
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: MiR-206 reduced the malignancy of hepatocellular carcinoma cells in vitro by inhibiting MET and CTNNB1 gene expressions
Qiang Hea,
Haiyan Dub and
Yundong Li *c
*c
aDepartment of Hepatobiliary Surgery, Linyi People's Hospital, Linyi, Shandong, China
bPediatric Intensive Care Unit, Linyi People's Hospital, Linyi, Shandong, China
cDepartment of Oncology, Jining No. 1 People's Hospital, No. 6, Jiankang Road, Rencheng, 272111, Jining, Shandong, China. E-mail: fhbdgjgcfth@163.com; Tel: +86-0537-6056-666
First published on 14th January 2019
Abstract
The anti-cancer role of miR-206 in hepatocellular carcinoma (HCC) cells has been reported, but its mechanism of action remains poorly understood. This research aimed to investigate the anti-HCC mechanism of miR-206. We analyzed 25 pairs of HCC and adjacent tissue specimens from HCC patients. Two patient-derived HCC cell lines were established. MiR-206 levels in tissue specimens and cell lines were detected by qRT-PCR. MiR-206 overexpression was mimicked by miR-206 mimic transfection. MET or CTNNB1 gene was overexpressed by transient transfection. Protein and protein phosphorylation levels of interests were assessed by western blotting. HCC cell malignancy in vitro was evaluated by cell proliferation, apoptosis, colony formation, trans-well invasion assays as well as western blotting assessing the marker proteins of epithelial or mesenchymal phenotype. We found that miR-206 level was significantly lower in HCC tissue specimens in comparison to adjacent counterparts. Two patient-derived HCC cell lines showed lower miR-206 level than L02 human hepatocytes. MiR-206 mimic transfection significantly reduced phosphorylation levels of pan-Akt Ser9, Erk1 Thr202/Tyr204 and Gsk-3beta Ser308 as well as protein levels of beta-catenin and c-Met in primary HCC cells in vitro. Luciferase reported assay and AGO2-RNA co-immunoprecipitation assays results demonstrated that miR-206 reduced MET and CTNNB1 gene expressions in HCC cells by interacting with the 3′ UTR of their mRNAs. Restoring c-Met or beta-catenin protein level by MET or CTNNB1 transient overexpression partially restored the malignancy of HCC cells in vitro. We concluded that miR-206 might inhibit HCC development by targeting MET and CTNNB1 gene expression.
Introduction
Liver cancer is currently the second deadliest cancer type worldwide, and hepatocellular carcinoma is the most frequently diagnosed liver cancer.1 Proliferation, survival, metastasis and therapeutic resistance of HCC cells are driven by oncogenic signaling pathways, such as MAPK, PI3K-Akt and Wnt/beta-catenin signaling pathways.2 Activation of these cancer-promoting pathways has been found to be regulated by microRNAs (miRNAs), and loss of expression of these miRNAs plays fundamental role in HCC progression with high prognostic value.3–5 miRNAs are non-coding RNAs that are often of about 22 nucleotides in length;6 these small RNAs mediate gene silencing by interacting with their target sequences on the 5′ or 3′ untranslated region (UTR) of their target mRNAs, resulting the association of these target mRNAs with RNA-induced silencing complex (RISC), which blocks mRNA translation and mediates mRNA degradation.7,8MiR-206 is a cancer-suppressive microRNA in colorectal cancer, non-small-cell lung cancer, ovarian cancer and neck squamous cell carcinoma by suppressing the activation of PI3K-Akt and MAPK signaling pathways.9–13 MiR-206 has also been described as a tumor-suppressive microRNA in HCC: miR-206 was found downregulated in HCC in association with advance in tumor stage and patients' worsened outcome,14 and the anti-proliferative/pro-apoptotic effects of overexpressing miR-206 in HCC cells in vitro were also reported.14–16 However, molecular mechanisms underlying the HCC-suppressive role of miR-206 remain largely underexplored.
In this research, after confirming the downregulation of miR-206 in HCC tissue specimens and HCC cells comparing to non-malignant counterparts, we explored the impact of miR-206 overexpression on the activation of key signaling pathways implicated in HCC progression, including MAPK, PI3K-Akt and Wnt/beta-catenin signaling pathways in HCC cells in vitro. Our data suggested that miR-206 could directly downregulate beta-catenin protein expression as well as inhibiting the activation of MAPK and PI3K-Akt signaling pathways by directly downregulating c-Met protein expression; restoring c-Met or beta-catenin protein expression by transient overexpression partially abrogated the anti-tumor effect of miR-206 overexpression on patient-derived HCC cells.
Materials and methods
HCC patients and tissue specimens
25 pairs of HCC tissue specimens and adjacent tissue specimens were collected from 25 HCC patients during surgery. The patients included 17 males and 8 females at age of 44.16 ± 8.04 who were diagnosed with HCC at the stage of II ∼ III B during 2015–2017. None of the patient received chemo-, radio- or immuno-therapy before surgery. Adjacent tissue specimens were obtained from non-malignant liver tissues 5–10 cm away from tumor margin at the minimal amount for histological examination. The tissue specimens were either stored in liquid nitrogen immediately after excision or used to establish patient-derived HCC cell lines (described later). This research was approved by the ethical review committee at Linyi People's Hospital and informed consent in written from each participant.Establishment of patient-derived HCC cell lines and cell culture
Establishment of the two patient-derived HCC cell line HCC1 and HCC2 was performed as described by Xin et al.17 HCC tissue samples were obtained during surgery from two patients and subcutaneously transplanted into 6 week-old female SCID Beige mice (Vital River Laboratory, Beijing, China). Two xenografts from the two patients were excised when the xenograft reached 500 mm3, and the xenograft was minced and cultured in RPMI-1640 medium with L-glutamine (11875119, Thermo Fisher Scientific) supplemented with 10% FBS (0500, Sciencell) and 1% of antibiotic solution (0513, Sciencell). Cells were cultured in vitro for 10 passages before liquid nitrogen storage. The cells were further analyzed after recovery. L-02 human hepatocytes were purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology and were cultured under the same condition described above. Animal experiments involved in this research was approved by the ethical review committee at Linyi People's Hospital.MiR-206 mimic and transient overexpression plasmid transfection
Transfection of miR-206 mimic and miR mimic negative control into HCC1 and HCC2 was performed as described by Liu et al.15 Briefly, the cells were cultured on 6-well-plate until 70% confluency, before they were rinsed with PBS and cultured using antibiotic-free culture medium for 24 hours, followed by transfection with 10 nM of miR-206 mimic or miRNA mimic negative control (miR10000462-1-5, Ribobio, Guangzhou, China) using 4‰ (v/v) of Lipofectamine 3000 (L3000150, Thermo Fisher Scientific). MET or CTNNB1 gene overexpressing plasmids were constructed by Genecopoeia. After the transfection of miR-206 mimic or miRNA mimic negative control, plasmid transfection was performed at 0.5 μg mL−1 following the product manual using 4‰ (v/v) of Lipofectamine 3000. Cells were assayed 24 hours after transfection, unless otherwise indicated.qRT-PCR
qRT-PCR was performed to detect miR-206 level in tissue homogenate or cell lysate samples. Total RNA was extracted using Trizol reagent (15596026, Thermo Fisher Scientific), and miRNAs in the total RNA were reversely-transcribed using miRNA first-strand cDNA synthesis kit (QP018, Genecopoeia). Semi-quantification of miR-206 was performed using miRNA qRT-PCR detection kit (QP016, Genecopoeia) with a thermocycler. Primer sequences were as follows: miR-206 forward, 5′-TGGAATGTAAGGAAGTGTGTGGAA-3′; U6 forward, 5′-GGGCAGGAAGAGGGCCTAT-3′. The reverse adapter primer was universal and was included in the miRNA qRT-PCR detection kit.Western blotting
Protein levels of interest was investigated by western blotting. Cells after transfection for 24 hours were lysed with pre-cooled cell lysis buffer for Western and IP (P0013, Beyotime) by harsh pipetting. The cell lysate samples were subject to bicinchoninic acid assay using the Enhanced BCA Protein Assay Kit (P0009, Beyotime), and cell lysate sample aliquots each containing 40 μg of total proteins were subject to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Cellular proteins separated by SDS-PAGE were transferred onto nitrocellulose membrane (1620115, Bio-Rad), and the membrane was incubated with primary antibodies probing the following proteins or protein modifications: pan-Akt pS308 (ab38449), pan-Akt (ab8805), Erk1 pT202/Y204 (ab24157), Erk1 (ab214168), Gsk-3 beta pS9 (ab214168), Gsk-3 beta (ab32391), beta-catenin (ab16051), GAPDH (ab8245), N-cadherin (ab18203), E-cadherin (ab1416), followed by incubation with HRP-conjugated secondary antibodies and imaging using ECL substrate (32106 or 34580, Thermo Fisher Scientific) and X-ray film (34089, Thermo Fisher Scientific). Primary and secondary antibodies were purchased from Abcam. The primary antibodies were used at a working concentration of 1 μg mL−1, while the secondary antibodies were used at 0.5 μg mL−1. Quantification of western blotting results was performed by gray scale analysis using Image J software. Integrated density of each band of protein of interest was first normalized to that of GAPDH as housekeeping gene before normalized to the mean value of the control group.Luciferase reporter assay
Luciferase reporter assay was performed to verify whether miR-206 would regulate c-Met and beta-catenin protein expression in HCC cells by mRNA targeting. To construct the reporter plasmid, wild type (WT) cDNA of 3′ UTR of MET or CTNNB1 mRNA was inserted downstream of Gaussia luciferase gene on the reporter plasmids. Scrambled cDNA of 3′ UTR of MET or CTNNB1 mRNA was inserted downstream of Gaussia luciferase gene as negative control (scrambled). The reporter plasmids also encoded secreted alkaline phosphatase (SEAP) gene as internal reference. These reporter plasmids were constructed by Genecopoeia. HCC cells cultured on 6-well-plate at 70% confluency were first transfected with reporter plasmids at 0.5 μg mL−1 using 4‰ (v/v) of Lipofectamine 3000; 24 hours later, the cells were transfected with miR mimic-NC or miR-206 mimic as described above. 48 hours later, activity of Gaussia luciferase and SEAP in the cell culture media were detected using Secrete-Pair Dual Luminescence Assay Kit (LF033, Genecopoeia) using a microplate reader. Gaussia luciferase activity was first normalized to that of SEAP activity before data analysis.AGO2-RNA co-immunoprecipitation (AGO2-RIP)
AGO2-RIP assay was performed to investigate whether miR-206 mimic transfection would influence MET or CTNNB1 mRNA silencing mediated by the RISC complex. Cells cultured on 6-well-plates at 70% confluency were first transfected with miR-206 mimic or miRNA mimic negative control and lysed as described above. The cell lysate samples were pre-cleared by centrifugation at 10000 × g for 3 minutes under 4 °C, and the supernatants were subject to AGO2-RIP using a customized Imprint RNA Immunoprecipitation Kit (Sigma Aldrich). Briefly, the cell lysate samples were incubated under 4 °C overnight with magnetic protein A beads, which was pre-ligated with rabbit anti-rat IgG antibody and rat anti-AGO2 antibodies that were associated with the kit. After incubation, the beads were collected using a magnetic stand and washed with PBS, and RNAs associated with the beads were detached using the kit following the product manual. The amount of mRNA of MET or CTNNB1 in the beads-associated RNAs was evaluated by qRT-PCR by 2−ΔCt method. AGO2 protein in the cell lysate and precipitation product was evaluated by western blotting.Cell proliferation, apoptosis, colony formation and trans-well invasion assay
Cell proliferation was evaluated by EdU assay. Cells after transfection were harvested by trypsin (59427C, Sigma Aldrich) digestion and inoculated on 96-well-plate at a density of 104 cells per well. Cells were allowed to settle overnight, and cell proliferation was assessed using EdU microplate assay kit (C10214, Thermo Fisher Scientific) with a microplate reader. Fluorescent intensity at 568/585 nm was considered positively correlated with the amount of newly synthesized DNA.Cell apoptosis was evaluated by flow cytometry. Cells after transfection were cultured in 6-well-plate at 3 × 104 per well in serum-free medium for 12 hours to induce apoptosis. Cells were then harvested and labeled with FITC-Annexin V and PI (V13242, Thermo Fisher Scientific), followed by flow cytometry evaluating the percentage of FITC-Annexin V+ PI− cells in the total cells.
Colony formation assay was performed as described by Franken et al. with minor modifications.18 Cells after transfection were harvested and inoculated on 6-well-plate at a density of 103 cells per well. After cell culture for 1 week, the cells were fixed with acetic acid/methanol at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 ratio for 5 minutes at 25 °C and stained with 0.5% crystal violet solution for 2 hours. Clonogenicity was assessed by counting the number of colonies with 50 or more cells in each well under microscope.
:7 ratio for 5 minutes at 25 °C and stained with 0.5% crystal violet solution for 2 hours. Clonogenicity was assessed by counting the number of colonies with 50 or more cells in each well under microscope.
Trans-well invasion assay was performed using matrigel-coated trans-well chamber (08-774-122, Fisher Scientific). Trans-well with 8 μm pores were placed into a 24-well plate. Cells after transfection were harvested and re-suspended in serum-free medium. 5 × 103 cells were inoculated into the trans-well chamber, which was inserted into cell culture medium supplemented with 5% FBS. Cells were incubated for 16 hours, and cells migrated to the underside of the trans-well chamber were fixed, stained and counted as described above. Trans-well invasion capacity was assessed by counting the number of cells migrated.
Statistical analysis
All data were presented as fold induction when applicable. Each dot represented one data point. Student's t test was performed for comparison between two groups, while one-way analysis of variance with Tukey's test as the post-hot test was performed for multiple comparison. A difference was considered statistically significant when p < 0.05.Results
MiR-206 expression is downregulated in HCC tissue specimens and HCC cells comparing to non-malignant counterparts
In this research we first investigated the expression of miR-206 in tissue specimens from 25 HCC patients by qRT-PCR. We found a significant downregulation of miR-206 in HCC pathological tissue specimens comparing to non-malignant adjacent ones (Fig. 1A and B). We next established two patient-derived HCC cell lines, HCC1 and HCC2, using HCC pathologic tissue samples from two patients. qRT-PCR results indicated that miR-206 levels in these two patient-derived HCC cells were significantly lower than that in L02 human hepatocytes (Fig. 1C). These data suggested that miR-206 was frequently downregulated during HCC development.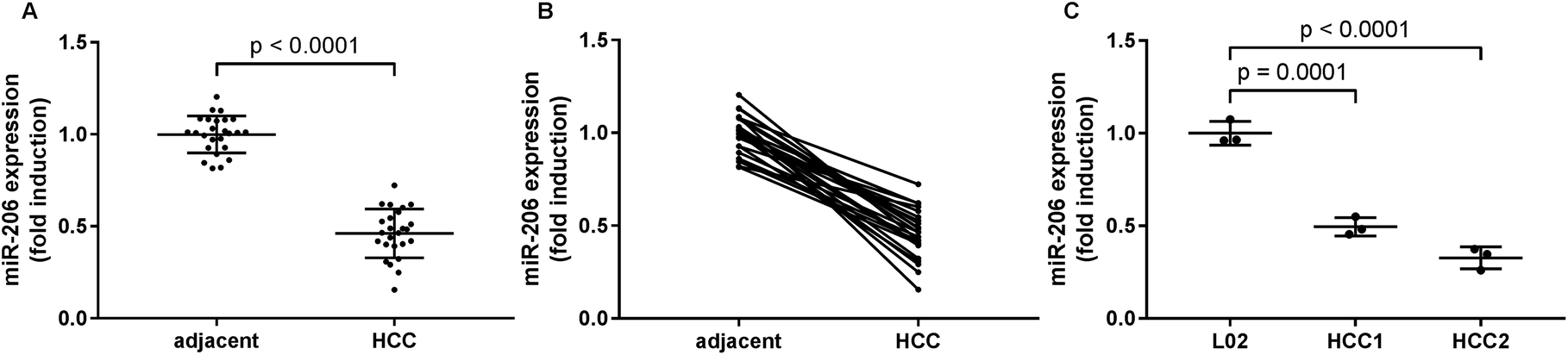 | ||
| Fig. 1 MiR-206 was downregulated in HCC tissue specimens and primary HCC cells, comparing to non-malignant counterparts. (A and B) miR-206 levels in 25 pairs of HCC tissue specimens and adjacent counterparts were detected by qRT-PCR. HCC and adjacent tissue specimens from the same patient were connected with strait line in (B). (C) miR-206 levels were detected by qRT-PCR in L02 non-malignant hepatocytes and two primary HCC cells (HCC1 and HCC2) derived from two HCC patients' tumor tissue specimens. | ||
MiR-206 overexpression reduced the activation of MAPK, PI3K-Akt and Wnt/beta-catenin signaling pathways in HCC cells in vitro
To investigate the molecular mechanism underlying the anti-HCC role of miR-206, we analyzed the impact of miR-206 overexpression (mimicked by miR-206 mimic transfection) on the activation of three well-illustrated tumor promoting signaling pathways, namely the PI3K-Akt, MAPK and Wnt/beta-catenin signaling pathways in the two patient-derived HCC cells. Our western blotting results showed that Ser308 phosphorylation of Akt, Thr202/Tyr204 phosphorylation of Erk1 and Ser9 phosphorylation of Gsk-3beta were significantly reduced in miR-206 mimic-transfected (mimic) HCC1 or HCC2 cells comparing to miRNA mimic control-transfected (NC) ones (Fig. 2A–C and F), suggesting that the three cancer-promoting signaling pathways were inhibited in the two patient-derived HCC cells by miR-206 overexpression in vitro.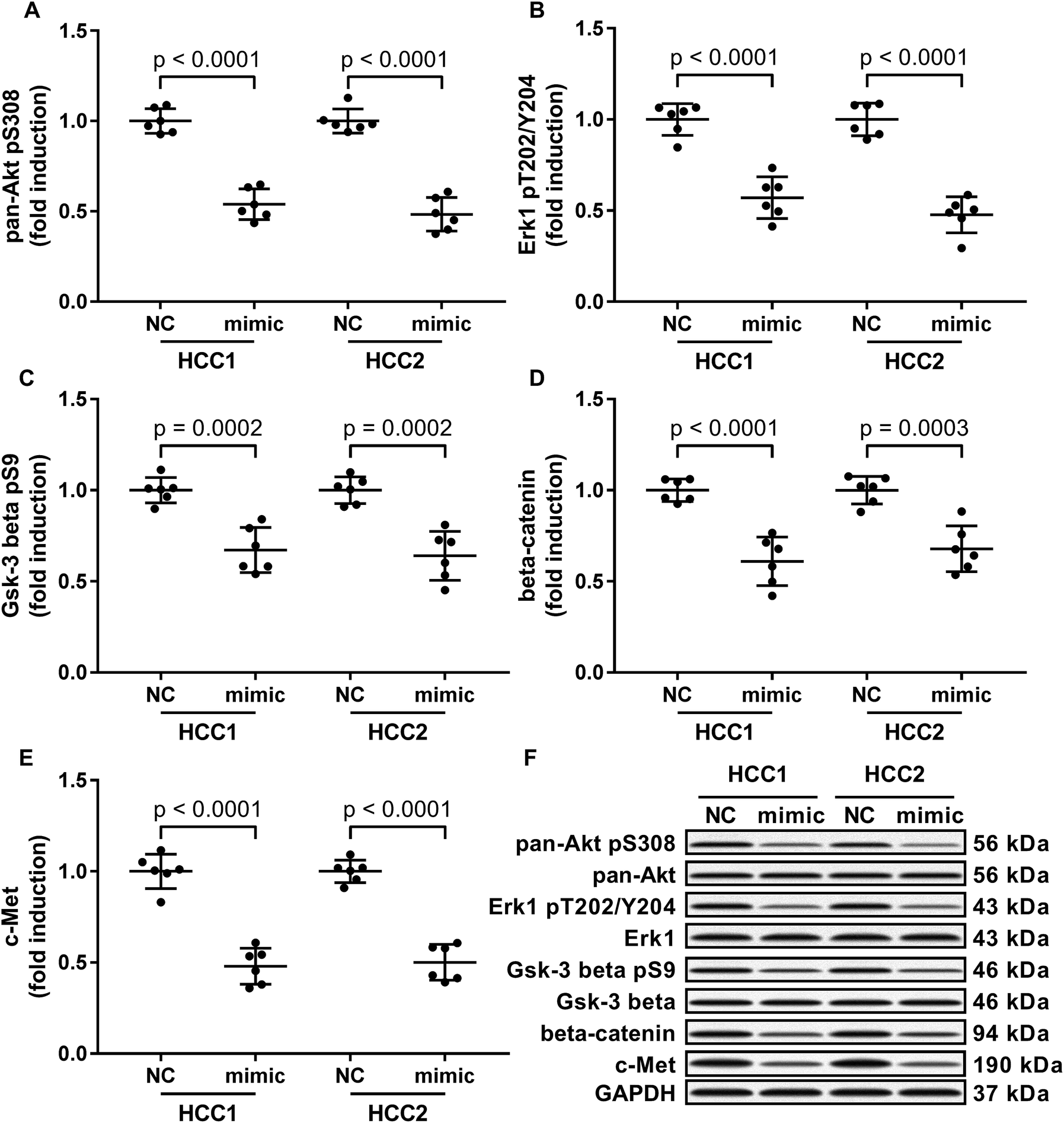 | ||
| Fig. 2 MiR-206 mimic transfection reduced the activation of tumor-promoting signaling pathways as well as the expression of beta-catenin and c-Met proteins in the two patient-derived HCC cell lines in vitro. Cells were transfected with miRNA mimic negative control (NC) or miR-206 mimic (mimic) for 24 hours before analysis. (A) Activation of PI3K-Akt pathway was represented by pan-Akt Ser308 phosphorylation level. (B) Activation of MAPK signaling pathway was represented by Erk1 Thr202/Tyr204 phosphorylation level. (C) Activation of Wnt/beta-catenin signaling pathway was represented by Gsk-3 beta Ser9 phosphorylation level. (D and E) Protein levels of beta-catenin and c-Met. (F) Representative western blotting images. | ||
MiR-206 overexpression reduced beta-catenin and c-Met protein expression in HCC cells by mRNA targeting
Previous researches have described MET as a miR-206 target gene,19 and the inverse correlation between miR-206 level and beta-catenin protein level has also been reported.20–22 Considering that c-Met activates both PI3K-Akt and MAPK signaling pathways, and that reduced Gsk-3beta Ser9 phosphorylation often leads to increased beta-catenin protein degradation, we thought to investigate whether miR-206 mimic transfection would affect c-Met and beta-catenin protein level in the two patient-derived HCC cells. Our western blotting results showed that both beta-catenin and c-Met proteins were downregulated in HCC1 and HCC2 cells upon miR-206 transfection (Fig. 2D–F). To verify whether miR-206 would reduce c-Met protein expression in HCC cells by targeting MET mRNA, and to investigate whether miR-206 would regulate beta-catenin protein expression through the same mechanism, we constructed luciferase reporter plasmid carrying Gaussia luciferase gene that was flanked at 3′ with cDNA of wild-type MET or CTNNB1 mRNA 3′ UTR (WT), while those flanked at 3′ by scrambled cDNAs with the same length was used as negative control (scrambled). After transfecting the luciferase reporter plasmids and miR-206 mimic successively into HCC1 cells, we found that miR-206 mimic transfection significantly reduced Gaussia luciferase activity in HCC1 cells that were transfected with WT luciferase reporter plasmid but not the control ones (Fig. 3A and B), suggesting that both MET and CTNNB1 mRNA were targeted by miR-206 in HCC cells. To further verify this mechanism, we performed AGO2-RNA co-immunoprecipitation in HCC1 cells with or without miR-206 mimic transfection. Out data suggested that transfection with miR-206 significantly increase the association of both MET and CTNNB1 mRNA with AGO2 protein (Fig. 3C). These data suggested that miR-206 could reduce MET and CTNNB1 gene expression in HCC cells by targeting their mRNA.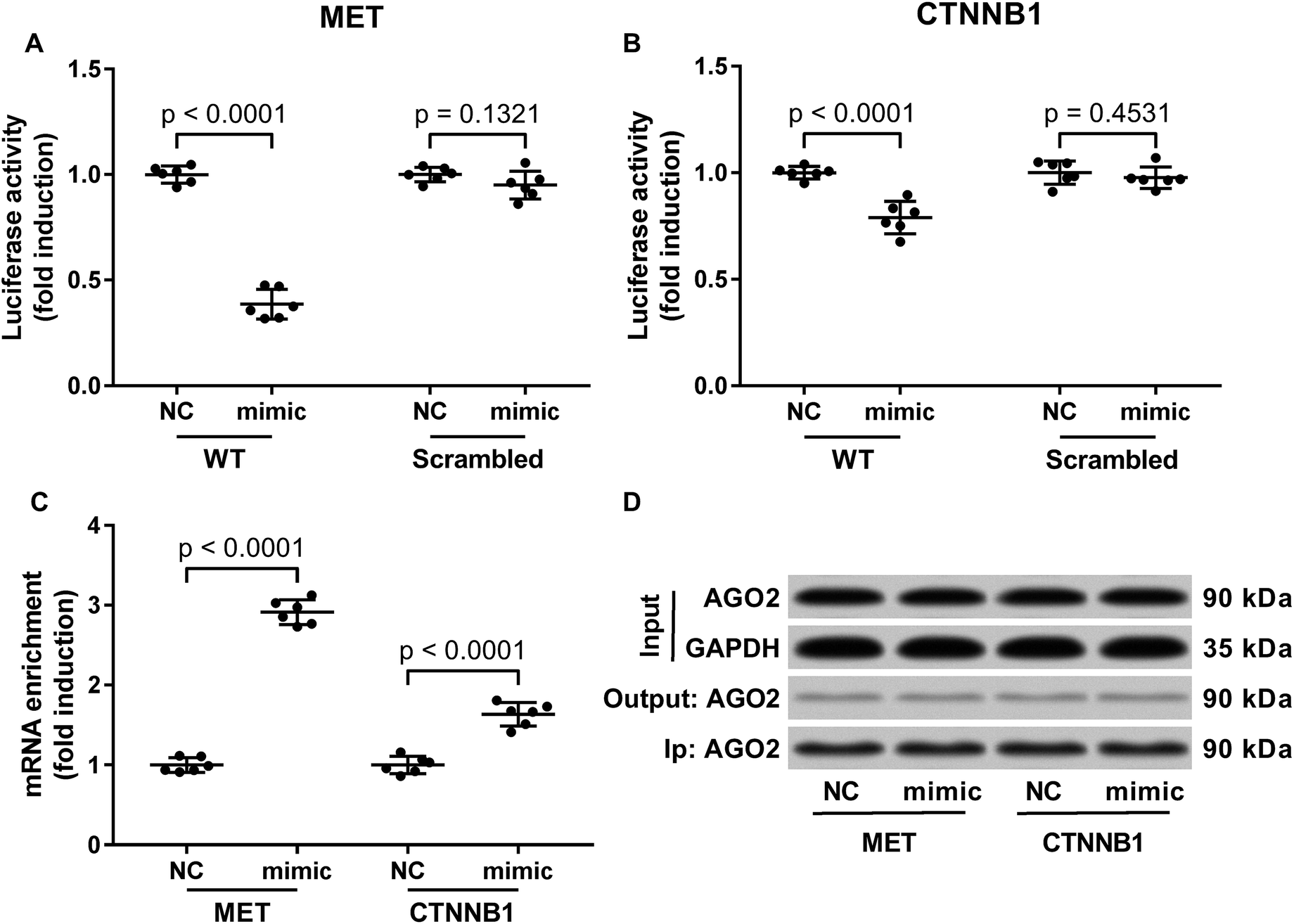 | ||
| Fig. 3 MiR-206 inhibited MET and CTNNB1 gene expressions in primary HCC cells by targeting the 3′ UTR of their mRNAs. (A and B) Luciferase reporter assay evaluating the interaction between miR-206 and 3′ UTR of MET or CTNNB1 mRNA in patient-derived HCC cells. Cells were first transfected with wildtype (WT) or scrambled reporter plasmids for 24 hours before miRNA mimic negative control (NC) or miR-206 mimic (mimic) transfection. Activity of Gaussia luciferase was assessed in the cell culture media 48 hours after transfection using secreted alkaline phosphatase activity as internal reference. (C) AGO2-RNA co-immunoprecipitation evaluating the association between RISC complex and MET or CTNNB1 mRNA in patient derived HCC cells with or without miR-206 mimic transfection. (D) Western blotting detecting whether AGO2 protein was equally precipitated in the four experimental groups in (C). Cells were transfected with miRNA mimic negative control (NC) or miR-206 mimic (mimic) for 24 hours before analysis. Input, the cell lysate before AGO2-RIP; output, the cell lysate after AGO2-RIP; Ip, magnetic beads were subject to western blotting after AGO2-RIP. | ||
Restoring c-Met and beta-catenin protein expression partially antagonized the tumor suppressive effect of miR-206 overexpression on HCC cells
To verify whether miR-206 would inhibit HCC development by targeting MET and CTNNB1 gene expression, we transfected HCC1 and HCC2 cells with MET or CTNNB1 gene overexpressing plasmid to restore c-Met or beta-catenin protein expression, respectively, after miR-206 mimic transfection (Fig. 4A and B). Our cell functional assay results showed that restoring c-Met or beta-catenin protein expression in miR-206 mimic-transfected HCC1 cells significantly restored its cell proliferation, colony formation, trans-well invasion capacity and epithelial-to-mesenchymal transition while reducing their apoptosis in vitro (Fig. 4C–G and 5), suggesting that miR-206 would reduce the malignancy of HCC cells in vitro by reducing c-Met and beta-catenin protein expression via mRNA targeting.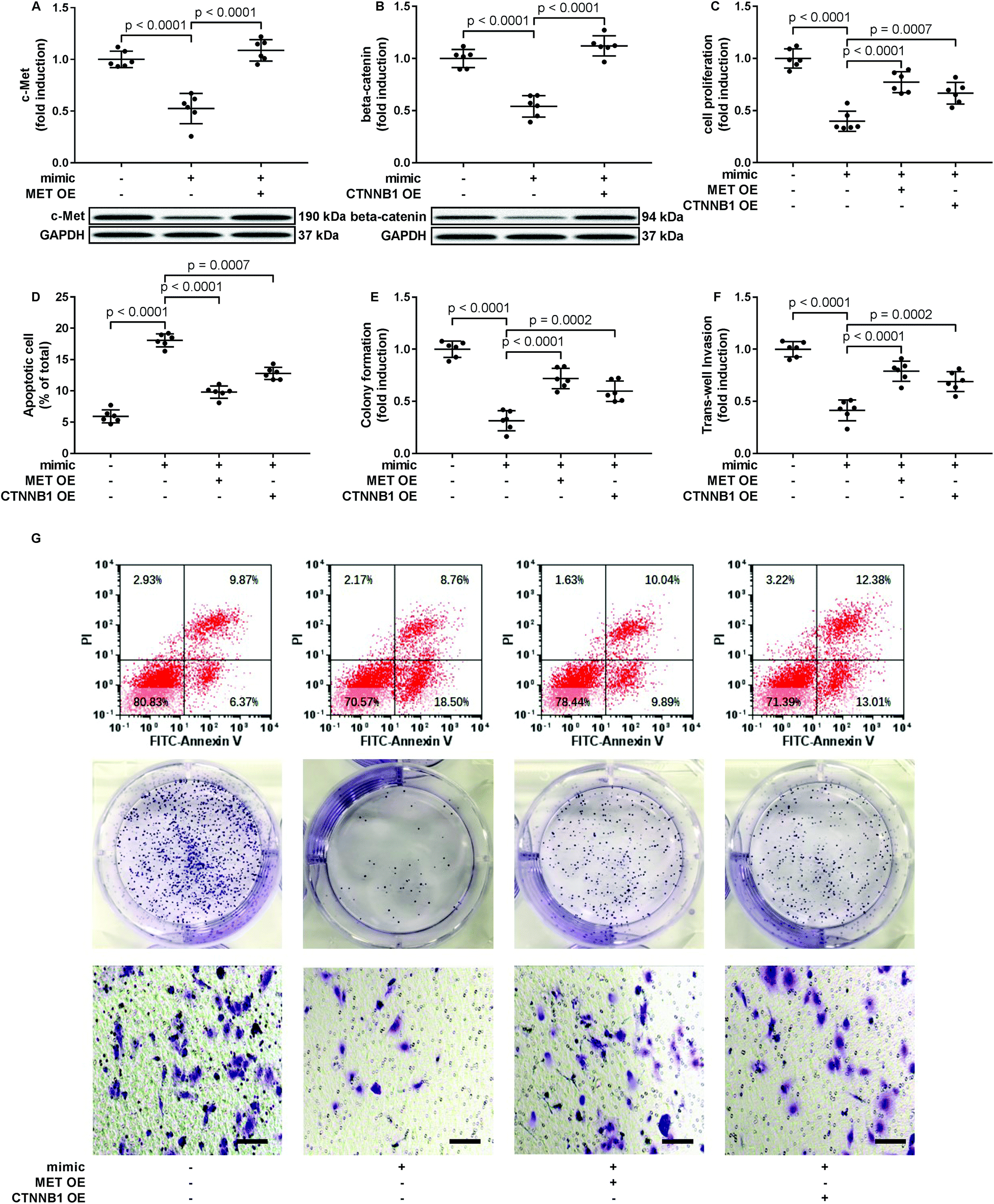 | ||
| Fig. 4 Restoring c-Met of beta-catenin protein expression partially rescued the malignancy of patient-derived HCC cells that were downregulated by miR-206 mimic transfection. After miRNA mimic negative control (NC) or miR-206 mimic transfection, primary HCC cells were further transfected with MET or CTNNB1 overexpressing (OE) plasmids (+) or empty vectors (−). Cells were assessed 24 hours after transfection. (A and B) c-Met or beta-catenin protein level in miR-206 mimic transfected HCC cells was restored by transient transfection. (C–F) HCC cell malignancy was evaluated by cell proliferation, apoptosis, colony formation and trans-well invasion assays. (G) Is representative results of cell apoptosis, colony formation and trans-well invasion assays. | ||
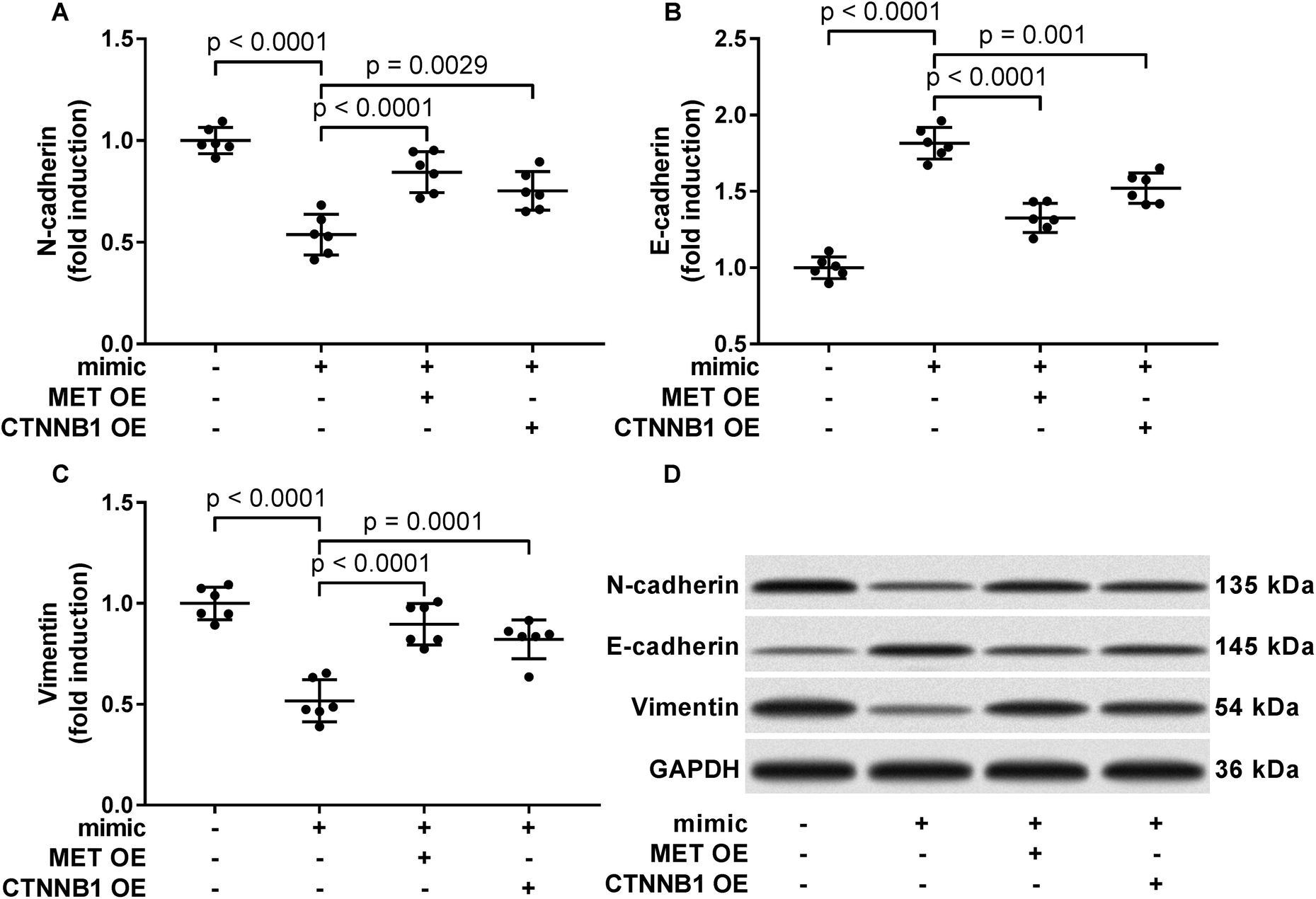 | ||
| Fig. 5 Restoring c-Met of beta-catenin protein expression partially rescued epithelial-to-mesenchymal transition of patient-derived HCC cells that were downregulated by miR-206 mimic transfection. After miRNA mimic negative control (NC) or miR-206 mimic transfection, primary HCC cells were further transfected with MET or CTNNB1 overexpressing (OE) plasmids (+) or empty vectors (−). Epithelial and mesenchymal phenotype of HCC cells was assessed by western blotting evaluating the protein levels of N-cadherin, E-cadherin and vimentin. | ||
Activation of PI3K-Akt led to Gsk-3beta Ser 9 phosphorylation, thus inhibiting beta-catenin degradation and increasing its protein level. To investigate to what extent the reduce in beta-catenin protein level led by miR-206 mimic transfection is due to CTNNB1 mRNA targeting, we analyzed beta-catenin protein level in miR-206 mimic-transfected HCC1 with or without MET overexpression. Our western blotting results showed that MET overexpression indeed partially recovered beta-catenin protein level in miR-206 mimic-transfected HCC1 cells (Fig. 6A), suggesting that beta-catenin protein level in HCC cells was regulated by both mIR-206 and c-Met-PI3K-Akt axis. On the other hand, overexpressing CTNNB1 showed minimal impact on c-Met protein level in miR-206 mimic-transfected HCC1 cells (Fig. 6B).
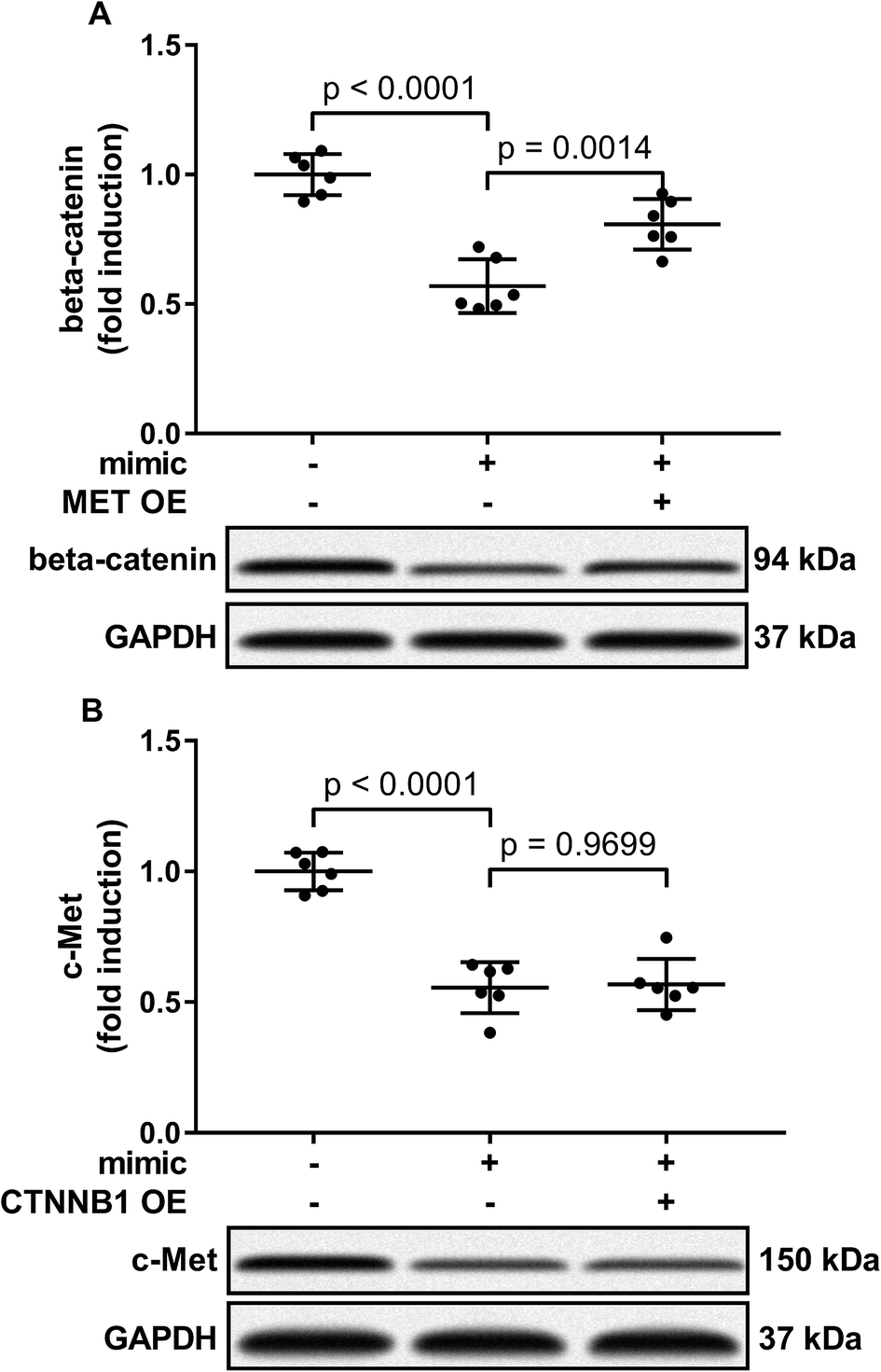 | ||
| Fig. 6 Restoring c-Met protein level in miR-206-transfected HCC cells partially recovered beta-catenin protein level, but restoring beta-catenin protein level showed minimal influence on that of c-Met in miR-206-transfected HCC cells. After miRNA mimic negative control (NC) or miR-206 mimic transfection, primary HCC cells were further transfected with MET or CTNNB1 overexpressing (OE) plasmids (+) or empty vectors (−). Protein level of c-Met or beta-catenin was determined by western blotting. | ||
Discussion
The present research aimed to verify the anti-HCC role of miR-206 and investigate its anti-cancer mechanism in HCC. Our data suggested that miR-206 could reduce HCC cell proliferation, survival, colony formation, trans-well invasion and EMT in vitro by reducing c-Met and beta-catenin protein, possibly by targeting their mRNAs.The anti-HCC role of miR-206 was first described by Liu et al., who reported that transfection with miR-206 mimic significantly reduce HepG2 cell growth and increased cell apoptosis. They proposed that this effect of miR-206 was due to NOTCH3 targeting, since previous researchers have identified NOTCH3, a tumor promoting gene in HCC,23,24 as a target gene of miR-206,25 and Liu et al. indeed demonstrated a significant decrease in Notch3 protein level in HepG2 cells after miR-206 transfection. However, their conclusion is undermined by the lack of rescue assay verifying the involvement of NOTCH3 inhibition in miR-206-mediated HCC inhibition. Expression level change of miR-206 in HCC comparing to non-malignant tissue remains in controversial. Tan et al. reported a significant upregulation of miR-206 level in HCC serum samples comparing to non-cancerous ones,26 while Oksuz et al. reported a mild (yet not statistically significant) upregulation of miR-206 in HCC serum comparing to non-cancerous counterparts.27 However, Yunqiao et al. later reported a significant downregulation of miR-206 in HCC, which was correlated with HCC progression in patients whose tissue specimens were examined.14 They also confirmed that transfection with miR-206 mimic would significantly inhibit HepG2 cell growth, survival and invasion in vitro. Similar results were also reported by Pang et al., who further identified CDK9 gene as a target of miR-206 in regulating HCC cell proliferation and apoptosis.16 We hypothesized that the controversial expression of miR-206 might be caused by the varying source of isolated RNA, such as cancer tissue, cell line or serum. However, the expression and function of serum miRNA might not be in accordant with that in tissue or cell.
In the present research, we observed a significant decrease in miR-206 level in 25 HCC tissue specimens comparing to their adjacent counterparts. We also found that miR-206 levels in the two patient-derived HCC cells were lower than that in L02 cells, an immortalized liver cell line frequently used as non-malignant control in HCC research. It should be noted that our observation might be biased by the insufficiency of sample size, and it's possible for a tumor suppressive miRNA, lncRNA or protein to be upregulated during tumor development. Nevertheless, in the present research we observed that transfection with miR-206 could significantly reduce HCC cell proliferation and promote apoptosis in vitro, which is in consistent with previous findings.14–16 We also found that miR-206 mimic transfection would inhibit trans-well invasion and EMT of HCC cells in vitro.
To investigate the molecular mechanism underlying the anti-HCC role of miR-206, we first monitored the activation of three important tumor promoting signaling pathways, namely the PI3k-Akt, MAPK and Wnt/beta-catenin signaling pathways, in HCC cells with or without miR-206 mimic transfection. Our data suggested that activation of these pathways in HCC could be inhibited by miR-206 transfection. PI3k-Akt and MAPK signaling in HCC cells are primarily activated downstream of Epidermal growth factor receptor (EGFR), insulin-like growth factor 1 receptor (IGF1R) and c-Met receptor (MET),28–30 among these three is MET that have been identified as a target gene of miR-206.19,31 Inhibition of MET gene expression by miR-206 has been suggested to limit the development of various types of cancers.9,11,31–35 We found that miR-206 mimic transfection significantly reduced c-Met protein level in patient-derived HCC cells, and restoring c-Met protein level by transient overexpression could partially restore the malignancy of HCC cells that were reduced by miR-206 mimic transfection in vitro, suggesting that inhibiting c-Met protein expression is implicated in the HCC-suppressive role of miR-206. We observed no significant influence on EGFR or IGF1R protein level in HCC cells by miR-206 mimic transfection (data not shown), although several previous reports have described the inhibition of EGFR gene expression by miR-206 via mRNA targeting in lung squamous cell carcinoma,36 head and neck squamous cell carcinoma37 and ovarian carcinoma cells.38 We haven't figured out if this is due to the heterogeneity of cancer cells of different tissue origin, because we are actually able to reproduce the inhibitive effect on EGFR gene expression using siRNA or miR-128b mimic in HCC cells, which was reported in lung cancer cells.39 It might worth investigating whether the interaction between mRNA and miRNA can be influenced by intracellular space in cancer cells.
We also found that beta-catenin protein level and Gsk-3beta Ser9 phosphorylation level were both decreased in HCC after miR-206 transfection. The inverse correlation between miR-206 level and beta-catenin protein or mRNA level has been noticed,20–22 but whether CTNNB1 mRNA was a direct target of miR-206 hasn't been investigated before. In this research we performed luciferase reporter assay and AGO2-RNA co-immunoprecipitation and verified the 3′ UTR of CTNNB1 mRNA as a direct target of miR-206, although miR-206-induced CTNNB1 gene silencing seemed less potent than that of MET gene. Bioinformatic analysis using microT-CDS algorithm revealed that 3′ UTR of MET gene contain more binding sites for miR-206 in comparison to that of CTNNB1, which might explain why miR-206 mimic transfection inhibited MET gene expression more potently that CTNNB1 gene. Cell functional assay further showed that restoring c-Met and beta-catenin protein levels in HCC could partially rescue HCC cell malignancy that was reduced by miR-206 mimic transfection, suggesting that inhibition of both c-met and beta-catenin protein expressions was involved in the anti-HCC role of miR-206. Besides, we found that restoration of MET attenuated miR-206 overexpression-induced inhibition of beta-catenin, but addition of CTNNB1 showed little influence on c-Met level, suggesting that c-Met is the upstream of Wnt/beta-catenin signaling pathways.
In conclusion, in this research we verified the downregulation of miR-206 in HCC. Our data suggested that miR-206 would significantly reduce the malignancy of HCC cells in vitro by targeting MET and CTNNB1 gene expression. Restoring endogenous miR-206 expression or delivering this anti-HCC miRNA into HCC cells might be valuable for HCC management.
Ethics approval and consent to participate
This study was performed in accordance with NIH guidelines. This research was approved by the ethical review committee of Linyi People's Hospital and informed consent in written from each participant.Conflicts of interest
The authors have no conflict of interest to declare.References
- D. Sia, A. Villanueva, S. L. Friedman and J. M. Llovet, Gastroenterology, 2017, 152, 745–761 CrossRef CAS PubMed.
- J. Zucman-Rossi, A. Villanueva, J. C. Nault and J. M. Llovet, Gastroenterology, 2015, 149, 1226–1239 CrossRef CAS PubMed.
- C. M. Wong, F. H. Tsang and I. O. Ng, Nat. Rev. Gastroenterol. Hepatol., 2018, 15, 137–151 CrossRef CAS PubMed.
- M. Klingenberg, A. Matsuda, S. Diederichs and T. Patel, J. Hepatol., 2017, 67, 603–618 CrossRef CAS PubMed.
- N. Yang, N. R. Ekanem, C. A. Sakyi and S. D. Ray, Adv. Drug Delivery Rev., 2015, 81, 62–74 CrossRef CAS PubMed.
- M. Ha and V. N. Kim, Nat. Rev. Mol. Cell Biol., 2014, 15, 509–524 CrossRef CAS PubMed.
- H. O. Iwakawa and Y. Tomari, Trends Cell Biol., 2015, 25, 651–665 CrossRef CAS PubMed.
- I. Daugaard and T. B. Hansen, Trends Genet., 2017, 33, 208–219 CrossRef CAS PubMed.
- Z. Xu, C. Zhu, C. Chen, Y. Zong, H. Feng, D. Liu, W. Feng, J. Zhao and A. Lu, Cell Death Dis., 2018, 9, 974 CrossRef PubMed.
- Y. Tang, G. Xiao, Y. Chen and Y. Deng, Anticancer Drugs, 2018, 29, 725–735 CrossRef CAS PubMed.
- C. Dai, Y. Xie, X. Zhuang and Z. Yuan, Biomed. Pharmacother., 2018, 104, 763–770 CrossRef CAS PubMed.
- F. Liu, X. Zhao, Y. Qian, J. Zhang, Y. Zhang and R. Yin, Biomed. Pharmacother., 2017, 96, 229–237 CrossRef CAS PubMed.
- Q. Y. Chen, D. M. Jiao, Y. Q. Wu, J. Chen, J. Wang, X. L. Tang, H. Mou, H. Z. Hu, J. Song, J. Yan, L. J. Wu, J. Chen and Z. Wang, Oncotarget, 2016, 7, 18247–18261 Search PubMed.
- L. Yunqiao, H. Vanke, X. Jun and G. Tangmeng, Hepatogastroenterology, 2014, 61, 1302–1307 Search PubMed.
- W. Liu, C. Xu, H. Wan, C. Liu, C. Wen, H. Lu and F. Wan, Int. J. Mol. Med., 2014, 34, 420–428 CrossRef CAS PubMed.
- C. Pang, G. Huang, K. Luo, Y. Dong, F. He, G. Du, M. Xiao and W. Cai, Cancer Med., 2017, 6, 2398–2409 CrossRef CAS PubMed.
- H. Xin, K. Wang, G. Hu, F. Xie, K. Ouyang, X. Tang, M. Wang, D. Wen, Y. Zhu and X. Qin, PLoS One, 2014, 9, e85308 CrossRef PubMed.
- N. A. Franken, H. M. Rodermond, J. Stap, J. Haveman and C. van Bree, Nat. Protoc., 2006, 1, 2315–2319 CrossRef CAS PubMed.
- X. L. Ren, G. Y. He, X. M. Li, H. Men, L. Z. Yi, G. F. Lu, S. N. Xin, P. X. Wu, Y. L. Li, W. T. Liao, Y. Q. Ding and L. Liang, J. Cancer Res. Clin. Oncol., 2016, 142, 581–592 CrossRef CAS PubMed.
- Y. R. Park, S. Y. Seo, S. L. Kim, S. M. Zhu, S. Chun, J. M. Oh, M. R. Lee, S. H. Kim, I. H. Kim, S. O. Lee, S. T. Lee and S. W. Kim, Biosci. Rep., 2018, 38, BSR20180664 CrossRef PubMed.
- G. Liu, G. Luo, Z. Bo, X. Liang, J. Huang and D. Li, Exp. Mol. Pathol., 2016, 101, 89–99 CrossRef CAS PubMed.
- B. D. Adams, K. P. Claffey and B. A. White, Endocrinology, 2009, 150, 14–23 CrossRef CAS PubMed.
- L. Gramantieri, C. Giovannini, A. Lanzi, P. Chieco, M. Ravaioli, A. Venturi, G. L. Grazi and L. Bolondi, Liver Int., 2007, 27, 997–1007 CrossRef CAS PubMed.
- C. Giovannini, L. Bolondi and L. Gramantieri, Int. J. Mol. Sci., 2016, 18, 56 CrossRef PubMed.
- G. Song, Y. Zhang and L. Wang, J. Biol. Chem., 2009, 284, 31921–31927 CrossRef CAS PubMed.
- Y. Tan, G. Ge, T. Pan, D. Wen, L. Chen, X. Yu, X. Zhou and J. Gan, PLoS One, 2014, 9, e107986 CrossRef PubMed.
- Z. Oksuz, M. S. Serin, E. Kaplan, A. Dogen, S. Tezcan, G. Aslan, G. Emekdas, O. Sezgin, E. Altintas and E. N. Tiftik, Mol. Biol. Rep., 2015, 42, 713–720 CrossRef CAS PubMed.
- S. Whittaker, R. Marais and A. X. Zhu, Oncogene, 2010, 29, 4989–5005 CrossRef CAS PubMed.
- A. Villanueva, P. Newell, D. Y. Chiang, S. L. Friedman and J. M. Llovet, Semin. Liver Dis., 2007, 27, 55–76 CrossRef CAS PubMed.
- K. Breuhahn, T. Longerich and P. Schirmacher, Oncogene, 2006, 25, 3787–3800 CrossRef CAS PubMed.
- D. Yan, E. Dong Xda, X. Chen, L. Wang, C. Lu, J. Wang, J. Qu and L. Tu, J. Biol. Chem., 2009, 284, 29596–29604 CrossRef CAS PubMed.
- R. Tang, F. Ma, W. Li, S. Ouyang, Z. Liu and J. Wu, Int. J. Mol. Sci., 2017, 18, 1510 CrossRef PubMed.
- L. Zhang, L. Xia, L. Zhao, Z. Chen, X. Shang, J. Xin, M. Liu, X. Guo, K. Wu, Y. Pan and D. Fan, Carcinogenesis, 2015, 36, 390–399 CrossRef CAS PubMed.
- Q. Y. Chen, D. M. Jiao, L. Yan, Y. Q. Wu, H. Z. Hu, J. Song, J. Yan, L. J. Wu, L. Q. Xu and J. G. Shi, Mol. BioSyst., 2015, 11, 2290–2302 RSC.
- Z. Zheng, D. Yan, X. Chen, H. Huang, K. Chen, G. Li, L. Zhou, D. Zheng, L. Tu and X. D. Dong, PLoS One, 2015, 10, e0128751 CrossRef PubMed.
- H. Mataki, N. Seki, T. Chiyomaru, H. Enokida, Y. Goto, T. Kumamoto, K. Machida, K. Mizuno, M. Nakagawa and H. Inoue, Int. J. Oncol., 2015, 46, 1039–1050 CrossRef CAS PubMed.
- K. Koshizuka, T. Hanazawa, I. Fukumoto, N. Kikkawa, R. Matsushita, H. Mataki, K. Mizuno, Y. Okamoto and N. Seki, J. Hum. Genet., 2017, 62, 113–121 CrossRef CAS PubMed.
- B. H. Choi, D. Y. Ryu, I. G. Ryoo and M. K. Kwak, Oncotarget, 2017, 8, 107188–107205 Search PubMed.
- G. J. Weiss, L. T. Bemis, E. Nakajima, M. Sugita, D. K. Birks, W. A. Robinson, M. Varella-Garcia, P. A. Bunn Jr, J. Haney, B. A. Helfrich, H. Kato, F. R. Hirsch and W. A. Franklin, Ann. Oncol., 2008, 19, 1053–1059 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |