments were run at 0.5 mL min−1 and continued for 2 CV. Elution was monitored by the eluate UV absorbance at 254 nm. Experiments were recorded and analyzed using the provided Unicorn 4.11 software package (GE Healthcare).Aptamer samples were prepared by diluting the appropriate aptamer(s) in running buffer to a final concentration of 20 μM and immediately heated in a boiling water bath for 3 minutes and cooled in an ice bath to allow the aptamer to anneal. If quinine was to be added to the samples, quinine would be diluted in the sample to a final concentration of 0.1 mM prior to the annealing step.
2.4 Deprotection of 5′ thiol modified OR6
The day prior to aptameric probe immobilization, the thiol-protecting group (–S–(CH2)6–O–DMT) was cleaved from the 5′-thiol modification by incubating the purified aptamer samples in a dithiothreitol (DTT)-containing buffer (20 mM borate, 100 mM DTT, pH 8.5) for 1 h. Following deprotection, aptamer samples were dialyzed overnight into ddH2O using a 2 kDa cutoff Slide-A-Lyzer dialysis cassette (Thermo Scientific). Following dialysis the deprotected aptamer was used immediately.2.5 AuNP-OR6 preparation
AuNPs were functionalized with 5′ thiol modified OR6 DNA, freshly deprotected SH–OR6 was added to 15 nm gold nanoparticles (AuNPs), obtained from Sigma-Aldrich, to a final concentration of 20 μM and 0.5 OD per mL, respectively, in functionalization buffer (10 mM sodium phosphate, 0.01% SDS, pH 7.4). The oligonucleotide/AuNP solution was incubated at room temperature for 20 min. The mixture was then salt aged by increasing the concentration of NaCl to 0.05 M using 4 M NaCl in functionalization buffer. The oligonucleotide/AuNP solution was then incubated under agitation for 20 min. This process was repeated for another increment of 0.05 M NaCl and for every 0.1 M NaCl increment thereafter until a final NaCl concentration of 1.0 M was reached. The salting process was followed by incubation overnight at room temperature under agitation. To remove excess SH–OR6, the AuNP solution was centrifuged (10000 × g for 20 min) pelleting the AuNPs, the supernatant was removed and the pellet resuspended in functionalization buffer. The washing process was repeated 3 times.
To prevent OR6 AuNP surface interactions,25 the OR6 functionalized particles were reacted with mercaptohexanol (MCH), forming a MCH self-assembling monolayer between the immobilized oligo nucleotides. MCH and OR6-AuNPs were taken up in functionalization buffer to a final concentration of 100 μM and ∼0.5 OD per mL, respectively. The mixture was incubated at room temperature for 2 hours, then washed using centrifugation as described above. The OR6-AuNPs were taken up in AuNP buffer (10 mM sodium phosphate, 100 mM NaCl, 0.01% SDS, pH 7.4) to a final concentration of ∼1 × 1012 AuNP per mL.
2.6 UV-VIS AuNP-OR6 binding experiments
Experiments were carried out using a Cary Bio 100 UV-VIS spectrophotometer (Agilent Technologies) with a Peltier temperature controller and 12-cuvette sample holder. The data was recorded and analyzed using the provided WinUV 5.0 software package (Agilent Technologies). Experiments were performed by acquiring UV-VIS traces from 400–750 nm at set time intervals (15 min to 1 h) for a set time period (2–12 h) at 20 °C. Samples were prepared by diluting 100 μL of OR6-AuNP (∼1 × 1012 AuNP per mL) in a binding mixture containing 20 μM MS3, the appropriate concentration of quinine (0–250 μM) in binding buffer (10 mM sodium phosphate, 0.5 M NaCl, 0.01% SDS, pH 7.4) to a final volume of 500 μL in a UV disposable plastic cuvette (BrandTech Scientific). The concentration of OR6-AuNP used was chosen to be in the range of approximately 0.02–0.09 AU, well within the linear range for this instrument and an absorbance range often used in similar studies.26–28 The quinine and NaCl were added while the cuvettes were loaded in the sample holder so the experiment could be promptly started after the addition. Prior to the making of the samples OR6-AuNP and MS3 were mixed together in AuNP buffer and heated in a 65–70 °C water bath (a boiling water bath would strip the DNA from the surface of the AuNP) for 5 min and then allowed to cool to room temperature.2.7 AuNP-OR6 binding analysis
Absorbance ratios (A520 nm/A650 nm) after 2 h were plotted against quinine concentration and analyzed with the SigmaPlot software package (Systat Software). The Kd was determined by fitting the dose–response curve to a one-site saturation Langmuir ligand-binding model. The concentration limit of detection (CLoD) was determined from the standard deviation of the residuals obtained from the least-squares regression of the linear region (0–10 μM) of the dose–response curve.25,293. Results and discussion
3.1 Demonstration of ligand binding using isothermal titration calorimetry
In order to determine if the cocaine-binding aptamer is functional when split into the two overlapping strands, MS3 and OR6 (Fig. 1), we used ITC to monitor ligand binding. When the two strands OR6 and MS3 are mixed and annealed in a 1:1 molar ratio and then quinine is titrated into the DNA mixture, we observe ligand binding with a Kd value of (16 ± 2) μM (Fig. S2a†). With a small sequence optimization to make the overlapping oligo pair AC3 and AC4 (Fig. S1 in ESI†) the affinity tightened to (4.6 ± 0.4) μM (Fig. 2a). When compared with the intact MN4 aptamer, this value is approximately 23 fold weaker.14,15 A similar level of binding affinity weakening for split aptamers has been observed previously for the cocaine-binding aptamer when separated into 2 strands.8 Despite the reduction in affinity, the ITC data clearly confirms that the OR6/MS3 aptamer is functional in binding quinine and also demonstrates that it is possible to assemble three DNA strands into a binding-competent complex. We will note that at the NaCl concentration used in all the binding experiments in this study, only the high affinity site is populated.30
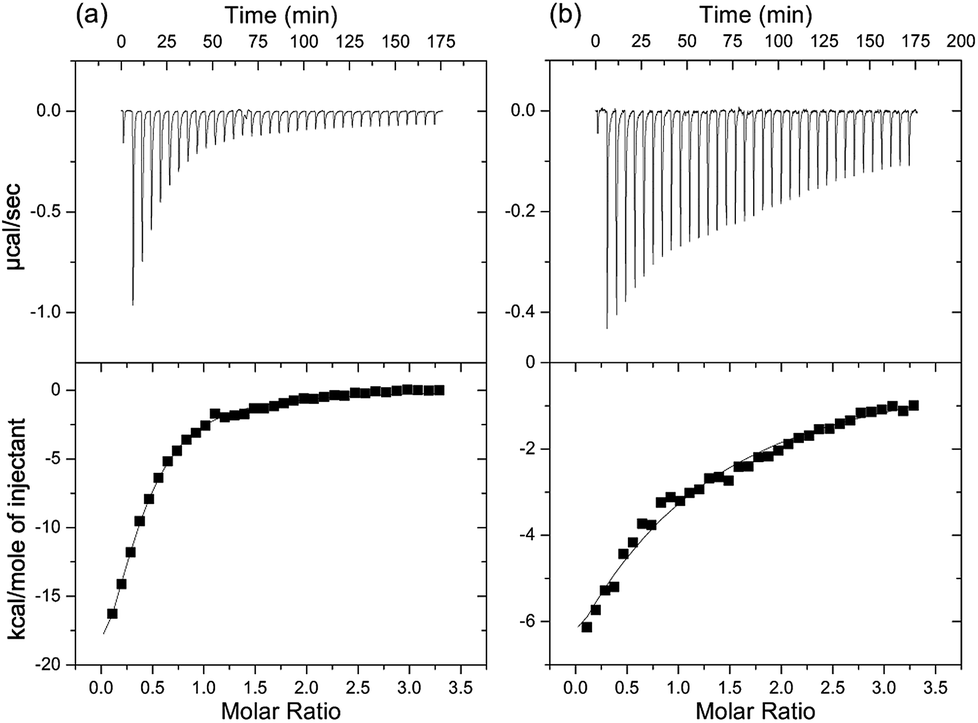 | ||
| Fig. 2 ITC data showing the interaction of (a) quinine with a 1:1 molar ratio mixture of the AC3 and AC4 DNA strands and (b) the interaction of the AC3 and AC4 strands. On top is the raw titration data showing the heat resulting from each injection of the titration. The bottom shows the integrated heat plot after correcting for the heat of dilution. These data provide a Kd value of (4.6 ± 0.4) μM for the AC3/AC4 combination binding quinine and a Kd value of (36 ± 7) μM for the two strands interacting. Data for both runs were acquired in 20 mM sodium phosphate (pH 7.4), 140 mM NaCl, 5 mM KCl at 15 °C. | ||
As the two DNA strands (OR6/MS3 or AC3/AC4) contain some complimentary regions, it is possible that these regions hybridize in the absence of ligand. To evaluate this, we first looked at strand association by ITC and titrated MS3 into OR6 (Fig. S2b†). We observed that the two strands do interact with a Kd value of (3.1 ± 0.2) μM. For the AC3/AC4 pair of strands, the affinity of one strand for the other is (36 ± 7) μM (Fig. 2b).
3.2 Oligomer formation shown by size-exclusion chromatography
We next employed size-exclusion chromatography (SEC) to investigate the size of the DNA complex (aptachain) formed in the presence and absence of the ligand quinine. The SEC chromatograms of both ligand-free and quinine-bound aptachain complexes are depicted in Fig. 3. Both bound and unbound curves contain two sharp peaks at retention times (tR) of 118 and 140 min that correspond to the migration of the OR6 and MS3 monomer oligonucleotides. Both chromatograms also contain a broad peak, corresponding to a DNA oligomeric complex, at a retention time (tR) of 62 min for the ligand-bound aptachain and 87 min in the absence of ligand. The ligand-bound aptachain complex begins to elute just before 30 min and the UV absorbance continues to increase until the tR is reached at which point the absorbance plateaus until ∼75 min, when the absorbance decreases markedly. For the ligand-free sample, the absorbance begins to increase at ∼45 min and increases until the tR of the unbound complex is reached at 87 min. We generated a SEC calibration curve (Fig. S3†) and extrapolated to estimate the molecular weight (MW) of the free and quinine-bound aptachain complexes. The quinine-bound tR of 62 min corresponds to ∼47 kDa (or 4 aptameric units), while the free quinine tR of 87 min corresponds to ∼23 kDa (or two aptameric units). The elution range of the quinine-bound aptachain (30–75 min) corresponds to a MW range of 117–32 kDa or nine to three aptameric units.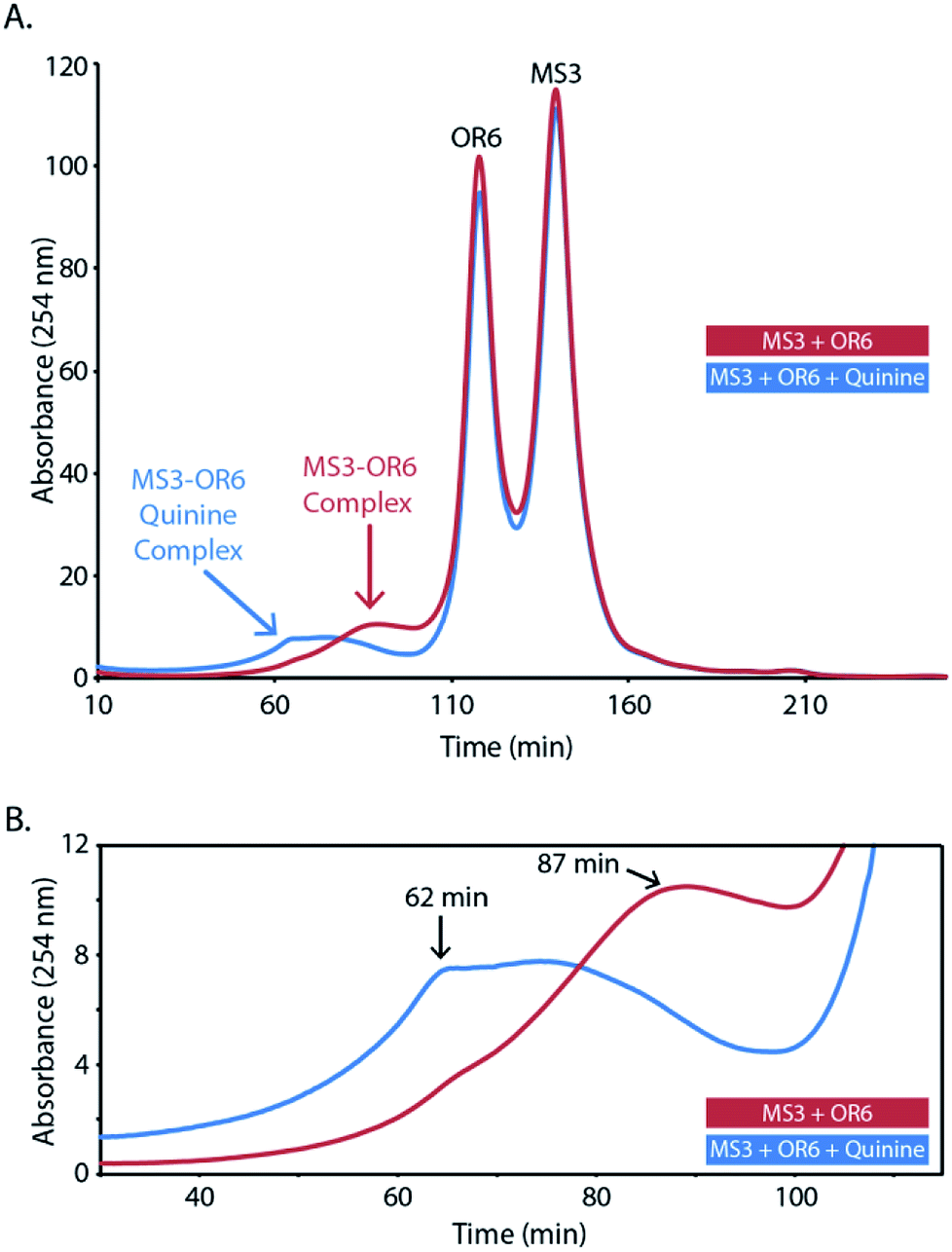 | ||
| Fig. 3 Size-exclusion chromatography analysis of the aptachain. (A) Overlaid chromatograms of the annealed mixture of MS3 + OR6 (red) and the aptachain (MS3 + OR6 + 100 μM quinine; blue). OR6 and MS3 monomer peaks are labelled and have retention times of 118 and 140 min, respectively. (B) Expanded region of the chromatogram between 30–110 min. | ||
The observed association of OR6 and MS3 by SEC in the absence of ligand agrees with the ITC data showing the two strands associating. In the presence of the quinine ligand, the peak due to aptachain formation elutes earlier than in the absence of ligand indicating that larger sized DNA complexes form in the presence of ligand. In both quinine-bound and unbound chromatograms, the sharp monomer peaks are higher in intensity than that of the DNA complexes. The relative abundances between aptachain complex and monomers in the SEC runs may not be representative of the solution abundance, as the extent of dissociation of the non-covalent aptachain complex caused by the FPLC and SEC column is not known.
3.3 Demonstration of aptachain formation in a biosensor application
To further confirm the formation of the aptachain complex and to demonstrate its usefulness in sensing applications, a 5′ thiol modified OR6 (5′–SH–(CH2)6–OR6) was immobilized onto the surface of 15 nm AuNPs. To prevent interactions between the oligonucleotide and the AuNP surface, mercaptohexanol was used to fill in the spaces between immobilized oligonucleotides.31,32 As multiple OR6 strands are immobilized on a single AuNP, upon ligand binding a cluster of branching linear aptachains from a single AuNP would presumably form (Fig. 4A). Binding assays were performed by adding quinine to AuNP-OR6 mixed with excess MS3 and binding was determined by monitoring the characteristic plasmonic absorbance of the AuNPs between 400 nm and 750 nm (Fig. 4B). In the absence of quinine, the maximal absorbance is observed at 522 nm. Although 15 nm AuNP should have a peak absorbance of 520 nm, this discrepancy is consistent with biomolecule conjugation to AuNPs.33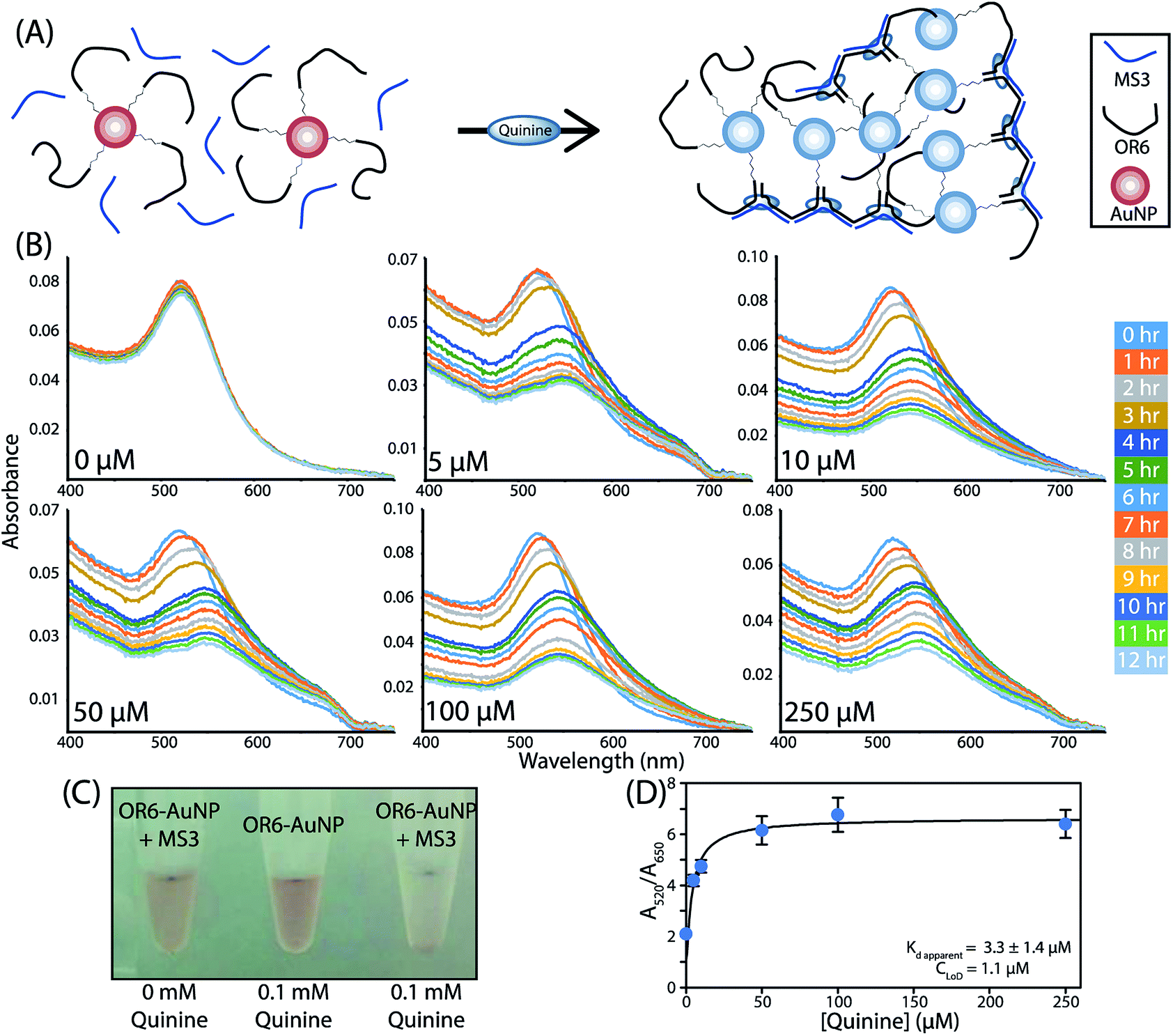 | ||
| Fig. 4 (A) Schematic depiction of the AuNP functionalized aptachain free and quinine-bound. (B) Absorbance traces of AuNP-functionalized aptachain assembly induced with varying concentrations of quinine. (C) The visible colour change induced with aptachain assembly bound AuNP functionalized aptachain compared to free AuNP aptachain and OR6-AuNP with quinine. (D) Aptachain dose–response curve after two-hour incubation with quinine concentrations ranging from 0–250 μM (n = 3). | ||
The ligand-induced aptachain assembly of AuNPs was observed when quinine was added to the AuNP-OR6 and MS3 mixture at different concentrations ranging from 0–250 μM and the absorbance measured for 12 hours (Fig. 4B). All of the quinine-containing samples experience a maximum wavelength shift that saturates at 550 nm after approximately 5 hours. This was coupled with a visible colour change in the solution (Fig. 4C). The peak wavelength of the quinine-free sample, 522 nm, did not change through the duration of the experiment, indicating that when coupled to AuNPs the complex does not form in the absence of quinine. As a control, after every 12 hour binding experiment, quinine was added to the quinine-free sample and the absorbance monitored in 15 min intervals. The characteristic redshift of complex formation was observed (Fig. S4†) confirming that when the AuNP is conjugated to OR6, the aptachain does not form in the absence of quinine, likely due to repulsive forces between adjacent AuNPs.34
This ligand-induced aptachain formation and the resulting plasmonic shift can be used to analytically determine the amount of ligand present. The greater the concentration of quinine added, the faster the peak wavelength saturates. The greatest absorbance difference between different concentrations of quinine is observed after 2 hours (Fig. S5†). A dose–response curve was assembled by plotting the A520/A650 nm absorbance ratio after 2 hours against the concentration of quinine (Fig. 4D). This aptachain colorimetric sensor was found to have an apparent Kd of (3 ± 1) μM and a concentration limit of detection (CLoD) of 1.1 μM for quinine. We expect that this value will improve with further optimization of the aptachain sequences and buffer conditions used.
4. Conclusions
In summary, we have used ITC to show that the aptachain-forming DNA sequences engineered from the cocaine-binding aptamer are functional and bind quinine, one of the targets of this aptamer. Additionally, we show using SEC that a larger DNA complex forms in the presence of quinine than in the absence of ligand. Finally, we demonstrate that the aptachain is a viable method for biosensing applications, such as the colorimetric AuNP sensor demonstrated here.We propose that this demonstrated aptachain formation could be extended to other aptamers that can be split into overlapping strands and retain activity. Indeed, similar aptamer-based linear chain formation has been reported for both the theophylline RNA aptamer21 and the DNA ATP-binding aptamer.22,23 We also propose that aptachain formation can be used for forming the ligand-induced assembly of shapes other than a chain, though this likely depends on the three-dimensional structure formed by the aptamer. The three-way junction secondary structure of the cocaine-binding aptamer should be useful in assembling a variety of shapes. Aptachain formation may also be useful in the design of nanoscale devices for cargo delivery, sensing and for forming DNA assemblies on surfaces.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC). We thank Edward Johnson for technical assistance and members of the Johnson lab for useful discussions.References
- L. A. Lanier and H. Bermudez, Curr. Opin. Chem. Eng., 2015, 7, 93–100 CrossRef PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- P. Chidchob and H. F. Sleiman, Curr. Opin. Chem. Biol., 2018, 46, 63–70 CrossRef CAS PubMed.
- S. G. Harroun, C. Prevost-Tremblay, D. Lauzon, A. Desrosiers, X. Wang, L. Pedro and A. Vallee-Belisle, Nanoscale, 2018, 10, 4607–4641 RSC.
- Y. Zhang, J. Tu, D. Wang, H. Zhu, S. K. Maity, X. Qu, B. Bogaert, H. Pei and H. Zhang, Adv. Mater., 2018, 30, 1703658 CrossRef PubMed.
- L. R. Schoukroun-Barnes, F. C. Macazo, B. Gutierrez, J. Lottermoser, J. Liu and R. J. White, Annu. Rev. Anal. Chem., 2016, 9, 163–181 CrossRef CAS PubMed.
- P. Röthlisberger and M. Hollenstein, Adv. Drug Delivery Rev., 2018, 134, 3–21 CrossRef PubMed.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2000, 122, 11547–11548 Search PubMed.
- M. A. D. Neves, O. Reinstein and P. E. Johnson, Biochemistry, 2010, 49, 8478–8487 Search PubMed.
- M. A. D. Neves, O. Reinstein, M. Saad and P. E. Johnson, Biophys. Chem., 2010, 153, 9–16 CrossRef CAS PubMed.
- A. K. Sharma and J. M. Heemstra, J. Am. Chem. Soc., 2011, 133, 12426–12429 CrossRef CAS PubMed.
- R. Zou, X. Lou, H. Ou, Y. Zhang, W. Wang, M. Yuan, M. Guan, Z. Luo and Y. Liu, RSC Adv., 2012, 2, 4636–4638 RSC.
- R. Pei, A. Shen, M. J. Olah, D. Stefanovic, T. Worgall and M. N. Stojanovic, Chem. Commun., 2009, 3193–3195 RSC.
- O. Reinstein, M. Yoo, C. Han, T. Palmo, S. A. Beckham, M. C. J. Wilce and P. E. Johnson, Biochemistry, 2013, 52, 8652–8662 CrossRef CAS PubMed.
- S. Slavkovic, M. Altunisik, O. Reinstein and P. E. Johnson, Bioorg. Med. Chem., 2015, 23, 2593–2597 CrossRef CAS PubMed.
- R. W. Harkness V, S. Slavkovic, P. E. Johnson and A. K. Mittermaier, Chem. Commun., 2016, 52, 13471–13474 RSC.
- A. A. Shoara, S. Slavkovic, L. W. Donaldson and P. E. Johnson, Can. J. Chem., 2017, 95, 1253–1260 CrossRef CAS.
- B. Gülbakan, K. Barylyuk, P. Schneider, M. Pillong, G. Schneider and R. Zenobi, J. Am. Chem. Soc., 2018, 140, 7486–7497 CrossRef PubMed.
- Y. Qiu, C. Gu, B. Li and H. Shi, Anal. Methods, 2018, 10, 2931–2938 RSC.
- S. Slavkovic, Z. R. Churcher and P. E. Johnson, Bioorg. Med. Chem., 2018, 26, 5427–5434 CrossRef CAS PubMed.
- J. Wang, W. Cheng, F. Meng, M. Yang, Y. Pan and P. Miao, Biosens. Bioelectron., 2018, 101, 153–158 CrossRef CAS PubMed.
- C. Lu, C. Saint-Pierre, D. Gasparutto, Y. Roupioz, E. Peyrin and A. Buhot, Langmuir, 2017, 33, 12785–12792 CrossRef CAS PubMed.
- L. Azéma, S. Bonnet-Salomon, M. Endo, Y. Takeuchi, G. Durand, T. Emura, K. Hidaka, E. Dausse, H. Sugiyama and J.-J. Toulmé, Nucleic Acids Res., 2018, 46, 1052–1058 CrossRef PubMed.
- S. Slavkovic and P. E. Johnson, Aptamers, 2018, 2, 45–51 Search PubMed.
- I. Lavagnini and F. Magno, Mass Spectrom. Rev., 2007, 26, 1–18 CrossRef CAS PubMed.
- A. Zuber, M. Purdey, E. Schartner, C. Forbes, B. van der Hoek, D. Giles, A. Abell, T. Monro and H. Ebendorff-Heidepriem, Sens. Actuators, B, 2016, 227, 117–127 CrossRef CAS.
- Z. Guo, C. Gu, X. Fan, Z. Bian, H. Wu, D. Yang, N. Gu and J. Zhang, Nanoscale Res. Lett., 2009, 4, 1428–1433 CrossRef CAS PubMed.
- M. A. Vetten, N. Tlotleng, D. Tanner Rascher, A. Skepu, F. K. Keter, K. Boodhia, L.-A. Koekemoer, C. Andraos, R. Tshikhudo and M. Gulumian, Part. Fibre Toxicol., 2013, 10, 50 CrossRef PubMed.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson Education Limited, Harlow, England, 6th edn, 2010 Search PubMed.
- M. A. D. Neves, S. Slavkovic, Z. R. Churcher and P. E. Johnson, Nucleic Acids Res., 2017, 45, 1041–1048 CAS.
- J. Zhang, L. Wang, H. Zhang, F. Boey, S. Song and C. Fan, Small, 2010, 6, 201–204 CrossRef CAS PubMed.
- X. Zhang and V. K. Yadavalli, Biosens. Bioelectron., 2011, 26, 3142–3147 CrossRef CAS PubMed.
- S. Thobhani, S. Attree, R. Boyd, N. Kumarswami, J. Noble, M. Szymanski and R. A. Porter, J. Immunol. Methods, 2010, 356, 60–69 CrossRef CAS PubMed.
- S.-H. Wu, Y.-S. Wu and C.-h. Chen, Anal. Chem., 2008, 80, 6560–6566 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07462c |
| ‡ Department of Laboratory Medicine, Keenan Research Centre for Biomedical Science, St. Michael's Hospital, Toronto, Ontario, Canada, M5B 1W8 |
| This journal is © The Royal Society of Chemistry 2019 |