DOI:
10.1039/C9RA00577C
(Paper)
RSC Adv., 2019,
9, 8894-8904
Synthesis of C4-alkynylisoxazoles via a Pd-catalyzed Sonogashira cross-coupling reaction†
Received
23rd January 2019
, Accepted 6th March 2019
First published on 18th March 2019
Abstract
A Pd-catalyzed Sonogashira cross-coupling reaction for the synthesis of C4-alkynylisoxazoles from 3,5-disubsitituted-4-iodoisoxazoles and terminal alkynes was described, which could afford the corresponding products with high yield (up to 98%). The results indicated that the steric effect from the group at the C3 position of the isoxazole had greater influence on the cross-coupling reaction than that from the group at the C5 position. In addition, the group at the C3 position of the isoxazole showed negligible electronic effects on the cross-coupling reaction. Furthermore, a gram-scale reaction of the Sonogashira coupling reaction was also investigated. Finally, a plausible mechanism for the Sonogashira coupling reaction was proposed.
Introduction
Isoxazole and its derivatives are important structural units of many molecules of biological interest.1 As a result, numerous synthetic approaches have been made to access the isoxazole core, including the [3 + 2] cycloaddition between alkynes/alkenes and nitrile oxides,2 intramolecular cycloisomerization of α,β-unsaturated oximes,3 condensation between hydroxylamine and 1,3-dicarbonyl compounds or α,β-unsaturated carbonyls.4 Many of the reported methods have two main limitations, (i) limitation to the preparation of disubstituted isoxazoles, and (ii) low regioselectivity.5 In order to overcome these limitations, direct functionalization of isoxazoles or 4-haloisoxazoles has been developed by many research groups.6
Transition-metal-catalyzed cross-coupling reactions have become one of the most prominent and reliable methods for the formation of carbon–carbon bonds.7 Among them, the Pd-catalyzed Sonogashira cross-coupling reaction between aromatic halides and terminal alkynes is very useful in the synthesis of carbo- and heterocycles, natural products, polymers, and molecular nanostructures.8 Using this powerful reaction, some trisubstituted isoxazoles were synthesized. Yamanaka and co-workers first reported that the Pd(PPh3)Cl2 catalyzed Sonogashira cross-coupling reaction of 3-substituted-5-methyl-4-iodoisoxazoles with terminal alkynes, affording three examples of 3-substituted-5-methyl-4-alkynylisoxazole products in good yields (up to 86% yield).6a In 2001, Kromann and co-workers explored that Pd(PPh3)Cl2 catalyzed Sonogashira cross-coupling reaction of 3-ethoxy-5-methyl-4-iodoisoxazole with phenylacetylene, giving product of 3-ethoxy-5-methyl-4-phenylethynylisoxazole for only one example in 58% yield.6c Larock and co-workers chose 3-(3,4,5-trimethoxyphenyl)isoxazole-5-methyl-4-iodoisoxazole as a substrate to react with seven terminal alkynes, giving the targeted products with the yields between 30% and 83%.6e In 2005, a series of 3-carboxamide-5-(2,4-dihydroxy-5-isopropylphenyl)-4-alkynylisoxazoles were synthesized through Pd(PPh3)Cl2 catalyzed Sonogashira cross-coupling reaction of an alkynyl moiety and an isoxazole scaffold as novel HSP90 inhibitors by Zhang and co-works.6l Recently, Guo and co-workers reported that Sonogashira cross-coupling reaction of 3-trifluoromethyl-5-phenyl-4-iodoisoxazoles with phenylacetylene in the presence of Pd(PPh3)2Cl2, affording the targeted product in 80% yield.9 However, none of the above researches studied the impact of the groups at C3 position and C5 position of isoxazole core on the Sonogashira cross-coupling reaction. Because of the decreased symmetry in isoxazole, when compared to furan, pyrrole, and benzene, reaction channels which are equivalent in furan, pyrrole, and benzene will lead to the different results in isoxazole. For examples, Fall and co-workers discovered that 3-carboxylate-5-methylisoxazole reacted with 1-(4-bromophenyl)ethan-1-one to afford the targeted product, but 4-carboxylate-5-methylisoxazole did not afford desired product.6f Coffman and co-workers investigated the effects of the groups of 3,5-, 4,5-, and 3,4-bis(2-nitrophenyl)isoxazoles on the reaction.10 Lately, Morita and co-workers reported intramolecular electrophilic aromatic substitution reaction only occurred at the 5-position of isoxazole rather than at the 3-position.11
Herein, we investigated the effect of the groups at C3 and C5 positions of the isoxazole on the Sonogashira cross-coupling reaction. Using 3,5-disubstituted-4-iodoisoxazoles and terminal alkynes as the substrates, we synthesized a series of 3,5-disubstituted-4-alkylnylisoxazole products.
Results and discussion
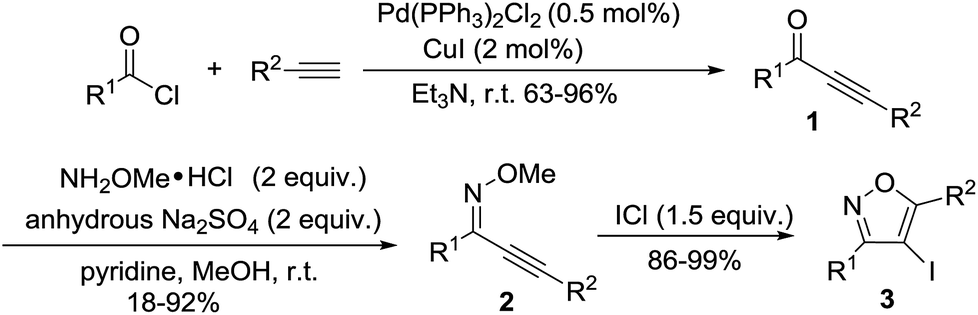
During our investigation, intermediate ynones 1 were prepared by the Sonogashira cross-coupling of an acid chloride and a terminal alkyne in the presence of Pd(PPh3)2Cl2. The O-methyl oximes 2 were prepared by the reaction of ynones 1 with methoxylamine hydrochloride in the presence of pyridine and anhydrous Na2SO4. The substrates of 3,5-disubstituted-4-iodoisoxazoles 3 were prepared through the electrophilic cyclization of O-methyl oxime 2 in the presence of ICl (Scheme 1).
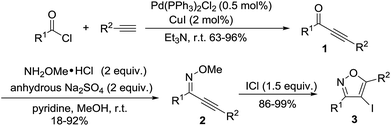 |
| | Scheme 1 Preparation of 3,5-disubstituted-4-iodoisoxazoles 3. | |
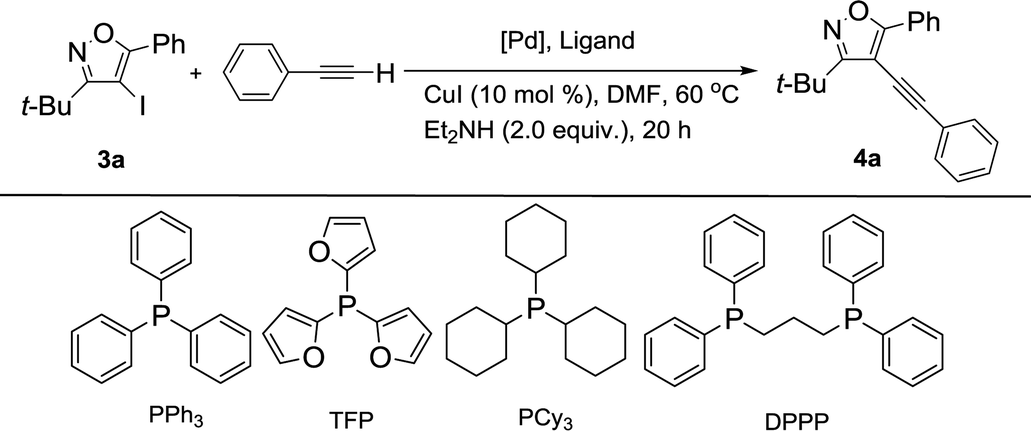
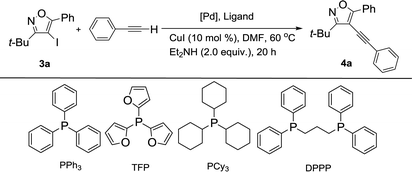
We chose 3-(tert-butyl)-4-iodo-5-phenylisoxazole (3a) with phenylacetylene as a model substrate to identify the feasibility of our process. When 3a reacted with phenylacetylene in anhydrous DMF at 60 °C in the presences of Pd(PPh3)2Cl2 (5 mol%), CuI (10 mol%), and Et2NH (2 equiv.) under nitrogen atmosphere, the desired product 4a was obtained in 26% yield after 20 h and the conversion of starting material 3a was 47% (Table 1, entry 1). To improve the reaction efficiency, three kinds of phosphine ligands, such as PPh3, PCy3 and DPPP, were examined for this reaction under similar condition (Table 1 entries 2–4). Unfortunately, the yield of targeted product 4a could only be up to 30% (Table 1, entry 2). Then, different palladium catalysts were screened for the reaction (Table 1, entries 5–9). Among them, 3a reacted with phenylacetylene to afford the corresponding product 4a in 60% yield and the conversion of 3a could be increased to 66% in the presence of Pd(acac)2 (Table 1, entry 9). Encouraged by this result, a series of phosphine ligands, such as PPh3, TFP, PCy3 and DPPP, were tested for this reaction under similar condition (Table 1, entries 9–12). The general trends in yields of product 4a related to different phosphine ligands were as followed: PPh3 > TFP > PCy3 > DPPP. The impact of the catalyst loading on the reaction was also investigated, and the yield of 4a was strongly influenced by the amount of catalyst loading (Table 1, entries 13 and 14). The yield of 4a was decreased with the amount of catalyst loading decreased to 3.5 mol% (Table 1, entry 13). However, a further increase the amount of catalyst loading did not improve the yield of 4a (Table 1, entry 14).
Table 1 Effects of catalyst precursors, ligands and catalyst loadingsa
|
| Entry |
Catalyst (mol%) |
Ligand (mol%) |
3a conv.b (%) |
4a yieldb (%) |
| Reaction condition: 3a (0.3 mmol, 1 equiv.), phenylacetylene (0.6 mmol, 2 equiv.), [Pd], ligand, CuI (10 mol%), Et2NH (2 equiv.), DMF (3 mL), 60 °C under N2 protection. Isolated yield. |
| 1 |
Pd(PPh3)2Cl2 (5) |
— |
47 |
26 |
| 2 |
Pd(PPh3)2Cl2 (5) |
PPh3 (10) |
49 |
30 |
| 3 |
Pd(PPh3)2Cl2 (5) |
PCy3 (10) |
48 |
28 |
| 4 |
Pd(PPh3)2Cl2 (5) |
DPPP (5) |
35 |
20 |
| 5 |
Pd(PPh3)4 (5) |
— |
43 |
26 |
| 6 |
Pd(OAc)2 (5) |
PPh3 (10) |
52 |
31 |
| 7 |
Pd(TFA)2 (5) |
PPh3 (10) |
24 |
18 |
| 8 |
Pd(MeCN)2Cl2 (5) |
PPh3 (10) |
50 |
29 |
| 9 |
Pd(acac)2 (5) |
PPh3 (10) |
66 |
60 |
| 10 |
Pd(acac)2 (5) |
PCy3 (10) |
42 |
26 |
| 11 |
Pd(acac)2 (5) |
TFP (10) |
56 |
46 |
| 12 |
Pd(acac)2 (5) |
DPPP (5) |
36 |
22 |
| 13 |
Pd(acac)2 (3.5) |
PPh3 (7) |
48 |
35 |
| 14 |
Pd(acac)2 (7.5) |
PPh3 (15) |
64 |
58 |
The effects of bases, solvents and temperatures were then examined (Table 2). No reaction took place without bases (not shown in Table 2). Moderate yields were obtained when Et2NH and Et3N were used as bases (Table 2, entries 1 and 2). As we added the base n-butylamine or DIPEA (N,N-diisopropylethylamine) to this reaction system, the reaction proceeded to give the targeted product 4a with low yields (25% and 18%, respectively) (Table 2, entries 3 and 4). The selection of solvents proved to have a dramatic influence on the reaction (Table 2, entries 1, 5–8). When the reaction was carried out in anhydrous DMF, a dipolar aprotic solvent, targeted product 4a could be obtained in 60% yield (Table 2, entry 1). Other solvents, such as anhydrous THF, MeCN, toluene and DCE, resulted in low yields (30%, 25%, 22% and 20%, respectively) (Table 2, entries 5–8). At last, the effects of temperatures on the reaction were investigated (Table 2, entries 1 and 9 and 10). The results showed that the optimum reaction temperature was 60 °C in terms of yield (Table 2, entry 1).
Table 2 Effects of bases, solvents and temperaturesa

|
| Entry |
Base |
Solvent |
Temp. (°C) |
3a conv.b (%) |
4a yieldb (%) |
| The reaction was carried out with 3a (0.3 mmol, 1 equiv.), phenylacetylene (0.6 mmol, 2 equiv.), CuI (10 mol%), base (2.0 equiv.) in the solvent at the stated temperature in the presence of Pd(acac)2 (5 mol%) and PPh3 (10 mol%) under N2 atmosphere. Isolated yield. |
| 1 |
Et2NH |
DMF |
60 |
66 |
60 |
| 2 |
Et3N |
DMF |
60 |
63 |
58 |
| 3 |
n-Butylamine |
DMF |
60 |
38 |
25 |
| 4 |
DIPEA |
DMF |
60 |
25 |
18 |
| 5 |
Et2NH |
THF |
60 |
41 |
30 |
| 6 |
Et2NH |
MeCN |
60 |
46 |
35 |
| 7 |
Et2NH |
Toluene |
60 |
33 |
22 |
| 8 |
Et2NH |
DCE |
60 |
26 |
20 |
| 9 |
Et2NH |
DMF |
30 |
38 |
22 |
| 10 |
Et2NH |
DMF |
90 |
82 |
40 |
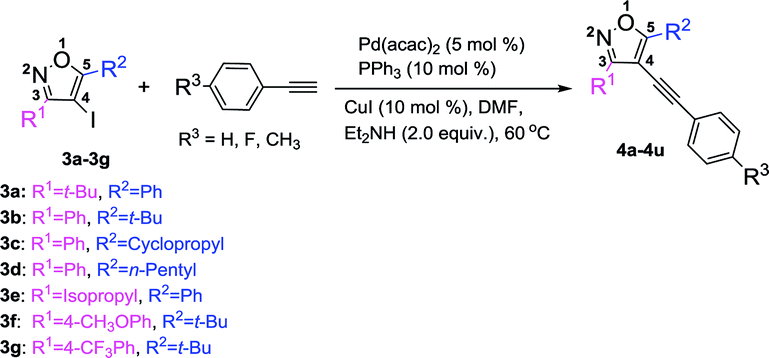
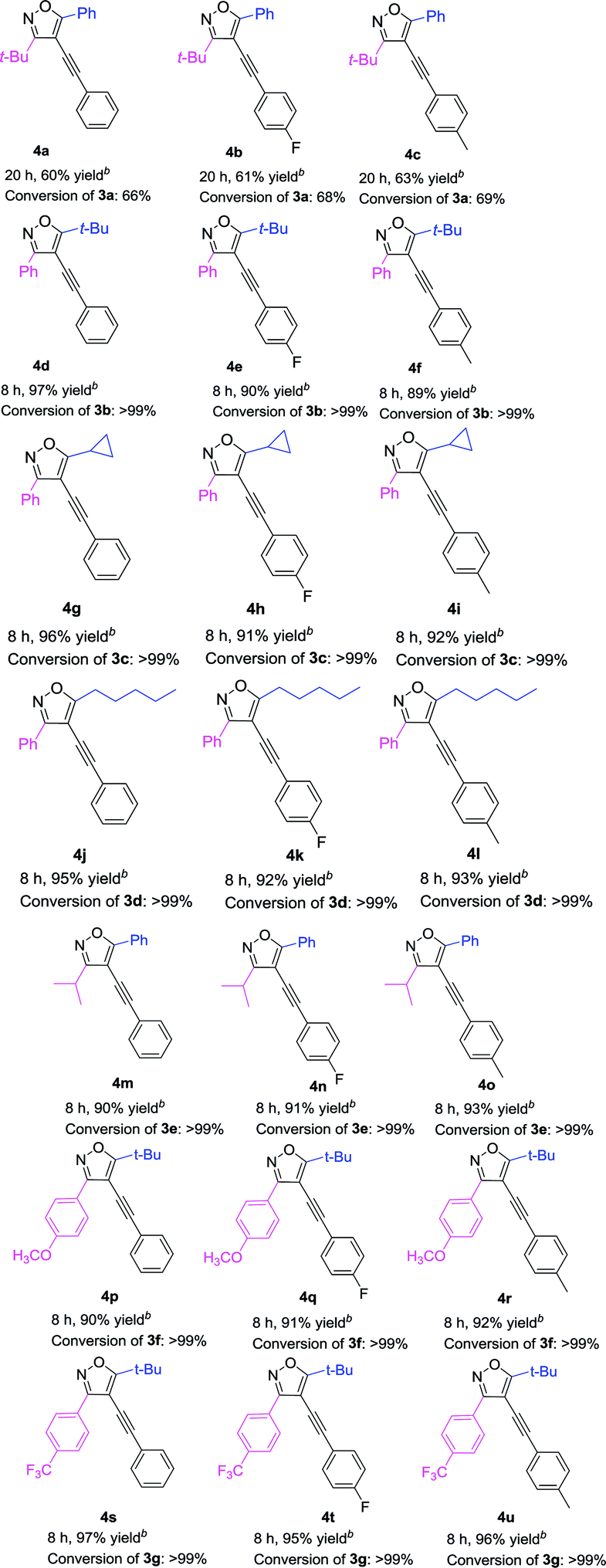
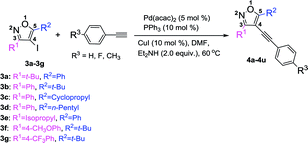
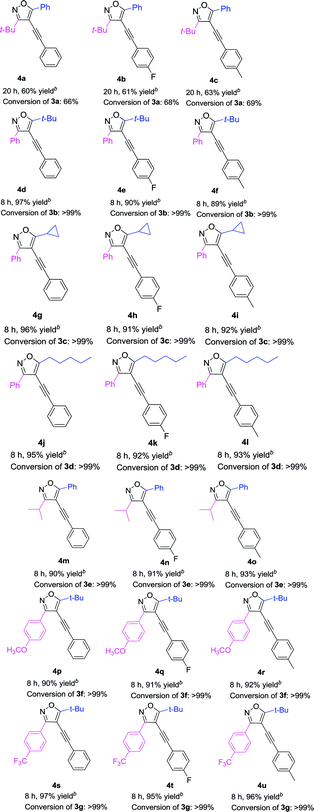
With the above optimized reaction condition in hand (5 mol% Pd(acac)2, 10 mol% PPh3, 10 mol% CuI, and 2 equiv. Et2NH in anhydrous DMF at 60 °C under N2 atmosphere), the effects of the substituent at 3 and 5 position of the isoxazole were examined (Table 3). We used the substrate 3a to react with three different kinds of terminal alkynes, affording the corresponding targeted products (4a, 4b, and 4c) with the yields of 60%, 61%, and 63%, respectively (Table 3). For these reactions, the substrate 3a couldn't be completely transferred, and its conversion was 66%, 68%, and 69%, respectively. When we used 3b as the substrate, with a phenyl group at the 3 position and a tert-butyl group at the 5 position to react with three kinds of terminal alkynes, the targeted products (4d–4f) were obtained in high yields (up to 97%) and almost full conversion of 3b was gained within 8 h. Such phenomena were very interesting, which encouraged us to investigate the impact of the groups at the 3 and 5 positions of 4-iodoisoxazoles on the reaction. First, we studied the impact of the group at the 5 position of 4-iodoisoxazoles on the reaction. When we used the substrate 3c (5 position was cyclopropyl group) or 3d (5 position was n-pentyl group) to react with the three kinds of terminal alkynes, all of the products (4g–4l) could be obtained in high yields, accompanied by almost full conversation of 3c or 3d within 8 h. Second, we examined the effect of the group at the 3 position of 4-iodoisoxazole on the reaction. We chose the substrate 3e, with an isopropyl group at the 3 position, to react with the three kinds of terminal alkynes, affording the targeted products (4m, 4n, and 4o) in high yields (90%, 91%, and 93%, respectively), and 3e was almost transferred in the three reactions. As the steric hindrance from the tert-butyl group is greater than that from the isopropyl group, this implied that 4-iodoisoxazoles containing large steric hindrance functional groups at the 3 position was poor for this type Sonogashira cross-coupling reaction. Then, we further investigated the electronic effects of the group at the 3 position of 4-iodoisoxazoles. We chose 3f containing strong electron donor group (OCH3) and 3g containing strong electron-withdrawing group (CF3) to test the electronic effects on the reaction. To our delight, all of the targeted products (4p–4u) could be obtained with high yields and the starting materials (3f and 3g) could be completely transferred with 8 h. The results implied that the steric effect rather than the electronic effect of the group at the 3 position affected the efficiency of the cross-coupling reactions.
Table 3 Pd-catalyzed Sonogashira cross-coupling between 3,5-disubstituted-4-iodoisoxazoles 3a–3g and terminal alkynesa
|
| Reaction condition: 3 (0.3 mmol, 1 equiv.), terminal alkynes (0.6 mmol, 2 equiv.), Pd(acac) (5 mol%), PPh3 (10 mol%), CuI (10 mol%), Et2NH (2 equiv.), and DMF (3 mL) under N2 protection. Isolated yield. |
 |
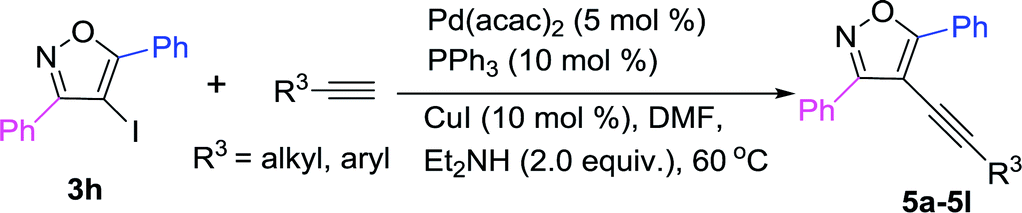
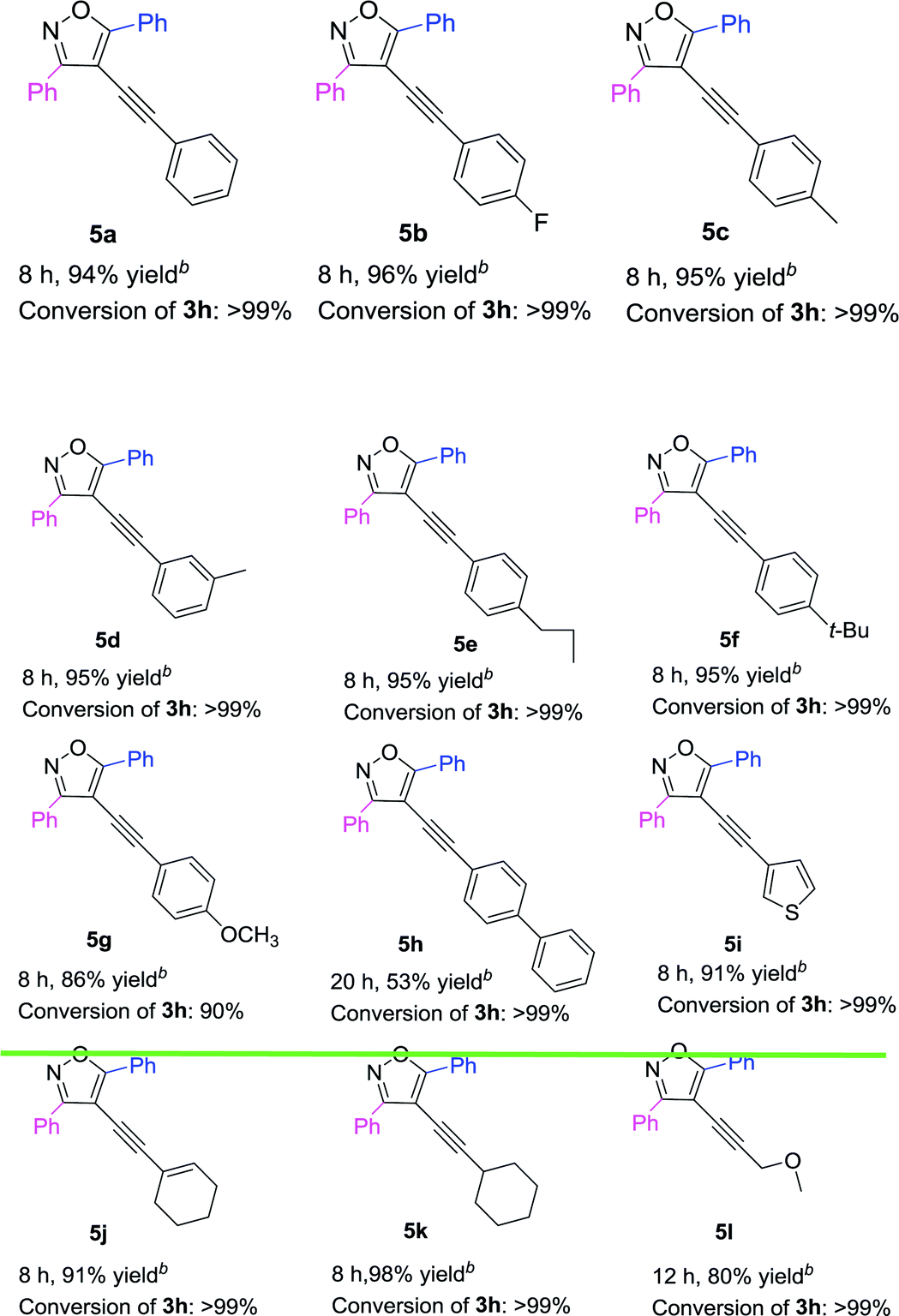
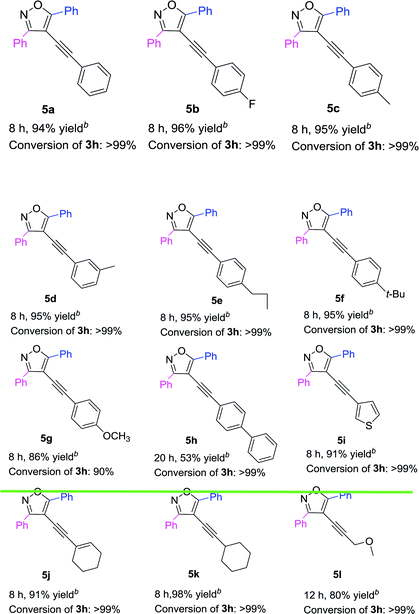
Based on the above results (Table 3), 3,5-diphenyl-4-iodoisoxazole 3h was chosen as a substrate to react with a variety of terminal alkynes in the presence of Pd(acac)2/PPh3 complex (Table 4). The results showed that both electron-donating and electron-withdrawing substituents on the phenyl groups of the terminal alkynes were well accommodated, achieving the desired products (5a–5g) in high yields. The 4-ethynyl-1,1′-biphenyl could also be converted into 5h in 53% yield. Furthermore, R3 = thiophenyl was also afforded the targeted product 5i in 91% yield. Both 1-ethynylcyclohexene and 1-ethynylcyclohexane could be component coupling partner for this transformation (5j–5k). And 3-methoxyprop-1-yne could achieve the desired product 5l in 80% yield as well.
Table 4 Pd-catalyzed Sonogashira cross-coupling between 3,5-diphenyl-4-iodoisoxazole 3h and various terminal alkynesa

|
| Reaction condition: 3h (0.3 mmol, 1 equiv.), terminal alkynes (0.6 mmol, 2 equiv.), Pd(acac)2 (5 mol%), PPh3 (10 mol%), CuI (10 mol%), Et2NH (2 equiv.), and anhydrous DMF (3 mL) under N2 protection. Isolated yield. |
 |
To further demonstrate the synthetic application of the developed protocol, a gram-scale reaction was tested using substrates 3h to react with phenylacetylene, and it could proceed smoothly, affording the targeted product 5a in high yields (Scheme 2).
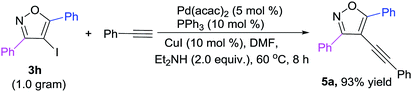 |
| | Scheme 2 A gram-scale reaction. | |
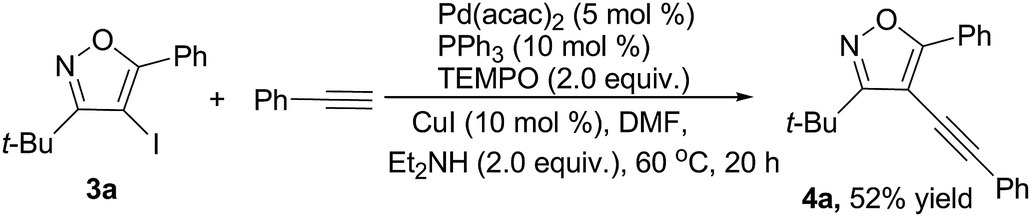
In addition, to gain insight into the mechanism of the Sonogashira coupling reaction, a control experiment had been conducted (Scheme 3). The yield of targeted product 4a was slightly decreased when run in the presence of radical inhibitor (2.0 equiv. TEMPO). The result excluded the possible involvement of radical species.
 |
| | Scheme 3 The control experiment. | |
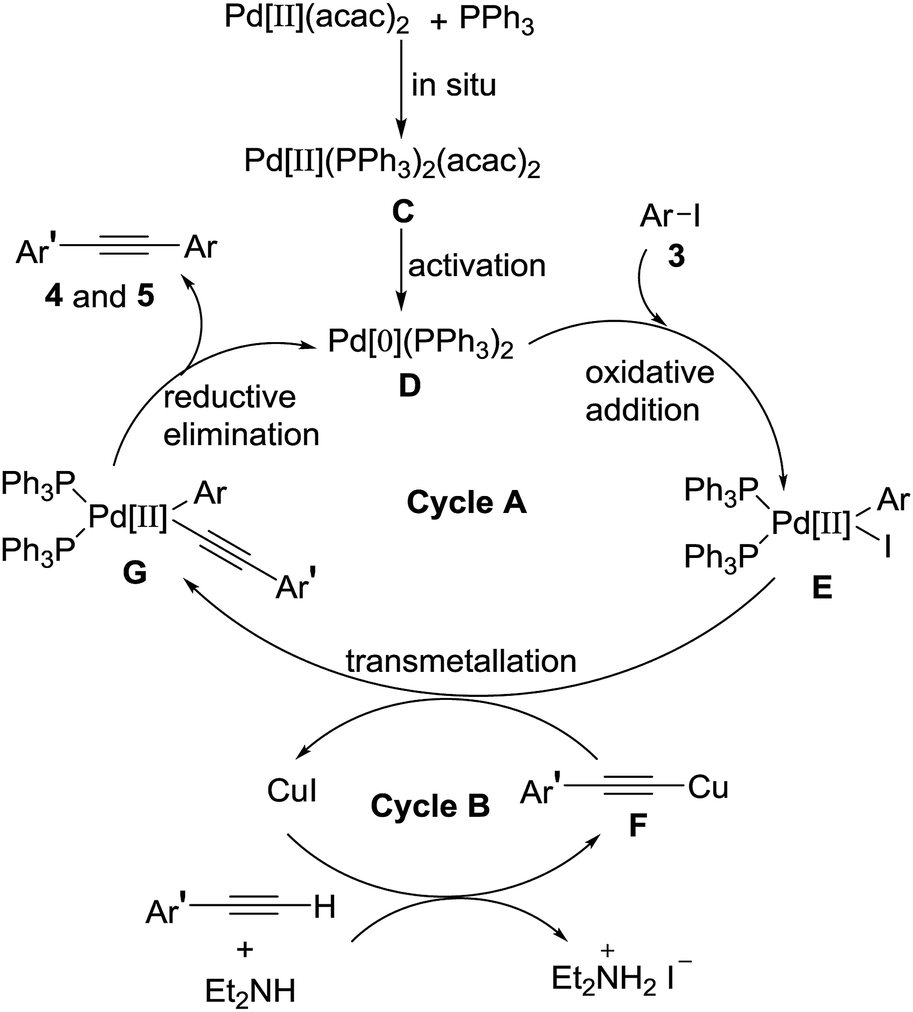
Based on the above experiments and classical description of the Sonogashira coupling reaction, a plausible mechanism for the reaction was depicted in Scheme 4, involving two connected catalytic cycles (cycle A and cycle B).8,12 In the cycle A, the catalytically active species Pd[0](PPh3)2 D was formed by reduction of Pd[II](PPh3)2(acac)2 C (activation) which was produced in situ by combination two equivalents of triphenylphosphine with a Pd(II)(acac)2. Oxidative addition of palladium[0] catalyst D to 4-iodoisoxazoles 3 produced intermediate E, followed by a transmetallation reaction of intermediate E with alkynyl copper F to give intermediate G. Reductive elimination of intermediate G to give the targeted product 4 and 5. Meanwhile, the generating Pd[0](PPh3)2 D promoted the next catalytic cycle. In the cycle B, terminal alkyne reacted with Et2NH and CuI to form alkynyl copper F, followed via transmetallation with intermediate E to afford intermediate G and regenerate CuI for the next catalytic cycle.
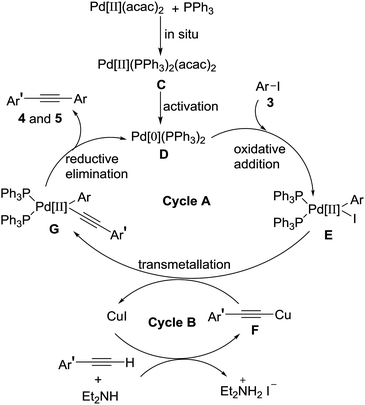 |
| | Scheme 4 Plausible reaction mechanism. | |
Conclusions
In summary, we report herein the Pd(acac)2/PPh3 complex catalyzed Sonogashira cross-coupling reaction of 3,5-disubstituted-4-iodoisoxazoles with terminal alkynes, which affords 3,5-disubstituted-4-alkynylisoxazole products in excellent yields. The effects of the groups at C3 and C5 positions of the isoxazole on the Sonogashira cross-coupling reaction were investigated. The group at C3 position of the isoxazole has great influence on the reaction due to steric effect, whereas the electric effect of the group at C3 position was negligible. Furthermore, a gram-scale reaction demonstrated potential synthetic application of this protocol.
Experimental section
General procedure (I) for the preparation of intermediate ynones (1)
Intermediates 1 were prepared according to a literature.3d To a 50 mL round-bottomed flask were added CuI (19 mg, 0.1 mmol), Pd(PPh3)2Cl2 (14 mg, 0.02 mmol), and triethylamine (10 mL). The flask was flushed with nitrogen atmosphere, and the terminal acetylene (5.0 mmol) was added to the stirred suspension, followed by immediately dropwise addition of acyl chloride (6.5 mmol). The resulting mixture was allowed to stir at room temperature overnight. Water (10 mL) was added to the reaction mixture to work up. The resulting mixture was extracted with ethyl acetate (3 × 20 mL). The organic layers were combined and dried over anhydrous MgSO4. The solvent was removed under a vacuum, and the residue was purified by column chromatography on silica gel (200–300 mesh) using ethyl acetate/petroleum ether as the eluent to afford the desired products 1.
4,4-Dimethyl-1-phenylpent-1-yn-3-one (1a). Prepared according to general procedure (I). Intermediate 1a was obtained as yellow oil (753.7 mg, 81% yield). The 1H NMR spectral data are in good agreement with the literature data.13
4,4-Dimethyl-1-phenylpent-2-yn-1-one (1b). Prepared according to general procedure (I). Intermediate 1b was obtained as yellow oil (770.8 mg, 77% yield). The 1H NMR spectral data are in good agreement with the literature data.14
3-Cyclopropyl-1-phenylprop-2-yn-1-one (1c). Prepared according to general procedure (I). Intermediate 1c was obtained as yellow oil (646.4 mg, 76% yield). The 1H NMR spectral data are in good agreement with the literature data.15
1-Phenyloct-2-yn-1-one (1d). Prepared according to general procedure (I). Intermediate 1d was obtained as yellow oil (900.5 mg, 90% yield). The 1H NMR spectral data are in good agreement with the literature data.16
4-Methyl-1-phenylpent-1-yn-3-one (1e). Prepared according to general procedure (I). Intermediate 1e was obtained as yellow oil (602.3 mg, 70% yield). The 1H NMR spectral data are in good agreement with the literature data.17
1-(4-Methoxyphenyl)-4,4-dimethylpent-2-yn-1-one (1f). Prepared according to general procedure (I). Intermediate 1f was obtained as a yellow oil (780.9 mg, 73% yield). The 1H NMR spectral data are in good agreement with the literature data.18
4,4-Dimethyl-1-(4-(trifluoromethyl)phenyl)pent-2-yn-1-one (1g). Prepared according to general procedure (I). Intermediate 1g was obtained as yellow oil (889.4 mg, 70% yield). 1H NMR (600 MHz, CDCl3) δ 8.21 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 8.3 Hz, 2H), 1.39 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 177.2, 139.7, 135.5, 133.7 (q, J = 31.5 Hz), 129.9, 125.8 (q, J = 3.0 Hz), 123.8 (q, J = 271.5 Hz), 105.6, 78.1, 30.2, 28.3. 19F NMR (564 MHz, CDCl3) δ −63.10. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C14H14F3O: 255.0997; found: 255.0995.
1,3-Diphenylprop-2-yn-1-one (1h). Prepared according to general procedure (I). Intermediate 1h was obtained as yellow oil (927.3 mg, 90% yield). The 1H NMR spectral data are in good agreement with the literature data.16
General procedure (II) for the preparation of intermediate ynone O-methyl oximes (2)
Intermediates 2 were prepared according to a literature procedure.3d To a 50 mL round-bottomed flask were added the alkynones (3.5 mmol), methoxylamine hydrochloride (7.0 mmol, 579 mg), anhydrous Na2SO4 (7.0 mmol, 994 mg), pyridine (1.0 mL), and anhydrous methanol (10 mL). The reaction mixture was stirred at room temperature overnight. The mixture was diluted with water (25 mL) and extracted with ethyl acetate (3 × 25 mL). The organic layers were combined, washed with brine, and dried over anhydrous MgSO4. The solvent was removed under a vacuum, and the residue was purified by flash column chromatography on silica gel (200–300 mesh) using ethyl acetate/petroleum ether as the eluent to afford the desired products 2.
(Z)-4,4-Dimethyl-1-phenylpent-1-yn-3-one O-methyl oxime (2a). Prepared according to general procedure (II). Intermediate 2a was obtained as yellow oil (617.5 mg, 82% yield). The 1H NMR spectral data are in good agreement with the literature data.19
(Z)-4,4-Dimethyl-1-phenylpent-2-yn-1-one O-methyl oxime (2b). Prepared according to general procedure (II). Intermediate 2b was obtained as yellow oil (492.7 mg, 70% yield). The 1H NMR spectral data are in good agreement with the literature data.19
(Z)-3-Cyclopropyl-1-phenylprop-2-yn-1-one O-methyl oxime (2c). Prepared according to general procedure (II). Intermediate 2c was obtained as a yellow oil (383.3 mg, 55% yield). The 1H NMR spectral data are in good agreement with the literature data.20
(Z)-1-Phenyloct-2-yn-1-one O-methyl oxime (2d). Prepared according to general procedure (II). Intermediate 2d was obtained as yellow oil (641.7 mg, 80% yield). 1H NMR (500 MHz, CDCl3) δ 7.88–7.82 (m, 2H), 7.40–7.35 (m, 3H), 4.09 (s, 3H), 2.55 (t, J = 7.2 Hz, 2H), 1.72–1.65 (m, 2H), 1.51–1.34 (m, 4H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 140.4, 134.1, 129.6, 128.4, 126.6, 104.1, 71.6, 63.1, 31.2, 28.1, 22.3, 19.9, 14.1. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C15H20NO: 230.1545; found: 230.1540.
(Z)-4-Methyl-1-phenylpent-1-yn-3-one O-methyl oxime (2e). Prepared according to general procedure (II). Intermediate 2e was obtained as yellow oil (492.7 mg, 70% yield). The 1H NMR spectral data are in good agreement with the literature data.21
(Z)-1-(4-Methoxyphenyl)-4,4-dimethylpent-2-yn-1-one O-methyl oxime (2f). Prepared according to general procedure (II). Intermediate 2f was obtained as yellow oil (642.1 mg, 80% yield). 1H NMR (600 MHz, CDCl3) δ 7.77 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 4.05 (s, 3H), 3.82 (s, 3H), 1.38 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 160.8, 139.9, 128.0, 126.9, 113.8, 111.3, 70.2, 62.8, 55.4, 30.8, 28.6. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C15H20NO2: 246.1494; found: 246.1488.
(Z)-4,4-Dimethyl-1-(4-(trifluoromethyl)phenyl)pent-2-yn-1-one O-methyl oxime (2g). Prepared according to general procedure (II). Intermediate 2g was obtained as yellow oil (792.7 mg, 80% yield). 1H NMR (600 MHz, CDCl3) δ 7.95 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 4.11 (s, 3H), 1.39 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 139.1, 137.6, 131.3 (q, J = 31.5 Hz), 126.9, 125.4 (q, J = 3.0 Hz), 126.9, 124.3 (q, J = 270.0 Hz), 112.6, 69.8, 63.3, 30.8, 28.8. 19F NMR (564 MHz, CDCl3) δ −62.70. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C15H17F3NO: 284.1262; found: 284.1258.
(Z)-1,3-Diphenylprop-2-yn-1-one O-methyl oxime (2h). Prepared according to general procedure (II). Intermediate 2h was obtained as yellow oil (501.9 mg, 61% yield). The 1H NMR spectral data are in good agreement with the literature data.19
General procedure (III) for iodocyclization using ICl as a catalyst (3)
To a stirred solution of the appropriate O-methyl oxime (3.0 mmol) in CH2Cl2 (30 mL), ICl (1 M in CH2Cl2, 1.2 equiv.) was added by dropwise, and then the solution was allowed to stir at room temperature. The reaction was monitored by TLC to establish completion. The excess ICl was removed by washing with a saturated aqueous solution of Na2S2O3. The aqueous solution was then extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under a vacuum to yield the crude product, which was purified by flash chromatography on silica gel (200–300 mesh) using ethyl acetate/petroleum ether as the eluent to afford the desired products 3.
3-(tert-Butyl)-4-iodo-5-phenylisoxazole (3a). Following general procedure (III), the substrate 3a was obtained as a colourless solid (961.4 mg, 98% yield). Mp 80–82 °C. The 1H NMR spectral data are in good agreement with the literature data.19
5-(tert-Butyl)-4-iodo-3-phenylisoxazole (3b). Following general procedure (III), the substrate 3b was obtained as a yellow solid (971.3 mg, 99% yield). Mp 91–93 °C. The 1H NMR spectral data are in good agreement with the literature data.19
5-Cyclopropyl-4-iodo-3-phenylisoxazole (3c). Following general procedure (III), the substrate 3c was obtained as a yellow solid (914.3 mg, 98% yield). Mp 73–75 °C. 1H NMR (400 MHz, CDCl3) δ 7.76 (dt, J = 7.4, 3.2 Hz, 2H), 7.51–7.45 (m, 3H), 2.18 (dq, J = 8.4, 5.1 Hz, 1H), 1.25–1.20 (m, 2H), 1.14 (ddd, J = 11.5, 6.9, 4.6 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 174.7, 163.1, 130.1, 128.9, 128.8, 128.7, 56.5, 9.4, 8.8. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C12H11INO: 311.9885; found: 311.9880.
4-Iodo-5-pentyl-3-phenylisoxazole (3d). Following general procedure (III), the substrate 3d was obtained as yellow oil (992.4 mg, 97% yield). 1H NMR (500 MHz, CDCl3) δ 7.85–7.79 (m, 2H), 7.49–7.44 (m, 3H), 2.91–2.83 (m, 2H), 1.82–1.72 (m, 2H), 1.39 (td, J = 7.2, 3.6 Hz, 4H), 0.98–0.89 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 174.8, 162.6, 129.9, 128.9, 128.6, 128.5, 57.3, 31.2, 27.1, 26.9, 22.3, 13.9. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C14H17INO: 342.0355; found: 342.0341.
4-Iodo-3-isopropyl-5-phenylisoxazole (3e). Following general procedure (III), the substrate 3e was obtained as a white solid (901.5 mg, 96% yield). Mp 70–72 °C. 1H NMR (600 MHz, CDCl3) δ 8.01 (dd, J = 5.2, 2.8 Hz, 2H), 7.52–7.46 (m, 3H), 3.05 (dt, J = 13.9, 6.9 Hz, 1H), 1.40 (d, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3) δ 169.9, 167.6, 130.6, 128.8, 127.7, 127.5, 56.8, 28.1, 20.9. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C12H12INO: 314.0042; found: 314.0040.
5-(tert-Butyl)-4-iodo-3-(4-methoxyphenyl)isoxazole (3f). Following general procedure (III), the substrate 3f was obtained as a yellow solid (1017.6 mg, 95% yield). Mp 75–77 °C. 1H NMR (600 MHz, CDCl3) δ 7.66–7.61 (m, 2H), 7.02–6.98 (m, 2H), 3.85 (s, 3H), 1.54 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 177.8, 164.5, 160.9, 130.6, 121.4, 114.1, 55.4, 34.5, 28.4. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C14H17INO: 358.0304; found: 358.0304.
5-(tert-Butyl)-4-iodo-3-(4-(trifluoromethyl)phenyl)isoxazole (3g). Following general procedure (III), the substrate 3g was obtained as a yellow solid (1019.1 mg, 86% yield). Mp 99–101 °C. 1H NMR (600 MHz, CDCl3) δ 7.82 (d, J = 8.1 Hz, 2H), 7.75 (d, J = 8.2 Hz, 2H), 1.55 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 163.2, 132.9, 131.9 (q, J = 31.5 Hz), 129.8, 125.6 (q, J = 4.5 Hz), 126.9, 124.1 (q, J = 271.5 Hz), 53.4, 34.7, 28.5. 19F NMR (564 MHz, CDCl3) δ −62.80. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C14H14F3INO: 396.0072; found: 396.0070.
4-Iodo-3,5-diphenylisoxazole (3h). Following general procedure (III), the substrate 3h was obtained as a colourless solid (739.1 mg, 71% yield). Mp 176–178 °C. The 1H NMR spectral data are in good agreement with the literature data.22
General procedure (IV) for the synthesis of C4-alkynyl isoxazoles (4 or 5)
A 10 mL two-neck round-bottomed flask with a reflux condenser was flame-dried under a stream of nitrogen and cooled to room temperature. 3 (0.3 mmol, 1.0 equiv.), Pd(acac)2 (4.7 mg, 5 mol%), PPh3 (7.9 mg, 10 mol%), CuI (5.8 mg, 10 mol%), and Et2NH (43.9 mg, 2 equiv.) were added, followed by the addition of anhydrous DMF (2.5 mL). The flask was flushed with nitrogen and the terminal acetylene (1.0 M in DMF, 2.0 equiv.) was added gradually by a syringe. The resulting solution was allowed to stir at 60 °C until completion as monitored by thin layer chromatography. After cooling to room temperature, the reaction was poured into 10 mL ethyl acetate and washed three times (3 × 10 mL) with water. The organic layers were combined, dried with anhydrous MgSO4, and then filtered. The filtrate was concentrated in vacuum, and the resulting residue was purified on silica gel column (200–300 mesh) using ethyl acetate/petroleum ether as eluent to afford the desired products 4 or 5.
3-(tert-Butyl)-5-phenyl-4-(phenylethynyl)isoxazole (4a). Following general procedure (IV), the product 4a was obtained as yellow oil (54.2 mg, 60% yield). 1H NMR (400 MHz, d6-DMSO) δ 8.14–8.11 (m, 2H), 7.60 (dddd, J = 6.3, 5.7, 3.3, 1.7 Hz, 5H), 7.49–7.46 (m, 3H), 1.48 (s, 9H). 13C NMR (100 MHz, d6-DMSO) δ 170.7, 169.0, 131.2, 130.9, 129.3, 129.0, 126.5, 125.9, 121.7, 96.8, 96.2, 79.4, 33.0, 27.8. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C21H20NO: 302.1545; found: 302.1542.
3-(tert-Butyl)-4-((4-fluorophenyl)ethynyl)-5-phenylisoxazole (4b). Following general procedure (IV), the product 4b was obtained as a white solid (58.4 mg, 61% yield). Mp 81–83 °C. 1H NMR (500 MHz, CDCl3) δ 8.17 (d, J = 7.5 Hz, 2H), 7.50 (dt, J = 15.9, 7.1 Hz, 5H), 7.09 (t, J = 8.5 Hz, 2H), 1.54 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 171.5, 169.8, 163.9, 161.9, 133.3, 133.2, 130.7, 129.0, 127.7, 126.5, 119.3, 119.2, 116.2, 116.1, 96.9, 96.0, 80.2, 33.6, 28.3. 19F NMR (470 MHz, CDCl3) δ −109.90. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C21H19FNO: 320.1451; found: 320.1443.
3-(tert-Butyl)-5-phenyl-4-(p-tolylethynyl)isoxazole (4c). Following general procedure (IV), the product 4c was obtained as a white solid (59.6 mg, 63% yield). Mp 77–79 °C. 1H NMR (500 MHz, CDCl3) δ 8.21 (dd, J = 8.1, 0.8 Hz, 2H), 7.52–7.42 (m, 5H), 7.21 (d, J = 7.9 Hz, 2H), 2.40 (s, 3H), 1.56 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 171.6, 169.5, 139.2, 131.3, 130.6, 129.5, 128.9, 127.8, 126.5, 120.1, 97.4, 97.3, 79.8, 33.6, 28.4, 21.8. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H22NO: 316.1701; found: 316.1694.
5-(tert-Butyl)-3-phenyl-4-(phenylethynyl)isoxazole (4d). Following general procedure (IV), the product 4d was obtained as a white solid (87.6 mg, 97% yield). Mp 58–60 °C. 1H NMR (400 MHz, CDCl3) δ 8.08 (dd, J = 6.5, 3.1 Hz, 2H), 7.51–7.45 (m, 5H), 7.39–7.33 (m, 3H), 1.58 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 182.2, 162.3, 131.1, 130.1, 128.9, 128.8, 128.7, 128.6, 128.0, 123.2, 96.8, 95.9, 79.6, 34.8, 28.5. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C21H20NO: 302.1545; found: 302.1539.
5-(tert-Butyl)-4-((4-fluorophenyl)ethynyl)-3-phenylisoxazole (4e). Following general procedure (IV), the product 4e was obtained as yellow oil (86.2 mg, 90% yield). 1H NMR (600 MHz, CDCl3) δ 8.06–8.01 (m, 2H), 7.48 (d, J = 4.0 Hz, 3H), 7.43 (dd, J = 8.1, 5.7 Hz, 2H), 7.05 (t, J = 8.5 Hz, 2H), 1.56 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 182.2, 163.6, 162.4, 162.0, 133.1, 133.0, 132.7, 130.2, 129.4, 128.9, 128.7, 128.6, 128.0, 119.4, 119.3, 116.0, 115.9, 96.6, 94.8, 79.3, 34.8, 28.5. 19F NMR (564 MHz, CDCl3) δ −110.30. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C21H19FNO: 320.1451; found: 320.1449.
5-(tert-Butyl)-3-phenyl-4-(p-tolylethynyl)isoxazole (4f). Following general procedure (IV), the product 4f was obtained as a white solid (84.2 mg, 89% yield). Mp 81–83 °C. 1H NMR (600 MHz, CDCl3) δ 8.09–8.04 (m, 2H), 7.49–7.47 (m, 3H), 7.36 (d, J = 8.1 Hz, 2H), 7.17 (d, J = 7.9 Hz, 2H), 2.38 (s, 3H), 1.56 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 182.0, 162.3, 138.9, 132.7, 131.1, 130.1, 129.4, 129.0, 128.7, 128.6, 128.0, 120.2, 96.9, 96.1, 78.9, 34.8, 28.5, 21.7. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H22NO: 316.1701; found: 316.1694.
5-Cyclopropyl-3-phenyl-4-(phenylethynyl)isoxazole (4g). Following general procedure (IV), the product 4g was obtained as yellow oil (82.1 mg, 96% yield). 1H NMR (400 MHz, CDCl3) δ 8.07 (dt, J = 9.3, 2.8 Hz, 2H), 7.55–7.43 (m, 4H), 7.32 (dd, J = 5.0, 3.0 Hz, 1H), 7.18 (dd, J = 5.0, 1.0 Hz, 1H), 2.37–2.27 (m, 1H), 1.38–1.32 (m, 2H), 1.21–1.16 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 177.0, 161.6, 131.4, 130.2, 128.9, 128.8, 128.7, 128.6, 127.8, 123.1, 97.5, 94.8, 78.6, 9.0, 8.9. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C21H18NO: 300.1388; found: 300.1386.
5-Cyclopropyl-4-((4-fluorophenyl)ethynyl)-3-phenylisoxazole (4h). Following general procedure (IV), the product 4h was obtained as a yellow solid (82.8 mg, 91% yield). Mp 85–87 °C. 1H NMR (500 MHz, CDCl3) δ 8.11–8.03 (m, 2H), 7.48 (dd, J = 7.4, 6.0 Hz, 5H), 7.06 (t, J = 8.5 Hz, 2H), 2.37–2.27 (m, 1H), 1.40–1.34 (m, 2H), 1.22–1.15 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 177.0, 163.8, 161.8, 161.6, 133.4, 133.3, 130.2, 128.8, 128.7, 127.8, 119.3, 119.2, 116.0, 115.8, 97.4, 93.7, 78.3, 9.0, 8.9. 19F NMR (564 MHz, CDCl3) δ −109.90, −109.91. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C20H15FNO: 304.1138; found: 304.1130.
5-Cyclopropyl-3-phenyl-4-(p-tolylethynyl)isoxazole (4i). Following general procedure (IV), the product 4i was obtained as yellow oil (82.6 mg, 92% yield). 1H NMR (400 MHz, CDCl3) δ 8.15–8.06 (m, 2H), 7.53–7.45 (m, 3H), 7.41 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 7.9 Hz, 2H), 2.39 (s, 3H), 1.40–1.34 (m, 2H), 1.23–1.16 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 176.7, 161.5, 138.9, 131.3, 130.2, 129.3, 128.8, 128.7, 127.7, 120.0, 97.6, 94.9, 77.8, 21.7, 8.9, 8.8. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C21H18NO: 300.1388; found: 300.1386.
5-Pentyl-3-phenyl-4-(phenylethynyl)isoxazole (4j). Following general procedure (IV), the product 4j was obtained as yellow oil (89.9 mg, 95% yield). 1H NMR (500 MHz, CDCl3) δ 8.14–8.06 (m, 2H), 7.52–7.45 (m, 5H), 7.10–7.03 (m, 2H), 2.96 (t, J = 7.5 Hz, 2H), 1.86 (p, J = 7.3 Hz, 2H), 1.41 (dd, J = 7.2, 3.6 Hz, 4H), 0.93 (t, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 176.9, 163.8, 161.8, 161.3, 133.5, 133.4, 130.2, 128.8, 127.7, 119.1, 116.1, 115.9, 98.7, 93.9, 78.3, 31.3, 26.9, 26.7, 22.4, 14.1. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H22NO: 316.1701; found: 316.1690.
4-((4-Fluorophenyl)ethynyl)-5-pentyl-3-phenylisoxazole (4k). Following general procedure (IV), the product 4k was obtained as yellow oil (91.9 mg, 92% yield). 1H NMR (500 MHz, CDCl3) δ 8.13 (td, J = 4.2, 2.5 Hz, 2H), 7.51–7.48 (m, 4H), 7.37 (dd, J = 4.2, 2.3 Hz, 3H), 3.01–2.91 (m, 2H), 1.87 (p, J = 7.3 Hz, 2H), 1.42 (dd, J = 4.2, 3.1 Hz, 4H), 0.94 (dt, J = 7.0, 3.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 176.9, 161.3, 131.5, 130.2, 128.9, 128.8, 128.7, 128.6, 127.8, 123.1, 98.9, 95.0, 78.6, 31.4, 27.0, 26.7, 22.4, 14.1. 19F NMR (470 MHz, CDCl3) δ −110.30. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H21FNO: 334.1607; found: 334.1599.
5-Pentyl-3-phenyl-4-(p-tolylethynyl)isoxazole (4l). Following general procedure (IV), the product 4l was obtained as yellow oil (91.9 mg, 98% yield). 1H NMR (500 MHz, CDCl3) δ 8.18–8.11 (m, 2H), 7.53–7.46 (m, 3H), 7.42 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 7.9 Hz, 2H), 2.97 (t, J = 7.5 Hz, 2H), 2.39 (s, 3H), 1.86 (dd, J = 14.7, 7.3 Hz, 2H), 1.46–1.39 (m, 4H), 0.95 (dd, J = 10.0, 4.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 176.7, 161.2, 138.9, 131.4, 130.1, 129.3, 128.9, 128.7, 127.7, 120.0, 99.0, 95.2, 77.8, 31.3, 26.9, 26.6, 22.4, 21.6, 14.0. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C23H24NO: 330.1858; found: 330.1852.
3-Isopropyl-5-phenyl-4-(phenylethynyl)isoxazole (4m). Following general procedure (IV), the product 4m was obtained as yellow oil (77.6 mg, 90% yield). 1H NMR (600 MHz, CDCl3) δ 8.23–8.18 (m, 2H), 7.57–7.55 (m, 2H), 7.53–7.46 (m, 3H), 7.42–7.38 (m, 3H), 3.27 (dt, J = 14.0, 7.0 Hz, 1H), 1.49 (d, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3) δ 169.9, 168.8, 131.5, 130.7, 129.0, 128.9, 128.7, 127.6, 126.4, 123.0, 97.6, 96.6, 79.2, 27.1, 20.7. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C20H18NO: 288.1388; found: 288.1385.
4-((4-Fluorophenyl)ethynyl)-3-isopropyl-5-phenylisoxazole (4n). Following general procedure (IV), the product 4n was obtained as yellow oil (83.3 mg, 91% yield). 1H NMR (600 MHz, CDCl3) δ 8.20–8.14 (m, 2H), 7.54–7.47 (m, 5H), 7.12–7.06 (m, 2H), 3.25 (dt, J = 14.0, 7.0 Hz, 1H), 1.48 (d, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3) δ 169.8, 168.9, 163.7, 162.1, 133.5, 133.4, 130.7, 130.6, 129.0, 128.8, 127.6, 127.5, 126.4, 119.2, 119.1, 116.1, 116.0, 97.5, 95.4, 78.9, 27.1, 20.9, 20.6. 19F NMR (564 MHz, CDCl3) δ −109.90. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C20H17FNO: 306.1294; found: 306.1288.
3-Isopropyl-5-phenyl-4-(p-tolylethynyl)isoxazole (4o). Following general procedure (IV), the product 4o was obtained as yellow oil (84.1 mg, 93% yield). 1H NMR (600 MHz, CDCl3) δ 8.26–8.17 (m, 2H), 7.52–7.44 (m, 5H), 7.21 (d, J = 7.9 Hz, 2H), 3.27 (dt, J = 13.9, 7.0 Hz, 1H), 2.40 (s, 3H), 1.49 (dd, J = 7.0, 1.7 Hz, 6H). 13C NMR (150 MHz, CDCl3) δ 169.8, 168.6, 139.1, 131.4, 130.6, 129.4, 128.9, 127.68, 126.4, 119.9, 97.8, 96.8, 78.5, 27.1, 21.7, 20.6. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C21H20NO: 302.1545; found: 302.1539.
5-(tert-Butyl)-3-(4-methoxyphenyl)-4-(phenylethynyl)isoxazole (4p). Following general procedure (IV), the product 4p was obtained as yellow oil (89.4 mg, 90% yield). 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 8.8 Hz, 2H), 7.49–7.45 (m, 2H), 7.36 (dd, J = 5.1, 1.9 Hz, 3H), 7.01 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 1.56 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 182.1, 161.9, 161.1, 131.1, 129.4, 128.6, 123.3, 121.4, 114.1, 96.5, 95.8, 79.9, 55.5, 34.8, 28.4. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H22NO2: 332.1651; found: 332.1647.
5-(tert-Butyl)-4-((4-fluorophenyl)ethynyl)-3-(4-methoxyphenyl)isoxazole (4q). Following general procedure (IV), the product 4q was obtained as yellow oil (95.3 mg, 91% yield). 1H NMR (600 MHz, CDCl3) δ 8.00 (d, J = 8.7 Hz, 2H), 7.47–7.41 (m, 2H), 7.06 (t, J = 8.6 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H), 1.55 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 182.1, 163.6, 161.9, 161.1, 133.1, 133.0, 129.3, 121.3, 119.4, 119.3, 116.0, 115.9, 114.1, 96.3, 94.7, 79.6, 55.5, 34.8, 28.4. 19F NMR (564 MHz, CDCl3) δ −110.34, −110.35, −110.36, −110.37, −110.38, −110.39, −110.40. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H22FNO2: 350.1556; found: 350.1552.
5-(tert-Butyl)-3-(4-methoxyphenyl)-4-(p-tolylethynyl)isoxazole (4r). Following general procedure (IV), the product 4r was obtained as yellow oil (95.3 mg, 92% yield). 1H NMR (600 MHz, CDCl3) δ 8.07–8.01 (m, 2H), 7.37 (d, J = 8.1 Hz, 2H), 7.17 (d, J = 7.9 Hz, 2H), 7.01–6.98 (m, 2H), 3.86 (s, 3H), 2.38 (s, 3H), 1.55 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 181.8, 161.9, 161.1, 138.8, 131.1, 129.5, 129.4, 121.5, 120.2, 114.1, 96.6, 95.9, 79.2, 55.5, 34.7, 28.4, 21.7. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C23H24NO2: 346.1807; found: 346.1798.
5-(tert-Butyl)-4-(phenylethynyl)-3-(4-(trifluoromethyl)phenyl)isoxazole (4s). Following general procedure (IV), the product 4s was obtained as yellow oil (107.5 mg, 97% yield). 1H NMR (600 MHz, CDCl3) δ 8.21 (d, J = 8.2 Hz, 2H), 7.76 (d, J = 8.3 Hz, 2H), 7.51–7.45 (m, 2H), 7.41–7.34 (m, 3H), 1.58 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 182.8, 161.1, 132.5, 132.1, 131.8, 131.2, 129.7, 129.0, 128.7, 128.3, 125.7 (q, J = 4.5 Hz), 125.1, 123.2, 122.9, 96.9, 96.4, 78.9, 34.9, 28.5. 19F NMR (564 MHz, CDCl3) δ −62.80. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H19F3NO: 370.1419; found: 370.1416.
5-(tert-Butyl)-4-((4-fluorophenyl)ethynyl)-3-(4-(trifluoromethyl)phenyl)isoxazole (4t). Following general procedure (IV), the product 4t was obtained as a yellow solid (110.4 mg, 95% yield). Mp 77–79 °C. 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.2 Hz, 2H), 7.75 (d, J = 8.3 Hz, 2H), 7.44 (dd, J = 8.7, 5.4 Hz, 2H), 7.07 (t, J = 8.6 Hz, 2H), 1.57 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 182.8, 163.8, 162.2, 161.1, 133.2, 133.1, 132.4, 132.1, 131.9, 128.3, 127.2, 125.7 (q, J = 3 Hz), 125.1, 123.2, 119.0, 118.9, 116.2, 116.0, 96.7, 95.3, 78.7, 34.9, 29.0, 28.4. 19F NMR (564 MHz, CDCl3) δ −62.80, −109.80. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H19F4NO: 388.1325; found: 388.1319.
5-(tert-Butyl)-4-(p-tolylethynyl)-3-(4-(trifluoromethyl)phenyl)isoxazole (4u). Following general procedure (IV), the product 4u was obtained as a yellow solid (111.4 mg, 97% yield). Mp 83–85 °C. 1H NMR (600 MHz, CDCl3) δ 8.22 (d, J = 8.2 Hz, 2H), 7.75 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.1 Hz, 2H), 7.19 (d, J = 7.9 Hz, 2H), 2.39 (s, 3H), 1.58 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 182.5, 161.1, 139.2, 132.5, 132.2, 132.0, 131.8, 131.6, 131.1, 129.5, 128.3, 126.8, 125.7 (q, J = 3 Hz), 123.2, 121.4, 119.8, 97.1, 96.6, 78.3, 34.9, 28.4, 21.7. 19F NMR (564 MHz, CDCl3) δ −62.80. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C23H21F3NO: 384.1575; found: 384.1571.
3,5-Diphenyl-4-(phenylethynyl)isoxazole (5a). Following general procedure (IV), the product 5a was obtained as a yellow solid (90.6 mg, 94% yield). Mp 90–92 °C. 1H NMR (400 MHz, CDCl3) δ 8.30 (dd, J = 8.1, 1.4 Hz, 2H), 8.21–8.14 (m, 2H), 7.61–7.51 (m, 8H), 7.41 (dd, J = 6.4, 2.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.0, 162.6, 131.5, 130.9, 130.4, 129.1, 129.0, 128.8, 128.7, 128.6, 128.0, 127.4, 126.5, 122.8, 97.5, 96.6, 79.9. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C23H16NO: 322.1232; found: 322.1229.
4-((4-Fluorophenyl)ethynyl)-3,5-diphenylisoxazole (5b). Following general procedure (IV), the product 5b was obtained as a white solid (96.6 mg, 96% yield). Mp 100–102 °C. 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J = 7.3 Hz, 2H), 8.17–8.11 (m, 2H), 7.56–7.49 (m, 8H), 7.09 (t, J = 8.5 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 170.1, 163.9, 162.7, 162.0, 133.5, 133.4, 130.9, 130.4, 129.1, 128.8, 128.6, 128.0, 127.5, 126.6, 119.0, 118.9, 116.2, 116.0, 97.3, 95.5, 79.7. 19F NMR (470 MHz, CDCl3) δ −109.62. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C23H15FNO: 340.1138; found: 340.1130.
3,5-Diphenyl-4-(p-tolylethynyl)isoxazole (5c). Following general procedure (IV), the product 5c was obtained as a white solid (95.5 mg, 95% yield). Mp 106–108 °C. 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 6.9 Hz, 2H), 8.23–8.15 (m, 2H), 7.58–7.43 (m, 8H), 7.22 (d, J = 7.9 Hz, 2H), 2.41 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.8, 162.6, 139.2, 131.4, 130.8, 130.3, 129.4, 129.0, 128.8, 128.7, 128.0, 127.5, 126.5, 119.8, 97.6, 96.8, 79.2, 21.7. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C24H18NO: 336.1388; found: 336.1382.
3,5-Diphenyl-4-(m-tolylethynyl)isoxazole (5d). Following general procedure (IV), the product 5d was obtained as a yellow solid (92.5 mg, 92% yield). Mp 95–97 °C. 1H NMR (600 MHz, CDCl3) δ 8.29 (d, J = 7.5 Hz, 2H), 8.22–8.12 (m, 2H), 7.57–7.51 (m, 6H), 7.36 (d, J = 5.9 Hz, 2H), 7.29 (t, J = 7.8 Hz, 1H), 7.21 (d, J = 7.6 Hz, 1H), 2.39 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 170.1, 162.7, 138.5, 132.1, 130.9, 130.4, 129.9, 129.1, 128.8, 128.7, 128.6, 128.1, 127.5, 126.6, 122.7, 97.6, 96.9, 79.5, 21.5. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C24H18NO: 336.1388; found: 336.1381.
3,5-Diphenyl-4-((4-propylphenyl)ethynyl)isoxazole (5e). Following general procedure (IV), the product 5e was obtained as a yellow solid (102.4 mg, 94% yield). Mp 97–99 °C. 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 6.8 Hz, 2H), 8.19 (dd, J = 6.4, 2.8 Hz, 2H), 7.53 (ddd, J = 25.3, 12.3, 7.4 Hz, 8H), 7.22 (d, J = 8.0 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 1.68 (dd, J = 15.0, 7.5 Hz, 2H), 0.98 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 169.8, 162.6, 144.0, 131.4, 130.8, 130.3, 129.0, 128.9, 128.8, 128.7, 128.0, 127.5, 126.5, 120.1, 97.7, 96.9, 79.2, 38.1, 24.5, 13.9. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C26H22NO: 364.1701; found: 364.1690.
4-((4-(tert-Butyl)phenyl)ethynyl)-3,5-diphenylisoxazole (5f). Following general procedure (IV), the product 5f was obtained as a yellow solid (105.3 mg, 93% yield). Mp 115–117 °C. 1H NMR (600 MHz, CDCl3) δ 8.29 (d, J = 7.8 Hz, 2H), 8.17 (d, J = 7.6 Hz, 2H), 7.52 (dd, J = 14.2, 7.5 Hz, 8H), 7.43 (d, J = 8.2 Hz, 2H), 1.35 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 169.9, 162.7, 152.5, 131.4, 130.9, 130.8, 130.4, 130.3, 129.2, 129.1, 128.9, 128.8, 128.7, 128.1, 127.9, 127.6, 126.6, 125.8, 119.9, 97.7, 96.9, 79.2, 35.1, 31.4. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C27H24NO: 378.1858; found: 378.1849.
4-((4-Methoxyphenyl)ethynyl)-3,5-diphenylisoxazole (5g). Following general procedure (IV), the product 5g was obtained as a white solid (90.6 mg, 89% yield). Mp 114–116 °C. 1H NMR (600 MHz, CDCl3) δ 8.32–8.26 (m, 2H), 8.21–8.13 (m, 2H), 7.56–7.45 (m, 8H), 6.92 (d, J = 8.7 Hz, 2H), 3.85 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 169.6, 162.6, 160.2, 133.1, 130.8, 130.4, 129.1, 128.9, 128.8, 128.1, 127.6, 126.5, 115.1, 114.4, 97.8, 96.7, 78.6, 55.6. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C22H19F4NO: 352.1338; found: 352.1334.
4-([1,1′-Biphenyl]-4-ylethynyl)-3,5-diphenylisoxazole (5h). Following general procedure (IV), the product 5h was obtained as a white solid (59.6 mg, 50% yield). Mp 120–122 °C. 1H NMR (600 MHz, CDCl3) δ 8.33–8.29 (m, 2H), 8.18 (dd, J = 7.7, 1.8 Hz, 2H), 7.65–7.60 (m, 6H), 7.55 (qdd, J = 6.6, 4.2, 2.2 Hz, 6H), 7.48 (dd, J = 10.5, 4.9 Hz, 2H), 7.41–7.38 (m, 1H). 13C NMR (150 MHz, CDCl3) δ 170.1, 162.7, 141.8, 140.3, 132.1, 131.0, 130.4, 129.2, 129.1, 128.9, 128.7, 128.1, 128.0, 127.5, 127.4, 127.2, 126.6, 121.7, 97.6, 96.5, 80.6. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C29H20NO: 398.1545; found: 398.1540.
3,5-Diphenyl-4-(thiophen-3-ylethynyl)isoxazole (5i). Following general procedure (IV), the product 5i was obtained as a white solid (89.3 mg, 91% yield). Mp 133–135 °C. 1H NMR (400 MHz, CDCl3) δ 8.27 (dd, J = 7.9, 1.4 Hz, 2H), 8.15 (dd, J = 6.6, 3.1 Hz, 2H), 7.60–7.48 (m, 7H), 7.36 (dd, J = 4.9, 3.0 Hz, 1H), 7.23 (dd, J = 5.0, 0.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 170.1, 162.7, 130.9, 130.3, 129.7, 129.3, 129.2, 129.1, 128.9, 128.6, 128.0, 127.4, 126.9, 126.5, 125.9, 121.8, 97.4, 91.9, 79.3. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C21H14NOS: 328.0796; found: 328.0790.
4-(Cyclohex-1-en-1-ylethynyl)-3,5-diphenylisoxazole (5j). Following general procedure (IV), the product 5j was obtained as yellow oil (88.8 mg, 91% yield). 1H NMR (400 MHz, CDCl3) δ 8.27–8.20 (m, 2H), 8.12 (dd, J = 6.2, 2.7 Hz, 2H), 7.56–7.44 (m, 6H), 6.28–6.21 (m, 1H), 2.30–2.24 (m, 2H), 2.21–2.15 (m, 2H), 1.74–1.63 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 169.3, 162.5, 136.1, 130.6, 130.2, 128.9, 128.8, 128.7, 127.9, 127.6, 126.4, 120.7, 98.7, 97.9, 28.8, 25.9, 22.3, 21.6. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C23H20NO: 326.1545; found: 326.1540.
4-(Cyclohexylethynyl)-3,5-diphenylisoxazole (5k). Following general procedure (IV), the product 5k was obtained as a white solid (96.2 mg, 98% yield). Mp 97–99 °C. 1H NMR (600 MHz, CDCl3) δ 8.27–8.23 (m, 2H), 8.12 (dd, J = 6.6, 3.0 Hz, 2H), 7.50 (dd, J = 8.2, 5.1 Hz, 5H), 2.83–2.63 (m, 1H), 1.97–1.90 (m, 2H), 1.78 (dt, J = 12.8, 5.2 Hz, 2H), 1.65–1.57 (m, 3H), 1.45–1.37 (m, 3H). 13C NMR (150 MHz, CDCl3) δ 169.4, 162.7, 130.6, 130.2, 129.1, 128.8, 128.6, 127.9, 127.7, 126.3, 102.2, 98.0, 71.0, 32.4, 30.2, 26.0, 24.9. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C23H22NO: 328.1701; found: 328.1696.
4-(3-Methoxyprop-1-yn-1-yl)-3,5-diphenylisoxazole (5l). Following general procedure (IV), the product 5l was obtained as a white solid (69.4 mg, 80% yield). Mp 68–70 °C. 1H NMR (600 MHz, CDCl3) δ 8.20 (dd, J = 7.9, 1.5 Hz, 2H), 8.09–8.03 (m, 2H), 7.56–7.46 (m, 6H), 4.41 (s, 2H), 3.46 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 170.7, 162.9, 131.1, 130.4, 129.1, 128.8, 128.5, 128.1, 127.3, 126.6, 96.8, 92.8, 76.9, 60.7, 58.1. HRMS (ESI-ion trap): m/z [M + H]+ calcd for C19H16NO2: 290.1181; found: 290.1180.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully thank the National Natural Science Foundation of China (21172081, 21372090), the Natural Science Foundation of Guangdong Province (S2013020013091) and the City of Guangzhou Science and Technology Plan Projects (201510010054) for financial support.
References
-
(a) R. G. Micetich and R. Raap, J. Med. Chem., 1968, 11, 159 CrossRef CAS PubMed;
(b) J. J. Talley, Prog. Med. Chem., 1999, 36, 201 CrossRef CAS PubMed;
(c) M. P. Giovannoni, C. Vergelli, C. Ghelardini, N. Galeotti, A. Bartolini and V. Dal Piaz, J. Med. Chem., 2003, 46, 1055 CrossRef CAS PubMed;
(d) M. Dougados, P. Emery, E. M. Lemmel, A. F. Zerbini, S. Brin and P. Van Rie, Ann. Rheum. Dis., 2005, 64, 44 CrossRef CAS PubMed;
(e) S. Naud, I. M. Westwood, A. Faisal, P. Sheldrake, V. Bavetsias, B. Atrash, K. M. J. Cheung, M. J. Liu, A. Hayes, J. Schmmitt, A. Wood, V. Chi, K. Boxall, J. Mak, M. Gurden, M. Valenti, A. D. H. Brandon, A. Henley, R. Baker, C. McAndrew, B. Matijssen, R. Burker, S. Hoelder, S. A. Eccles, F. I. Raynaud, S. Linardopoulos, R. L. M. V. Montfort and J. L. Blagg, J. Med. Chem., 2013, 56, 10045 CrossRef CAS PubMed;
(f) J.-J. Feng and J. Zhang, ACS Catal., 2016, 6, 6651 CrossRef CAS;
(g) Q.-Q. Cheng, Y. Yu, J. Yedoyan and M. P. Doyle, ChemCatChem, 2018, 10, 488 CrossRef CAS.
-
(a) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Org. Chem., 2005, 70, 7761 CrossRef PubMed;
(b) T. V. Hansen, P. Wu and V. V. Fokin, J. Org. Chem., 2005, 70, 7761 CrossRef CAS PubMed;
(c) H. Li, L. Yu, X. Zhang, W. L. Johnson, R. Figueroa and R. P. Hsung, Heterocycles, 2007, 74, 553 CrossRef CAS;
(d) M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed;
(e) S. Grecian and V. V. Fokin, Angew. Chem., Int. Ed., 2008, 47, 8285 CrossRef CAS PubMed;
(f) J. S. Oakdale, R. K. Sit and V. V. Fokin, Chem.–Eur. J., 2014, 20, 11101 CrossRef CAS PubMed;
(g) J.-X. Li, M. Hu, C.-S. Li, C. Li, J.-W. Li, W.-Q. Wu and H.-F. Jiang, Adv. Synth. Catal., 2018, 360, 2707 CrossRef CAS.
-
(a) K. Maeda, T. Hosokawa, S. Murahashi and I. Moritani, Tetrahedron Lett., 1973, 5075 CrossRef CAS;
(b) T. Hosokawa, N. Shimo, K. Maeda, A. Sonoda and S. Murahashi, Tetrahedron Lett., 1976, 383 CrossRef CAS;
(c) J. P. Waldo and R. C. Lorock, Org. Lett., 2005, 7, 5203 CrossRef CAS PubMed;
(d) J. P. Waldo and R. C. Lorock, J. Org. Chem., 2007, 72, 9643 CrossRef CAS PubMed;
(e) T. Okitsu, K. Sato, T. M. Potewar and A. Wada, J. Org. Chem., 2011, 76, 3438 CrossRef CAS PubMed;
(f) I. Cikotiene, Eur. J. Org. Chem., 2012, 2766 CrossRef CAS;
(g) C. R. Reddy, J. Vijaykumar, E. Jithender, G. P. K. Reddy and R. Gree, Eur. J. Org. Chem., 2012, 5767 CrossRef;
(h) L. Zhang, Q. Zeng, A. Mao, Z. Wu, T. Luo, Y. Xiao and J. Zhang, J. Org. Biomol. Chem., 2014, 12, 8942 RSC.
-
(a) T. T. Dang, U. Albrecht and P. Langer, Synthesis, 2006, 2515 CAS;
(b) S. Tang, J. He, Y. Sun, L. He and X. She, Org. Lett., 2009, 11, 3982 CrossRef CAS PubMed;
(c) S. Tang, J. He, Y. Sun, L. He and X. She, J. Org. Chem., 2010, 75, 1961 CrossRef CAS PubMed;
(d) D. Xiang, X. Xin, X. Liu, R. Zhang, J. Yang and D. Dong, Org. Lett., 2012, 14, 644 CrossRef CAS PubMed;
(e) S. Samai, T. Chanda, H. Ila and M. S. Singh, Eur. J. Org. Chem., 2013, 4026 CrossRef CAS;
(f) B. Raghava, G. Parameshwarappa, Y. Acharya, T. R. Swaroop, K. S. Rangappa and H. Ila, Eur. J. Org. Chem., 2014, 1882 CrossRef CAS.
- F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583 CrossRef CAS.
-
(a) H. Yamanaka, M. Shiraiwa, E. Yamamoto and T. Sakamoto, Chem. Pharm. Bull., 1981, 29, 3543 CrossRef CAS;
(b) N. Naksmura, Y. Tajima and K. Saki, Heterocycles, 1982, 17, 235 CrossRef;
(c) H. Kromann, F. A. Slok, T. N. Johansen and P. Krogsgaard-Larsen, Tetrahedron, 2001, 57, 2195 CrossRef CAS;
(d) H. A. Chiong and O. Daugulis, Org. Lett., 2007, 9, 1449 CrossRef CAS PubMed;
(e) J. P. Waldo, S. Mehta, B. Neuenswander, G. H. Lushington and R. C. Larock, J. Comb. Chem., 2008, 10, 658 CrossRef CAS PubMed;
(f) Y. Fall, C. Reynaud, H. Doucet and M. Santelli, Eur. J. Org. Chem., 2009, 4041 CrossRef CAS;
(g) I. I. F. Boogaerts and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 8858 CrossRef CAS PubMed;
(h) B. Chappell, N. Dedman and S. Wheeler, Tetrahedron Lett., 2011, 52, 3223 CrossRef CAS;
(i) D. Roy, S. Mom, S. Royer, D. Lucas, J. C. Hierso and H. Doucet, ACS Catal., 2012, 2, 1033 CrossRef CAS;
(j) D. Hay, O. Fedorov, P. Filippakopoulos, S. Martin, M. Philpott, D. S. Hewings, S. Uttakar, T. D. Heightman, S. Conway, S. Knapp and P. E. Brennan, Med. Chem. Commun., 2013, 4, 140 RSC;
(k) X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326 CrossRef CAS PubMed;
(l) J. Sun, C. Lin, X.-C. Qin, X.-P. Dong, Z.-C. Tu, F. Tang, C.-N. Chen and J.-C. Zhang, Bioorg. Med. Chem. Lett., 2015, 25, 3129 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. Q. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- Y.-W. Guo, X.-J. Wang, Z.-T. Zhu, J.-N. Zhang and Y.-M. Wu, Synlett, 2016, 27, 2259 CrossRef CAS.
- K. C. Coffman, T. A. Palazzo, P. Z. Hartley, J. C. Fettinger, D. J. Tantilo and M. Kurth, J. Org. Lett., 2013, 15, 2062 CrossRef CAS PubMed.
- T. Morita, S. Fukuhara, S. Fuse and H. Nakamura, Org. Lett., 2018, 20, 433 CrossRef CAS PubMed.
- A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- D. A. Alonso, C. Najera and M. C. Pacheco, J. Org. Chem., 2004, 69, 1615 CrossRef CAS PubMed.
- W. Kim, K. Park, A. Park, J. Choe and S. Lee, Org. Lett., 2013, 15, 1654 CrossRef CAS PubMed.
- S. Tang, L. Zeng, Y.-C. Liu and A.-W. Lei, Angew. Chem., Int. Ed., 2015, 54, 15850 CrossRef CAS PubMed.
- A. Park, K. Park, Y. Kim and S. Lee, Org. Lett., 2011, 13, 944 CrossRef CAS PubMed.
- Y. Zhu, B. Zhao and Y. Shi, Org. Lett., 2013, 15, 992 CrossRef CAS.
- B. Liang, M. Huang, Z. You, K. Lu, R. Fathi, J. Chen and Z. Yang, J. Org. Chem., 2005, 70, 6097 CrossRef CAS.
- J. P. Waldo and R. C. Larock, Org. Lett., 2005, 7, 5203 CrossRef CAS PubMed.
- D. D. Dolliver, B. T. Bhattarai, A. Pandey, M. L. Lanier, A. S. Bordelon, S. Adhikari, J. A. Dinser, P. F. Flowers, V. S. Wills and C. L. Scheier, J. Org. Chem., 2013, 78, 3676 CrossRef CAS PubMed.
- W. Kaewsri, C. Thongsornlleeb, J. Tummatorn and S. Ruchirawat, RSC Adv., 2016, 6, 48666 RSC.
- R. A. Day, J. A. Blake and C. E. Stephens, Synthesis, 2003, 1586 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00577c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*