
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrophase separation of a quadruple hydrogen bonding supramolecular polymer: effect of the steric hindrance of the ureido-pyrimidone on their viscoelasticity†
Lei Kana,
Peng Zhanga,
Hongkun Jianga,
Shuai Zhanga,
Zhengdao Liua,
Xinyue Zhanga,
Ning Ma *a,
Dengli Qiub and
Hao Wei*a
*a,
Dengli Qiub and
Hao Wei*a
aKey Laboratory of Superlight Materials and Surface Technology, Ministry of Education & College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, China. E-mail: weihao7512@126.com; nma@hrbeu.edu.cn
bBruker (Beijing) Scientific Technology Co., Ltd., Beijing 100081, China
First published on 18th March 2019
Abstract
Supramolecular polymers based on 2-ureido-4[1H]-pyrimidone (UPy) units with extremely high dimerization constants and adjustable properties have received significant attention. In this work, we attempt to discuss the relationship between the micro-phase separation and the viscoelastic properties of the supramolecular polymers. For this reason, polymers with different UPy moieties structures and different UPy moieties contents were prepared and studied. It was found that the UPy moiety with little hindrance at the six-position of the pyrimidone could self-assemble into a nano-fiber structure and the degree of the micro-phase separation increased with the content of the UPy moiety. With the enlargement of the steric hindrance of the six-position of the pyrimidone, the nano-fiber structure gradually disappeared, meaning the degree of the micro-phase separation decreased astonishingly. More importantly, with the degree of the micro-phase separation increased, the storage modulus or the elasticity modulus increased exponentially and the Tm and the loss modulus area increased linearly. These results would lead a new way to study and develop novel polymeric materials.
Introduction
Supramolecular polymers are linear chains or networks of low molar mass monomers connected together by non-covalent highly directional and reversible interactions.1–8 As a novel polymeric material, the supramolecular polymer has been studied intensively in the last 20 years and many different kinds of supramolecular self-assembly structures have been synthesized by chemists.9–18 The non-covalent interactions that can be reversibly broken and can be under thermodynamic equilibrium will bring additional features compared to normal polymers, potentially leading to new properties, such as improved processing,19–23 self-healing behavior24–28 or stimuli responsiveness29–34 and so on.Hydrogen bonding has been proven to be especially suitable as a noncovalent interaction for supramolecular synthesis because the directionality and saturation of hydrogen bonds allow the formation of strong and specific recognition structures which could perfectly control and simplify the self-assembly process of the monomers better than that of the other interactions.35–38 According to the kinetics and half-life study of the process of self-assembly, a high association constant is strongly required in preparing the polymer-like materials.39 2-ureido-4[1H]-pyrimidone (UPy) units with extremely high dimerization constants (6 × 107 M−1 in CHCl3) have the advantage both in commercial accessibility and the experimental availability in preparing new supramolecular structure. The high dimerization constant could turn the low-molecular-weight supramolecular monomer into those of a high-molecular-weight linear chains or network polymer-like materials which showed viscoelastic behavior and a glass transition temperature.40
The micro phase separation, which exists in most of the copolymer, is usually considered to have effect on the viscoelasticity properties and employed to describe the mechanical properties of the materials.41–43 But due to the uncontrollable and the nondeterminacy of the polymers chains, the microphase separation could not be formed controllably and quantitatively in the traditional polymer materials.41,42,44 Fortunately, with combination of the π–π interaction of the UPy ring and the additional lateral hydrogen bonding formed between the ureido, the simple UPy structure could stack into well-defined nanofibers which will result in obvious microphase separation and yield materials with sharp thermal transitions.45,46
Based on this phenomenon, a lot of attempts have been made on changing the substitution groups at six-position of the pyrimidone to regulate the aggregation of the nanofibers, and then to control the performance of the polymer. Meijer and his coworker prepared a series of UPy with different C6 position groups and coupled with the amorphous PEBs via ureido to get a viscoelastic material. It was found that all the polymer casted from CHCl3 showed the nanofiber structures under AFM though the groups at six-position of the pyrimidone were different, and the aggregation mechanism was discussed in details with a combination of characterization methods.45 Sijbesma and his coworker investigated the aggregation of the UPy dimer with bulky adamantyl substitution group in C6 position and the AFM and rheology tests confirmed the absence of the nanofiber structure in polymer.47 However, the effect of the C6 position groups on the viscoelastic property of UPys supramolecular polymer was still unclear. Inspired by the work studied before, in this paper, amorphous hydrogenated polybutylene telechelic polymer functionalized with different structure UPys was synthesized (Scheme 1) in which the substituent groups at six-position of pyrimidone were methyl (1), propyl (2), iso-propyl (3), tert-butyl (4) and heptyl (5) group with the steric hindrance gradually increased. Excluded the other influences, these selected groups permit us to investigate the influence of steric hindrance on the aggregation of bis-(UPy-urethan) polymers. The self-assembly of the UPy-urethan moieties and the viscoelastic properties of polymers 1–5 was investigated by FT-IR, 1H NMR, AFM, DSC, SAXS, dynamic mechanical analysis (DMA) and rheological test and showed that the aggregation behavior on the molecular scale can be directly linked to their macroscopic properties.
 | ||
| Scheme 1 The preparation process of the supramolecular polymer, details refer to the ESI.† Supramolecular polymer prepared with the same telechelic polymer but the different six-position. | ||
Experimental
Materials and methods
All reagents and solvents were purchased from commercial sources and used as received unless otherwise noted. 2-Amino-4-hydroxy-6-methylpyrimidine was purchased from Aldrich, United States. Hexyldiisocyanate was obtained from Bayer AG, Germany. Hydroxyl-terminated polybutadiene was purchased from QiLong Chemical Ltd., China. Hydroxyl-terminated hydrogenated polybutadiene was bought from Cray Valley, USA. Other reagents were bought from Aladdin chemical Ltd., China.Chloroform and ethyl acetate were dried over 4 Å mol sieves for at least 1 day. Triethylamine was dried over KOH pellets. Hydrogenated hydroxyl terminated polybutylene was heated at 100 °C in vacuum for dehydration.
Synthesis of linear supramolecular polymers was adapted as reported by Meijer et al. with a little modification.42 The supramolecular polymer solution was cast into a PTFE mould and dried at room temperature for 72 hours in dry atmosphere to obtain the thin film with the thickness of 1 mm. All the samples were annealed at 40 °C for 7 days before characterization.
Atomic Force Microscopy (AFM) measurements were performed on a Bruker Dimension Icon using RTESPA tips in the tapping mode. Polymer films were made by drop casting a 1 mg mL−1 solution in chloroform on pre-stripped mica disk and subsequent evaporation to air for at least 1 hour, followed by annealing in vacuo over night at 40 °C. The sample was then annealed at 40 °C for 7 days before characterization.
Fourier Transform Infrared (FTIR) spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR spectrometer. Polymer films were made by drop casting a 1 mg mL−1 solution in chloroform on ZnSe disk and subsequent evaporation to air for at least 1 hour.
1H-NMR spectra were recorded on a 500 MHz NMR (Bruker AVANCE, 500 MHz for 1H-NMR) under room temperature.
Dynamic Mechanical Analysis (DMA) tests were carried out on a TA Q800 instrument. The samples were cut into about 6 mm × 30 mm rectangular and fastened on the tension clamps. The tests were performed from −90 °C to 20 °C with a heating rate of 3 °C min−1.
Differential Scanning Calorimetry (DSC) measurements were performed on a TA Q200 apparatus between −80 and 150 °C at a rate of 10 °C min−1 with a sample weight of 5–10 mg. For monitoring the fibril formation, the samples were heated above the melting point and cooled back to −80 °C at 10 °C min−1 and followed by the reheating to 150 °C. Integration of the melting endotherm was performed with the TA Instruments Universal Analysis software.
Small angel X-ray scattering tests were carried out on an Anton Paar SAXS MC2 equipment. The supramolecular polymer about 1 mm thickness was placed on the sample holder and tested with a q value from 0.1 to 8 nm−1.
Rheology tests were conducted on a TA Discovery HR-2 rheometer. The strain sweep tests were carried out firstly to figure out the linear viscoelastic region. After that the frequency sweep tests were carried out for each sample from 0.1 to 200 Hz with a 0.3% strain. All the experiments were carried out on a 20 mm parallel plate with 1 mm gap at 75 °C.
Synthesis of the supramolecular polymers 1–5
The preparation procedures and details of the samples were listed in the ESI.†Result and discussion
According to the literature report,42 hydrogenated polybutylene was functionalized with both lateral urethane hydrogen bonding motifs and end-to-end functionalities UPy moieties, which were called polymer 1–5 in this paper (Scheme 1).The supramolecular polymers with the same telechelic polymers but different chemical structure of UPy moieties were prepared to investigate the effect of the steric hindrance of the UPy moieties on the viscoelastic properties. It was designed that the increase of the steric hindrance of the C6 position group will cause the change of the aggregation of the UPy dimers stack therefore affect the degree of the microphase separation which could affect the properties of the supramolecular polymer distinctly. The phase separation was observed via atomic force microscopy.
Fig. 1 showed the AFM image of the supramolecular polymer 1–5. Different from the previous report,46 polymer 1 exhibited a distinct, well defined nano-fibres structure in Fig. 1, indicating that the supramolecular polymer 1, where the methyl is at six-position, had obvious phase separation, which could also be verified by the transmission electron microscope (Fig. S1†) and small angle X-ray scattering (Fig. 2a). FT-IR and 1H-NMR data (ESI) indicated that the UPy moiety formed into quadruple hydrogen bonding system both in synthon and in polymer (solid film and in chloroform). Therefore, the formation of the nano-fibre structure of UPy dimer could be explained by the mechanism we investigated previously that the UPy-urethan moiety self-assembled into distinct nano-fibres with the π–π interaction of the conjugated structure and the lateral hydrogen bonding formed between urethan.48
 | ||
| Fig. 1 AFM phase images of supramolecular polymer 1–5 from (a) to (e). The nano-fibre structure disappeared gradually with the steric hindrance of the substitution at six-position of the pyrimidone increased. All the scale bar is 100 nm. | ||
 | ||
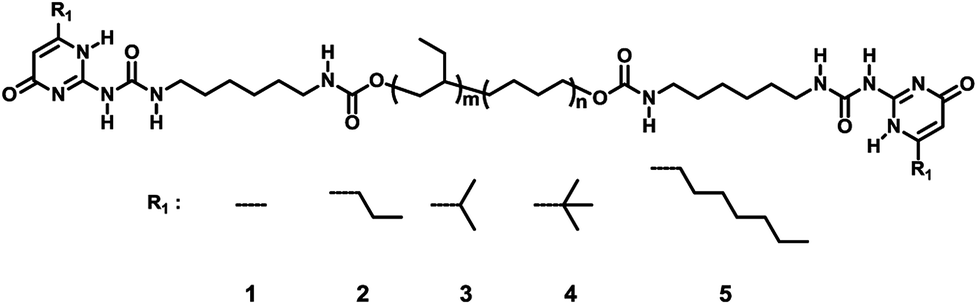
| Fig. 2 (a) The small angle X-ray scattering patterns and (b) DSC curves of supramolecular polymer 1 to 5, endo up. The peak of the SAXS patterns shifted to high scatter vector with the increase of steric hindrance increased. The melting peak of the supramolecular polymer shifted to lower temperature and the melting enthalpy decreased dramatically with the increase of the steric hindrance. | ||
The nanofiber hard domains of polymers 1 and 2 appeared more distinct and densely packed compared with the nanofibers of polymers 3–5. Fig. 1c–e showed the AFM images of supramolecular polymer 3–5. As the simplest substitution group, the methyl has little steric hindrance which would give no hinder to the formation of the nano-fibre stacks.48 With the increase of the steric hindrance of the substitution group at six-position, the distinct and well-defined boundary of the nano-fibre structure gradually became vague (Fig. 1c–e), which indicated that the degree of phase separation decreased dramatically. Meanwhile the amount and length of the fibrillar aggregates decreased, even the polymer 5 with the heptyl at six-position lack any distinct features in height or phase mode scan images. Apparently, the nanofiber formation is highly affected by changing the substituent at the six-position of the isocytosine.
Though the AFM could observe the microphase separation at the surface of the film, the structure inside of the materials was still unclear. Hence the small angle X-ray scattering (SAXS) and differential scanning calorimetry (DSC) tests were carried out to investigate the aggregation of the UPy dimer inside the solid polymer.
Fig. 2a demonstrated the SAXS patterns of the polymer 1 to 5. Polymer 1 had the lowest q peak value, and a clear asymmetric broadening of the peak was observed. This was a sign that motifs with larger diameter developed, which was related to the aggregation of the UPy-urea end groups into the nanofibers. The major difference between each polymer was the position and the intensity of the q peak, which was consistent with the different crystallization rates of the UPy moieties affected by the steric hindrance. The scattering peak shifted to the higher q value from polymer 1 to 5, meaning the π–π stack structure of the UPy moieties trended to be disordered and the d spacing decreased gradually. The absence of the scattering peak from polymer 4 and 5 indicated that the microphase separation disappeared strikingly. With the enlargement of the steric hindrance of the six-group in the UPy moiety, the large substitution would hinder the contact of the UPy dimers hence broaden the distance between the parallel pyrimidone and the spacing of the vertical π–π stack, which could lead to the loss of the nano-fibre structure in the solid state of the polymer. The disordered UPy moiety in the solid could induce the change both in thermal and mechanical properties of the material, which was discussed below.
The AFM images could demonstrate the phase-separation motif of the UPy stacks intuitively, the differential scanning calorimetry (DSC) could measure the degree of phase-separation quantitatively. The melting of the UPy nano-fibre in the heating process was recorded by DSC tests and the enthalpy-change of the melting peak was calculated. Fig. 2b showed melting peak of the DSC curves of different supramolecular polymer 1–5 and the thermal properties of polymers 1–5 were listed in Table S1 (ESI†). All polymers exhibited a well-defined Tg at about −48 °C. The endo peak of the DSC test was the character melting of the UPy stack hard-segment and the value of melting enthalpy could represent the content of the UPy stack hard-segment. Polymer 1 and 2 showed a distinct melting peak at about 63 °C in the first heating stage. There was not any crystallization exotherm appeared during cooling period, meanwhile all the polymers samples did not show an immediate reappearance of the melting endotherm upon reheating due to the high viscosity of the telechelic PEBs. Because of the annealing of the samples under 40 °C for 7 days (ESI†), the UPy-urethan end group could phase separated and crystallized into nanofibers within the hydrogenated polybutylene matrix completely. In order to determine the melting enthalpy, the melting transition of the first cycle of heating was employed. This crystallization, however, was highly affected by the substituent at the six-position of the UPy-unit. Integrate the peak area of the melting endotherm could represent the crystallization degree of the nano-fibre structure. As shown in Table S1 (ESI†), polymer 1 had a highest constant pressure melting enthalpy (7.8 J g−1), while the polymer 2 came the second (6.1 J g−1). Crystallization of the nanofibers showed a remarkable dependence on the substituent on the six-position. For polymer 3–5 it was almost impossible to crystallize. A very faint melting peak was observed of polymer 3 and for polymer 4 and 5 which was hardly to distinguish. In addition, in Table S1,† it was clearly demonstrated that the Tm fell gradually, indicating that the interaction of UPy moiety in the hard-segment vanished. Considering that the supramolecular polymer was connected via the noncovalent quadruple hydrogen bond, the degree of polymerization was dynamic in solution related to the concentration of the monomer which was investigated by Meijer et al.7 Because the supramolecular polymer had the similar molecular weights, the dynamic molecular weight of the supramolecular polymer in solid state was close to each other. So, the supramolecular polymer had the approximate contents of the UPy moiety and the effect of the molecule weight and viscous performance of the polymer chains could be neglected. These distinct changes should only be induced by the different molecule structure of the UPy end groups and hence by the way they aggregate.
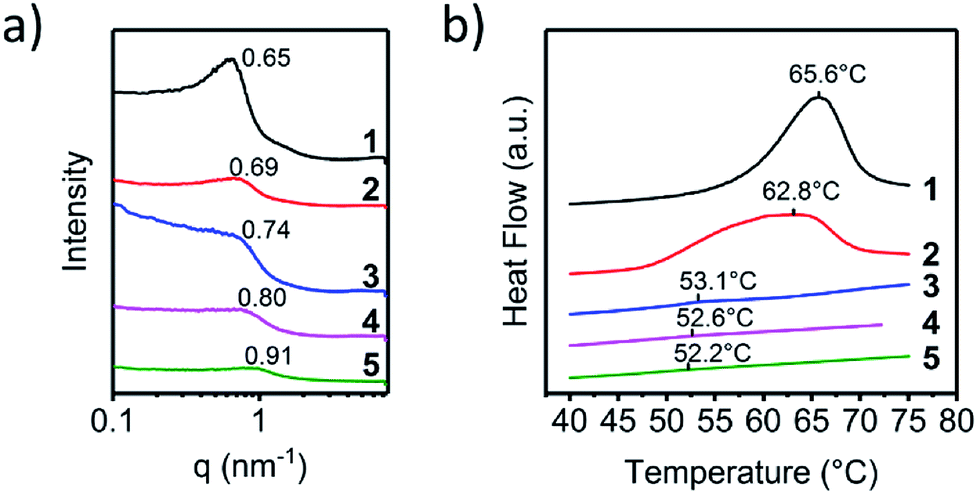
The dynamic mechanical analysis (DMA) and rheology tests were performed to characterize the effect caused by the molecular differences in the UPy end groups on the viscoelastic properties. Fig. 3a and b showed the character DMA curves of polymer 1–5. Obvious difference of the storage modulus could be observed in Fig. 3a. The storage modulus descended dramatically from polymer 1 to polymer 5. Because of the similar molecular weights of the polymers, this effect should originate from the changes of the formation of nano-fibre hard-segment induced by the molecular differences of the UPy end groups. The UPy with the pyrimidone structure acted as the hard-segment could enhance the strength of the material and hence promote the storage modulus. The storage modulus at −85 °C was selected and plotted in Fig. 3c. The black dot is the original data and the red line is the fitting curve. As shown in Fig. 3c, the relation between the storage modulus and melting enthalpy (in another way representing the amount of the hard-segment nano-fibres) was an exponentially growth curve. The storage modulus increased dramatically at the beginning with the increasement of the amount of the nano-fibres and gradually became gentle and tended to a certain asymptotic value.
 | ||
| Fig. 3 Viscoelasticity analysis of polymer 1–5. (a) E′ and (b) E′′ of polymer 1–5 from −90 °C to 20 °C. (c) The selected value at −85 °C were plotted and fitted. (d) The maximum values and (e) the peak areas of loss modulus of polymer 1–5 and (f) Tm versus melting enthalpy were plotted and fitted linearly. (g) G′ and (h) G′′ of the rheological test of supramolecular polymer 1–5. | ||
The loss modulus represented the viscous performance of the material, which could be affected by series ingredient such as the length, the flexibility the interaction of the polymer chains and so on. The differences could be obviously observed that loss modulus decreased gradually from polymer 1–5 (Fig. 3b). The maximum of the loss modulus increased linearly to the melting enthalpy as well as the content of UPy moiety hard-segment, (Fig. 3d) indicating that the UPy stack and the nano-fibre could provide additional inter-friction which could cause more internal friction to dissipate mechanical energy.
In order to determine the influence of UPy stack induced internal friction on the loss modulus in the whole process of the glass transition, the areas of the loss modulus peak between −60 °C to 20 °C were calculated and exhibited in Fig. 3e. Interesting result was discovered that the calculated peak area of loss modulus also increased linearly to the melting enthalpy just like the maximum of loss modulus did. The peak area of the loss modulus could represent the total energy dispersed in the test process. With the content of UPy hard-segment increased, there was more energy dissipated from the internal-friction raised from the interaction of the UPy nano-fibres. The rheology tests were carried out to determine the UPy interactions in the melting state of the supramolecular polymers. The small amplitude oscillatory shear tests, that is, frequency sweep, were carried out in the linear viscoelastic region. The ratios of the slope of G′ to G′′ of polymer 1 and polymer 2 were about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at low frequency, indicating that there were significant interactions between supramolecular polymer (Fig. S2, ESI†). With the steric hindrance of the six-position increased, the ratio of the slope of G′ to G′′ gradually changed to 1:2, indicating that the interaction between the UPy dimer strikingly weakened. Fig. 3g and h showed the storage modulus (G′) and loss modulus (G′′) against angular frequency for the different supramolecular polymer samples. The modulus exhibited strong frequency-dependent behaviour, indicating that the supramolecular polymers showed pronounced viscoelastic behaviour. Supramolecular polymers showed a broad rubber plateau with a storage modulus of about 106 Pa at high frequencies. At lower frequencies a sharp viscoelastic transition towards the terminal regime is observed.
:1 at low frequency, indicating that there were significant interactions between supramolecular polymer (Fig. S2, ESI†). With the steric hindrance of the six-position increased, the ratio of the slope of G′ to G′′ gradually changed to 1:2, indicating that the interaction between the UPy dimer strikingly weakened. Fig. 3g and h showed the storage modulus (G′) and loss modulus (G′′) against angular frequency for the different supramolecular polymer samples. The modulus exhibited strong frequency-dependent behaviour, indicating that the supramolecular polymers showed pronounced viscoelastic behaviour. Supramolecular polymers showed a broad rubber plateau with a storage modulus of about 106 Pa at high frequencies. At lower frequencies a sharp viscoelastic transition towards the terminal regime is observed.
A plateau in the storage modulus at low-frequency could be observed of polymer 1 and polymer 2, which was caused by the reversibility of the association of the quadruple hydrogen bonding units. However, the presence of a plateau could also be an indication that small clusters of hydrogen-bonded units were present. These clusters acted as hard domains and physical crosslinks in supramolecular polymers, which corresponding to the nano-fibres structures in the solid. As shown in Fig. 3g and h, both storage modulus and loss modulus enhanced with the increasing of the degree of microphase separation, which was agreed with the result of the dynamic mechanical analysis tests.
Table S2† listed the steric energy of the UPy moieties with different substitutions. The steric energy could represent the steric effect between the molecules, which could affect the aggregation of nano-fibre hence influence the properties of the polymers due to the repulsive forces between overlapping electron clouds. Steric hindrance is a consequence of steric effects. As expected, an increasement of steric energy versus the enlargement of the steric hindrance at six-position substitutions was observed, which meant the strong repulsive forces effect took place between UPy moieties. The steric hindrance could block nearby UPy dimers hence weaken the interaction between the UPy moieties, which could result in the disappearance of the nano-fibres structure and the reduction of the microphase separation.
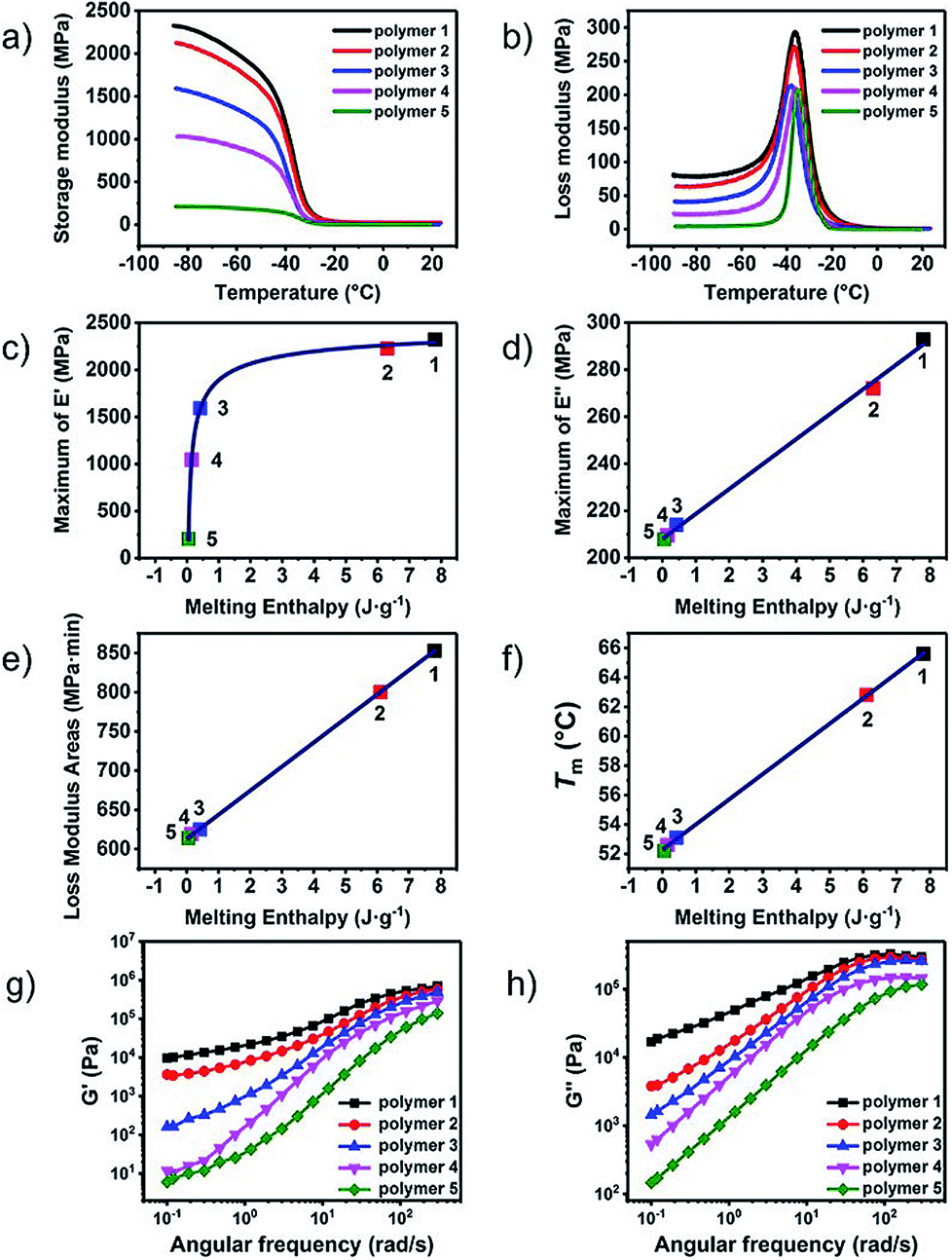
In order to study the influence of steric hindrance on the properties of the supramolecular polymers quantitatively, the thermal and mechanical properties were plotted against the steric energy in Fig. 4. An amazing discovery was found that the maximum of storage modulus, decreased with the steric energy linearly. Due to the same telechelic polymer but the different UPy moieties, the steric hindrance had great effect on the properties of the supramolecular polymers both in thermal and mechanical aspect. Lorentz growth curves were plotted for maximum of loss modulus, loss modulus areas and melting point. It could be found that when the steric energy was more than about 2.5 kcal mol−1, the curves started to descend dramatically until it reached about 4 kcal mol−1. The curves more than 4 kcal mol−1 showed an almost indiscernibly change, meaning that when the substitution is big enough or the steric hindrance is large enough, there would not be obvious effect on the performance of the supramolecular polymer.
 | ||
| Fig. 4 The steric energy against (a) maximum of E′, (b) maximum of E′′, (c) loss modulus areas and (d) melting point of supramolecular polymer 1 to 5. | ||
Based on the experimental and theoretical study on the supramolecular polymer with different substitutions at the six-position of the pyrimidone, the effect of the steric hindrance on the properties of the polymer was uncovered and discussed. The UPy moieties with little steric hindrance like methyl and propyl could assemble into nano-fibre structures hard-domains which could be observed and confirmed by series characterizations (polymer 1 and 2). The nano-fibres formed by the stack of the UPy moieties actually acted as the dynamic weak crosslinking structure which could turn the linear chains to the network polymeric materials. Here a tough polymeric material was obtained from a viscous liquid raw material.
The nano-fibres hard segment could not only enhance the strength of the materials but could also provide more internal-friction to dissipate the mechanical energy in the glass state. The external vibration applied on the materials could induce the relative sliding of the nano-fibres with each other, which could provide more internal friction. The internal friction provided by the relative motion of nano-fibres could dissipate more mechanical energy which could increase the areas of the loss modulus hence promoted the viscous performance of the supramolecular polymer.
Though the large steric hindrance induced by the big substitution at the six-position of the pyrimidone (iso-propyl, tert-butyl, and heptyl) would not affect the specific quadruple hydrogen bonding interaction, it could obstruct the UPy moieties and keep the UPy dimers from aggregating into the nano-fibre hard-domains (polymer 3 to 5). Though the polymer with the large steric hindrance showed a viscoelasticity behaviour, a strikingly decline of the performance both in thermal and mechanical properties was observed compared to polymer 1 and 2.
As a supplementary, the supramolecular polymers, bis-(UPy-urethan) functionalized amorphous polybutylene with different molecular weight, was synthesized to evaluate the effect of contents of the UPy moieties (Scheme S2,† polymer 6 to 9). The UPy moiety with the methyl at the six-position of the pyrimidone was employed to ensure the nano-fibre aggregation formed in the polymer system. The nano-fibre hard domains were confirmed via AFM (Fig. S3†), SAXS (Fig. S4†) and DSC (Fig. S5†) experiments. It could be observed that the amount of the nano-fibres hard domains increased with the contents of the UPy moieties, meaning that the rise of microphase separation degree. Furthermore, the DMA and rheology tests confirmed the effect of the UPy contents on the viscoelasticity performances. Be similar to polymer 1 to 5, the elasticity modulus increased exponentially and the loss modulus increased linearly versus the change of UPy contents (Fig. S6–S8†). In general, both of the thermal and mechanical properties of the supramolecular polymer enhanced with the increase of the degree of microphase separation.
Conclusions
In summary, a series of supramolecular polymers with different structure and different content of the UPy moieties were prepared. The effect of steric hindrance of C6 position groups of the pyrimidone on the aggregation of the UPy dimer was studied and the relationship between the UPy structure and the viscoelasticity was uncovered and discussed. The substitution group at six-position of the pyrimidone could affect the formation of the nano-fibers structure dramatically. The supramolecular polymer with little steric hindrance at the C6 position groups had the obvious nano-fiber structure which meant the obvious microphase separation formed in the polymer. The nano-fibers structures were the hard-segment in the polymer and the increase of the content of the nano-fibers structures meant the increase of the phase separation. The degree of the micro-phase separation in the polymer increased with the content of the UPy moieties. With the enlargement of the steric hindrance, the nano-fibers structure disappeared, indicating that the micro-phase separation vanished in the supramolecular polymer. The microphase separation could promote viscoelasticity performance of the supramolecular polymer. With the degree of the micro-phase separation increased, the hard-segment could enhance the storage modulus exponentially while the Tm and the loss modulus area increased linearly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21374009), and the Fundamental Research Funds of the Central University (HEUCFJ161003).References
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- M. Cheng, G. Zhu, L. Li, S. Zhang, D. Zhang, A. J. C. Kuehne and F. Shi, Angew. Chem., Int. Ed., 2018, 57, 14106–14110 CrossRef CAS.
- J. Liu, Y. Lan, Z. Yu, C. S. Y. Tan, R. M. Parker, C. Abell and O. A. Scherman, Acc. Chem. Res., 2017, 50, 208–217 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- W. Lu, X. Le, J. Zhang, Y. Huang and T. Chen, Chem. Soc. Rev., 2017, 46, 1284–1294 RSC.
- W. Zheng, L. Chen, G. Yang, B. Sun, X. Wang, B. Jiang, G. Yin, L. Zhang, X. Li, M. Liu, G. Chen and H. Yang, J. Am. Chem. Soc., 2016, 138, 4927–4937 CrossRef CAS PubMed.
- Z. Zhou, X. Yan, T. R. Cook, M. L. Saha and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 806–809 CrossRef CAS PubMed.
- D.-X. Chen, Y.-L. Sun, Y. Zhang, J.-Y. Cui, F.-Z. Shen and Y.-W. Yang, RSC Adv., 2013, 3, 5765–5768 RSC.
- G. Ju, F. Guo, Q. Zhang, A. J. C. Kuehne, S. Cui, M. Cheng and F. Shi, Adv. Mater., 2017, 29, 1702444 CrossRef PubMed.
- G. Ju, M. Cheng, F. Guo, Q. Zhang and F. Shi, Angew. Chem., Int. Ed., 2018, 57, 8963–8967 CrossRef CAS PubMed.
- L. R. Hart, S. Li, C. Sturgess, R. Wildman, J. R. Jones and W. Hayes, ACS Appl. Mater. Interfaces, 2016, 8, 3115–3122 CrossRef CAS PubMed.
- X. Zhai, Y. Ma, C. Hou, F. Gao, Y. Zhang, C. Ruan, H. Pan, W. W. Lu and W. Liu, ACS Biomater. Sci. Eng., 2017, 3, 1109–1118 CrossRef CAS.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- A. M. Pekkanen, R. J. Mondschein, C. B. Williams and T. E. Long, Biomacromolecules, 2017, 18, 2669–2687 CrossRef CAS PubMed.
- C. Loebel, C. B. Rodell, M. H. Chen and J. A. Burdick, Nat. Protoc., 2017, 12, 1521–1541 CrossRef CAS PubMed.
- G. M. L. van Gemert, J. W. Peeters, S. H. M. Sontjens, H. M. Janssen and A. W. Bosman, Macromol. Chem. Phys., 2012, 213, 234–242 CrossRef CAS.
- F. Herbst, D. Dohler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima and A. Harada, Macromol. Rapid Commun., 2016, 37, 86–92 CrossRef CAS PubMed.
- C. Li, C. Wang, C. Keplinger, J. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. You and Z. Bao, Nat. Chem., 2016, 8, 619–625 Search PubMed.
- E. Ogliani, L. Yu, I. Javakhishvili and A. L. Skov, RSC Adv., 2018, 8, 8285–8291 RSC.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- C. X. Xiong, M. R. Xie and R. Y. Sun, Macromol. Rapid Commun., 2017, 38, 8 CrossRef PubMed.
- O. Ikkala and G. ten Brinke, Science, 2002, 295, 2407–2409 CrossRef CAS PubMed.
- K. Shikinaka, S. Mori, K. Shigehara, H. Masunaga and T. Sakai, RSC Adv., 2016, 6, 52950–52956 RSC.
- H. K. Lee, H. Lee, Y. H. Ko, Y. J. Chang, N. K. Oh, W. C. Zin and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 2669–2671 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- T. Park, S. C. Zimmerman and S. Nakashima, J. Am. Chem. Soc., 2005, 127, 6520–6521 CrossRef CAS PubMed.
- J. Hirschberg, L. Brunsveld, A. Ramzi, J. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS PubMed.
- S. H. M. Sontjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- K. Yamauchi, A. Kanomata, T. Inoue and T. E. Long, Macromolecules, 2004, 37, 3519–3522 CrossRef CAS.
- G. H. Fredrickson and R. G. Larson, J. Chem. Phys., 1987, 86, 1553–1560 CrossRef CAS.
- B. M. Yavitt, Y. Gai, D.-P. Song, H. H. Winter and J. J. Watkins, Macromolecules, 2017, 50, 396–405 CrossRef CAS.
- C. D. Han and J. K. Kim, Polymer, 1993, 34, 2533–2539 CrossRef CAS.
- C. Y. Ryu, M. S. Lee, D. A. Hajduk and T. P. Lodge, J. Polym. Sci. B Polym. Phys., 1997, 35, 2811–2823 CrossRef CAS.
- W. P. J. Appel, G. Portale, E. Wisse, P. Y. W. Dankers and E. W. Meijer, Macromolecules, 2011, 44, 6776–6784 CrossRef CAS.
- H. Kautz, D. J. M. van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4265–4267 CrossRef CAS.
- D. J. M. van Beek, A. J. H. Spiering, G. W. M. Peters, K. te Nijenhuis and R. P. Sijbesma, Macromolecules, 2007, 40, 8464–8475 CrossRef CAS.
- B. Li, L. Kan, X. Zhang, J. Li, R. Li, Q. Gui, D. Qiu, F. He, N. Ma, Y. Wang and H. Wei, Langmuir, 2017, 33, 8493–8502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures of supramolecular polymer 1 to 9, morphology, thermal, dynamic mechanical analyses and rheology characterizations of polymer 5 to 9. See DOI: 10.1039/c8ra08861f |
| This journal is © The Royal Society of Chemistry 2019 |