
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA–affibody nanoparticle delivery system for cisplatin-based breast cancer chemotherapy†
Chao Zhanga,
HongLei Zhang *a,
MengNan Hana,
XueLi Yanga,
ChaoHong Peia,
ZhiDong Xuac,
Jie Du*a,
Wei Li*a and
Shengxi Chenb
*a,
MengNan Hana,
XueLi Yanga,
ChaoHong Peia,
ZhiDong Xuac,
Jie Du*a,
Wei Li*a and
Shengxi Chenb
aLaboratory of Medicinal Chemistry and Molecular Diagnosis of the Ministry of Education, College of Chemistry and Environmental Science, Hebei University, Baoding 071002, China. E-mail: liweihebeilab@163.com
bCenter for BioEnergetics, Biodesign Institute, Arizona State University, Tempe 85287, USA
cShijiazhuang Vince Pharmatech Co. Ltd., China
First published on 15th January 2019
Abstract
Cisplatin is the most widely used anticancer drug, but its side effects limit the maximum systemic dose. To circumvent the side effects, a DNA tetrahedron–affibody nanoparticle was prepared by combination of a DNA chain with cisplatin via interstrand crosslinks or adducts. Each nanocarrier can bind ∼68 molecules of cisplatin. This cisplatin nanoparticle exhibited high selectivity and inhibition for breast cancer HER2 overexpressing cells BT474 and lower toxicity in MCF-7 cells with low HER2 expression. The nano-drug inhibited the growth of BT474 cells by 94.57% at 512 nM (containing 33.3 μM cisplatin), which was higher than that of cisplatin (82.9%, 33.3 μM).
1. Introduction
Cisplatin and its derivatives are important drugs in the treatment of a variety of human cancers,1 including testicular cancer, breast cancer, head and neck cancer, and others.2 The mechanism of platinum chemotherapeutics involves DNA damage.3 Cisplatin could form interstrand crosslinks or adducts with the DNA strand.4 These adducts can cause the DNA duplex to bend, facilitating the binding of various proteins, and eventually leading to cell apoptosis. Cisplatin's interaction and reaction with other proteins has also been linked to cellular damage.5 Cisplatin drugs now account for almost 50% of clinically used anticancer therapeutic agents.6Despite its widespread clinical use, serious side effects are associated with the neurotoxicity of cisplatin and limit the maximum dose that can be administered.7 Moreover, cisplatin has a relatively short blood circulation time, resulting in suboptimal pharmacokinetics.8 Hence, there has been strong interest in developing novel platinum-based therapeutics, which can reduce the systemic toxicity of cisplatin and improve the efficacy of cancer treatments. Recently, much attention has been focused on creating drug delivery systems that can temporarily stabilize cisplatin and enable its transport to the tumor site. Candidate systems include liposomes, micelles, polymers, and inorganic nanoparticles.9–13 These systems enhance the permeability of cisplatin and its retention after deposition in tumor tissue. However, there are some obvious drawbacks, such as a lack of targeting, difficulty in releasing the drug, and unsatisfactory clinical outcomes. Consequently, more “active” delivery carriers are needed to improve tumor uptake.
DNA-based nanostructures as a drug delivery system have received more attention.14 DNA, as a genetic material, possesses high biocompatibility and low cytotoxicity. The ideal characteristics of DNA are convenient for application to the biomedical field.15 DNA also has excellent molecular properties, such as stability, mechanical rigidity, and nano-dimensions. It can be easily synthesized to a specified strand length to allow the formation of almost every shape of nanostructure.16 Moreover, it has a high drug loading efficiency and can be effectively internalized by cells.14 Thus, DNA nanoparticles represent a “smart” building block for the construction and development of versatile highly nontoxic drug nanocarriers. Kumar et al.17 reported an open-caged pyramidal DNA nanostructure for delivering doxorubicin (DOX), and this nanostructure significantly enhanced the cytotoxicity of the delivered doxorubicin to breast and liver cancer cells up to two-fold compared to free doxorubicin. Schüller et al.18 built a DNA origami tube decorated with 62-phosphate-(CpG) sequences that was both efficient and nontoxic, and it triggered higher levels of cellular immuno-stimulation than equal amounts of CpG. Furthermore, aptamers,19 antibody fragments,20 and affibodies21 have been linked to DNA nanostructures to provide active targeting to tumor tissues. In our previous work,21 we prepared a DNA–affibody nanoparticle that mimicked an antibody in its ability to specifically target the HER2 receptor. This nanoparticle could bind DOX to form a complex and exhibited greater selectivity for breast cancer cells overexpressing HER2 than DOX. Thus, considering the stability of cisplatin and DNA adduct, we aimed to use this DNA–affibody nanoparticle as a scaffold to deliver cisplatin drugs for treatment of HER2-overexpressing cancers.
2. Experimental section
2.1 Chemicals
The DNAs were synthesized from Sangon Biotech (Shanghai) Co., Ltd. N-(ε-maleimidocaproyloxy)succinimide ester (EMCS) and cis-platinum was purchased from Aladdin (Shanghai, China). Ni-NTA agarose and Sephadex G-25 were obtained from GE Healthcare (Piscataway, NJ, USA). Imidazole, sodium chloride, sodium acetate, polyacrylamide, trizma base, acetic acid, ethylenediaminetetraacetic acid (EDTA), magnesium chloride, ethanol, sodium diethyldithiocarbamate (DDTC) and DAPI were obtained from Sangon Biotech (Shanghai) Co., Ltd. Amicon® ultracentrifugal filters were purchased from Merck Millipore Ltd. (Darmstadt, Germany). Gibco® RPMI 1640 medium, trypsin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), dimethylsulfoxide (DMSO), antibiotic–antimycotic (100×), fetal bovine serum (FBS), and DAPI were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All chemicals were purchased and used without further purification.2.2 Preparation of DNA tetrahedron nanoparticle
The sequences of the four single-strand DNA were as shown in Table S1.† DNA1 (5.0 nmol), DNA2 (5.0 nmol), DNA3 (5.0 nmol), and DNA4 (5.0 nmol) were added to 4 mL of 0.9% NaCl. The reaction mixture was incubated at 95 °C for 10 min and then cooled to 4 °C over a period of 30 min. The obtained DNA tetrahedron nanoparticle was measured by the UV spectrometer (Nano Drop 2000c, Thermo Scientific, USA) and analyzed by 10% native polyacrylamide gel electrophoresis. The gel was run at 110 V for 1 h, and stained with Gel-Red (TransGene Biotech, Beijing, China).2.3 Determination of the stability of DNA nanoparticle in fetal bovine serum
Two samples in 100 μL of 0.9% NaCl were prepared as following: sample 1, 24 μM single strand DNA1; sample 2, 6 μM DNA tetrahedron. Each sample was added into 100 μL of fetal bovine serum and incubated at 37 °C. At the sampling time for every 2 h in 48 h, 5 μL of reaction mixture was taken out and added into 5 μL of loading buffer and then analyzed by 10% native polyacrylamide gel electrophoresis.2.4 Preparation of cisplatin–DNA tetrahedron nanoparticle
The DNA tetrahedron nanoparticle (1 μM) in 0.9% NaCl was treated with different concentration of cisplatin (17 mM, 33 mM, 66 mM, 132 mM) and incubated at 37 °C in dark for 2 h. Excess cisplatin was removed on a Sephadex G-25 column.2.5 Circular dichroism (CD) analysis
The structure of free DNA tetrahedron nanoparticle (0.1 μM) and cisplatin–DNA tetrahedron nanoparticle under same buffer condition (0.9% NaCl) was analyzed by circular dichroism (CD) spectroscopy (Bio-logic Mos-450, France). A 1 mm path length cuvette was used, which allowed a small volume of concentrated nanostructure solution to be measured. The CD spectra of nanostructure were also subtracted from background CD of cisplatin–DNA tetrahedron in same buffer.2.6 Preparation of DNA–affibody
The sequence of the affibody used in this study was MIHHHHHHLQVDNKFNKEMRNAYWEIALLPNLNNQQKRAFIRSLYDDPSQSANLLAEAKKLNDAQAPKVDC. The affibody was expressed in E. coli cells and purified using a Ni-NTA column.The process for preparing DNA tetrahedron–affibody nanoparticle was shown in Fig. S1.† DNA1 or DNA2 (200 μg, 10.3 nmol) was dissolved in 160 μL of 0.9% NaCl and treated with 40 μL of 10 mM Nε-malemidocaproyloxysuccinimide ester (EMCS) in dimethyl sulfoxide. The reaction mixture was incubated at room temperature for 3 h and stopped by adding of 20 μL of 3 M NaOAc. After the addition of 600 μL of ethanol and incubation at 4 °C for 30 min, the reaction mixture was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min. The DNA was dissolved in 50 μL of NaCl buffer and treated with 300 μg (38.1 nmol) of affibody in 300 μL of PBS buffer for incubation at room temperature for 5 h. Then, the reaction mixture was purified on a DEAE-Sepharose column, which was eluted with PBS buffer containing 0.2–0.9 M NaCl. The purified DNA–affibody chimera was analyzed by 10% denaturing polyacrylamide gel electrophoresis (PAGE). The elution from the previous step was continued by purification on a Ni-NTA chromatography column. The elution solution (900 μL) was loaded on a column containing 100 μL of Ni-NTA resin. Then the column was washed five times with 100 μL of 50 mM Tris·HCl, pH 8.0, containing 300 mM NaCl and 10 mM imidazole. Finally, the Ni-NTA column was eluted three times with 100 μL of 50 mM Tris·HCl, pH 8.0, containing 300 mM NaCl and 150 mM imidazole. Aliquots of each fraction were analyzed by 10% SDS-PAGE. The affibody–DNAs obtained were concentrated using Amicon® ultracentrifugal filters (MW cutoff 10 kDa).
000g for 30 min. The DNA was dissolved in 50 μL of NaCl buffer and treated with 300 μg (38.1 nmol) of affibody in 300 μL of PBS buffer for incubation at room temperature for 5 h. Then, the reaction mixture was purified on a DEAE-Sepharose column, which was eluted with PBS buffer containing 0.2–0.9 M NaCl. The purified DNA–affibody chimera was analyzed by 10% denaturing polyacrylamide gel electrophoresis (PAGE). The elution from the previous step was continued by purification on a Ni-NTA chromatography column. The elution solution (900 μL) was loaded on a column containing 100 μL of Ni-NTA resin. Then the column was washed five times with 100 μL of 50 mM Tris·HCl, pH 8.0, containing 300 mM NaCl and 10 mM imidazole. Finally, the Ni-NTA column was eluted three times with 100 μL of 50 mM Tris·HCl, pH 8.0, containing 300 mM NaCl and 150 mM imidazole. Aliquots of each fraction were analyzed by 10% SDS-PAGE. The affibody–DNAs obtained were concentrated using Amicon® ultracentrifugal filters (MW cutoff 10 kDa).
2.7 Preparation of cisplatin–DNA tetrahedron–affibody nanoparticle
DNA1–affibody (10.0 nmol), DNA2–affibody (10.0 nmol), DNA3 (10.0 nmol), and DNA4 (10.0 nmol) were added to 8 mL of NaCl buffer. The reaction mixture was incubated at 70 °C for 10 min then cooled to room temperature over a period of 30 min. Then, DNA tetrahedron–affibody nanoparticle was prepared based on DNA self-assembly. The obtained DNA tetrahedron–affibody nanoparticle was analyzed by 10% native polyacrylamide gel electrophoresis. The operation for preparation and analyzation process was carried out based on the previous studies.21 Finally, the DNA tetrahedron–affibody nanoparticle was concentrated using ultra centrifugal filters (MW cutoff 50 kDa). The concentrated DNA tetrahedron–affibody nanoparticle (1 μM) in 100 μL of 0.9% NaCl was treated with 10 μL of 66 mM cisplatin and incubated at 37 °C in dark for 2 h (Fig. S1†). Excess cisplatin was removed on a Sephadex G-25 column.2.8 Atomic force microscopy (AFM) characterization
For DNA tetrahedron–affibody and cisplatin–DNA tetrahedron–affibody nanoparticle imaging, 10 μL samples (10 nM) were deposited onto a freshly peeled mica surface for 5 min. Next, 40 mL of TAE/Mg2+ buffer (40 mM Tris, 20 mM acetic acid, 2 mM EDTA, 12 mM MgCl2, pH 8.0) was added in the mica and then dried in air at room temperature. The samples were imaged with AFM (Agilent Technologies, 5500 AFM/SPM System, USA).2.9 Quantification of the cisplatin/DNA tetrahedron–affibody ratio
Excess free cisplatin 100 μL in the previous step was added to 100 μL of DDTC in NaOH. Samples were incubated at 37 °C for 30 min and extracted with 500 μL of chloroform. The two layers were separated by centrifugation at 13000 rpm for 10 min. Finally, 400 μL of the chloroform layer was concentrated to dryness in vacuum (Eppendorf 5301, United Kingdom). The residue was dissolved in 200 μL of acetonitrile and 15 μL injected into the High-Performance Liquid Chromatography (Ultimate 3000, Thermo Scientific, USA) equipped with an Agilent Extend C-18 column (4.6 × 250 mm, 5.0 μM, Agilent) and a UV detector. A flow rate was adjusted to 1.0 mL min−1 with a gradient of pure water (Phase A) and acetonitrile (Phase B) by the following method: 0–15 min (75% B), 15–16 min (75–90% B), 16–21 min (90% B), 21–22 min (90–75% B). The amount of cisplatin binding to the DNA tetrahedron in the detection sample was calculated using the equation: the ratio of cisplatin/DNA = (nInitial cisplatin − nexcess free cisplatin)/npurified cisplatin–DNA tetrahedron.
2.10 Release assay of cisplatin
Three samples, 1 mL for each sample, were prepared as follows: sample 1, 300 μM cisplatin in 0.9% NaCl; sample 2, 4.6 μM cisplatin–DNA tetrahedron–affibody (containing 300 μM cisplatin) in 0.9% NaCl; sample 3, 4.6 μM cisplatin–DNA tetrahedron–affibody (containing 300 μM cisplatin) and 600 units of DNase I in 0.9% NaCl. Each sample was transferred into a dialysis tube (1 mL, MW cutoff 10 kDa), which was floated in 100 mL 1× phosphate-buffered saline (PBS, pH 7.4) and continuously stirred at room temperature. At the sampling time for every 2 h in 48 h, 100 μL of PBS buffer was taken out and the concentration of cisplatin was measured by HPLC.2.11 Detection of targeting of cisplatin–DNA tetrahedron–affibody
BT474 breast cancer cells (ATCC® HTB-20, overexpression of HER2) and MCF-7 breast cancer cells (ATCC® HTB-22, low expression of HER2 receptor) were cultured at 37 °C in a 5% CO2 atmosphere and grown in Gibco® RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic mix antibiotic supplement before use.BT474 cells and MCF-7 cells were grown on confocal small dish at a cell density of 1 × 105 cells per well at 37 °C for 24 h. When the cell confluency reached about 70%, the cells were treated with cisplatin and cisplatin–DNA tetrahedron–affibody at 1 μM concentration for 1 h. Then the cells were stained using 2.5 μg mL−1 DAPI for 30 min after the cells were rinsed with PBS for two times. Finally, the cells were fixed with 4% paraformaldehyde for 20 min. The fluorescent images were obtained using Olympus confocal laser scanning microscopy (Olympus Fluoview FV1000) with red and green filters. All images were recorded and the target cells counted using a 40× oil objective.
2.12 Biological activity of nanoparticles
Exponentially growing BT474 cells and MCF-7 cells were harvested and plated in 96-well plates at a concentration of 2 × 104 cells per well. After incubation at 37 °C for 24 h, the cells were treated with cisplatin and cisplatin–DNA tetrahedron–affibody nanoparticle at different concentrations for an additional 48 or 72 h. Then 20 μL of MTT (5 mg mL−1) was added to each well and the plates were incubated at 37 °C for 4 h. The supernatant was discarded, and 100 μL of DMSO was added to each well. The absorbance was recorded at 570 nm after 15 min. Inhibition of cell growth was obtained by the following formula: inhibition of cell growth (%) = (ODnegative control − ODtreatment) × 100%/(ODnegative control − ODbackground). Data are reported as the mean of three independent experiments, each run in quintuplicate.3. Results and discussion
3.1 Evaluation of DNA tetrahedron
A DNA tetrahedron was prepared and used as a vehicle to covalently bind multiple copies of cisplatin.21 This DNA tetrahedron was analyzed using UV-Vis spectrophotometry and native polyacrylamide gel electrophoresis (PAGE) (ESI, Fig. S2†). The stability of the DNA tetrahedron in fetal bovine serum (FBS) was determined. Gel electrophoresis showed that the band for the nanostructure remained almost unchanged up to 4 h of incubation, reflecting the presence of intact DNA nanoparticles in FBS (Fig. S2†). Extended incubation led to smeared bands, suggesting partial degradation of the nanostructure. However, the band for the nanostructure was still observed with attenuated intensity even after 24 h of incubation. On the contrary, single-stranded DNA was almost completely degraded by the strong degradation action of nucleases within only 2 h of incubation in FBS, which was consistent with the report by Li.22 Thus, these results indicated that the DNA tetrahedral structure as a drug carrier was relatively stable.3.2 Analysis of cisplatin–DNA tetrahedron complexes
The DNA tetrahedron was treated with 17 mM cisplatin and incubated at 37 °C in the dark for 2 h to form a cisplatin–DNA tetrahedron structure. The obtained cisplatin–DNA tetrahedron was purified using a Sephadex G-25 column. Circular dichroism (CD) spectroscopy, a powerful tool for monitoring DNA structure changes,23 was used to examine the structural integrity of cisplatin–DNA tetrahedron. As shown in Fig. 1b, the free DNA tetrahedron had three major peaks at 221 (positive), 248 (negative), and 277 (positive), which was almost consistent with the CD spectra of double-helical DNA.24 However, when cisplatin was added to the DNA solution, cisplatin underwent aquation to form more reactive [Pt(NH3)2Cl(OH2)]+ and [Pt(NH3)2(OH2)2]2+ species. The reactive platinum species then bound the DNA by forming coordination bonds with purine bases at the N-7 positions. This reaction resulted in primarily 1,2-intrastrand or 1,3-intrastrand crosslinks or adducts (Fig. 1a).4 Therefor, the binding of cisplatin to DNA changed the intensities of the original CD peaks, i.e., a decreased positive peak at 221 nm, an increased negative peak at 248 nm and a decreased positive peak at 277 nm, which indicated that cisplatin was intercalated into DNA tetrahedron.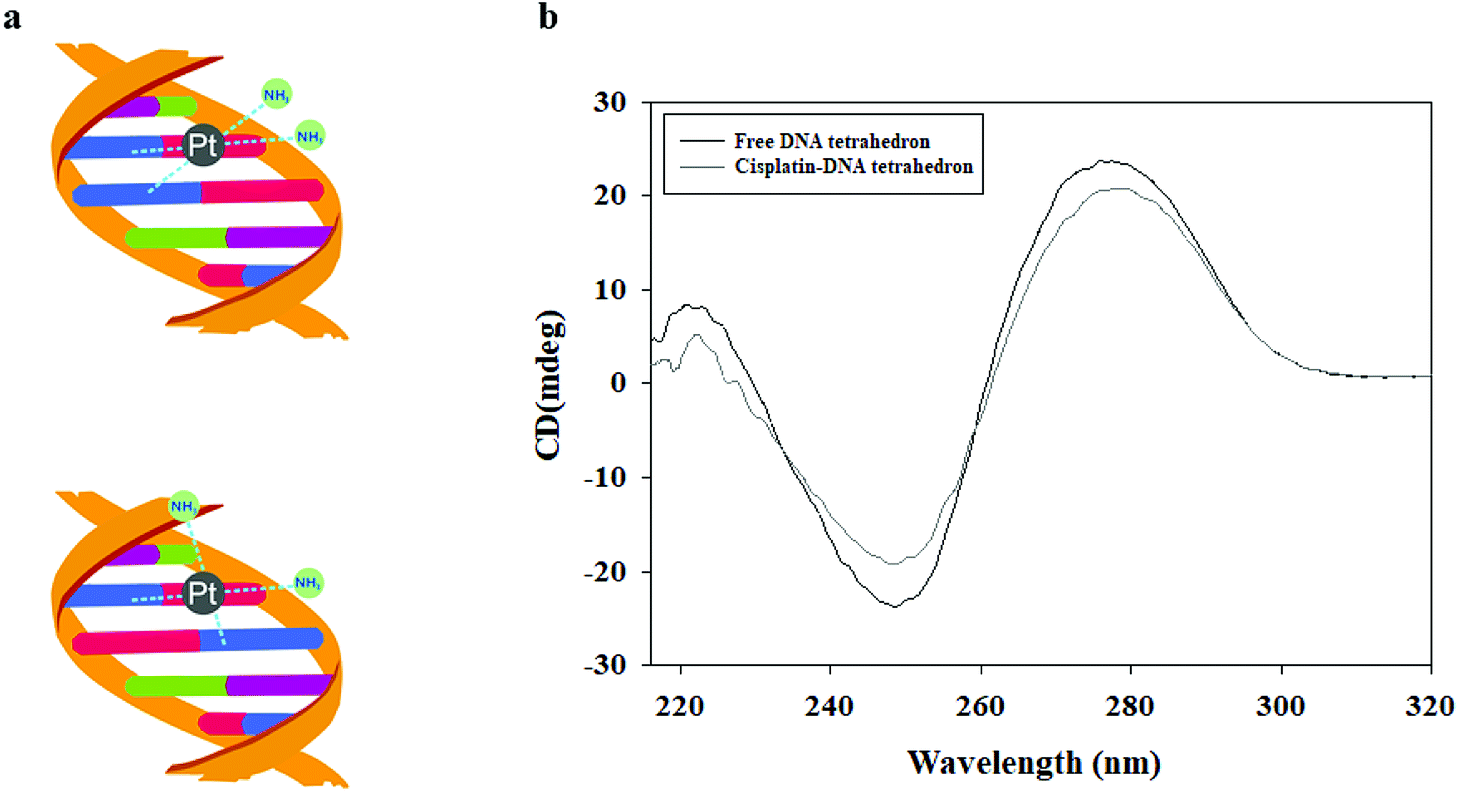 | ||
| Fig. 1 The interaction between DNA tetrahedron and cisplatin. (a) The predicted structure of DNA tetrahedron and cisplatin. (b) CD spectra of the free DNA tetrahedron and DNA tetrahedron grafted cisplatin. | ||
3.3 Structure of cisplatin–DNA tetrahedron–affibody nanoparticle
To impart targeting ability to this drug carrier, an DNA tetrahedron–affibody was prepared, which contained two affibody molecules and one DNA tetrahedron. The affibody was used to target antigen HER2 on the cancer cell surface. The structure of the cisplatin–DNA tetrahedron–affibody nanoparticle was characterized using 5% native PAGE (Fig. S3†) and atomic force microscopy (AFM) (Fig. 2). The apparent size of DNA tetrahedron–affibody and cisplatin–DNA tetrahedron–affibody were between 10 nm and 15 nm. The size of DNA tetrahedron–affibody nano-particle became slightly smaller after binding cisplatin, which was caused by cross-linking between DNA tetrahedron and cisplatin. The size of this nanometer particle is much smaller than that of the antibody (40 nm to 2 μm), which is more favourable for the drug to enter the cancer cells and take effect. | ||
| Fig. 2 AFM micrographs of nanoparticles. (a) Structure of DNA tetrahedron–affibody nanoparticle; (b) structure of cisplatin–DNA tetrahedron–affibody nanoparticle. Scale bars are 20 nm. | ||
3.4 Assay for binding of cisplatin with DNA and releasing of cisplatin from DNA
The ability of the DNA tetrahedron–affibody to deliver cisplatin was also determined. Since the absorption intensity of cisplatin at UV light was not strong enough, the amount/content of cisplatin could not be determined directly by its UV absorption. Thus, we developed an HPLC method for post-derivation of cisplatin by DDTC to increase its UV absorbance.25 The binding process of cisplatin with DNA was shown in Fig. S4.† The amount of cisplatin bound to the DNA tetrahedron–affibody in the detection sample was calculated, and the maximum ratio of cisplatin to DNA was 68.2 ± 1.4 based on triplicate assays (Fig. 3a). The drug loading capacity of DNA tetrahedron–affibody was higher than antibody molecule, which can deliver only a few molecules of the associated small molecule drug.26 The strong binding capability between cisplatin and DNA is reflected in the following two aspects. One is the coordination bond between the two molecules, making the stability strong enough to be unaffected by the solution. The other one is the easy access of cisplatin to DNA duplex and its combination with purine base due to the small steric hindrance of cisplatin. Besides, more cisplatin loading with DNA tetrahedron–affibody would avoid the degradation in vivo and increase the uptake of tumor cells. The toxicity of cisplatin would be reduced accordingly.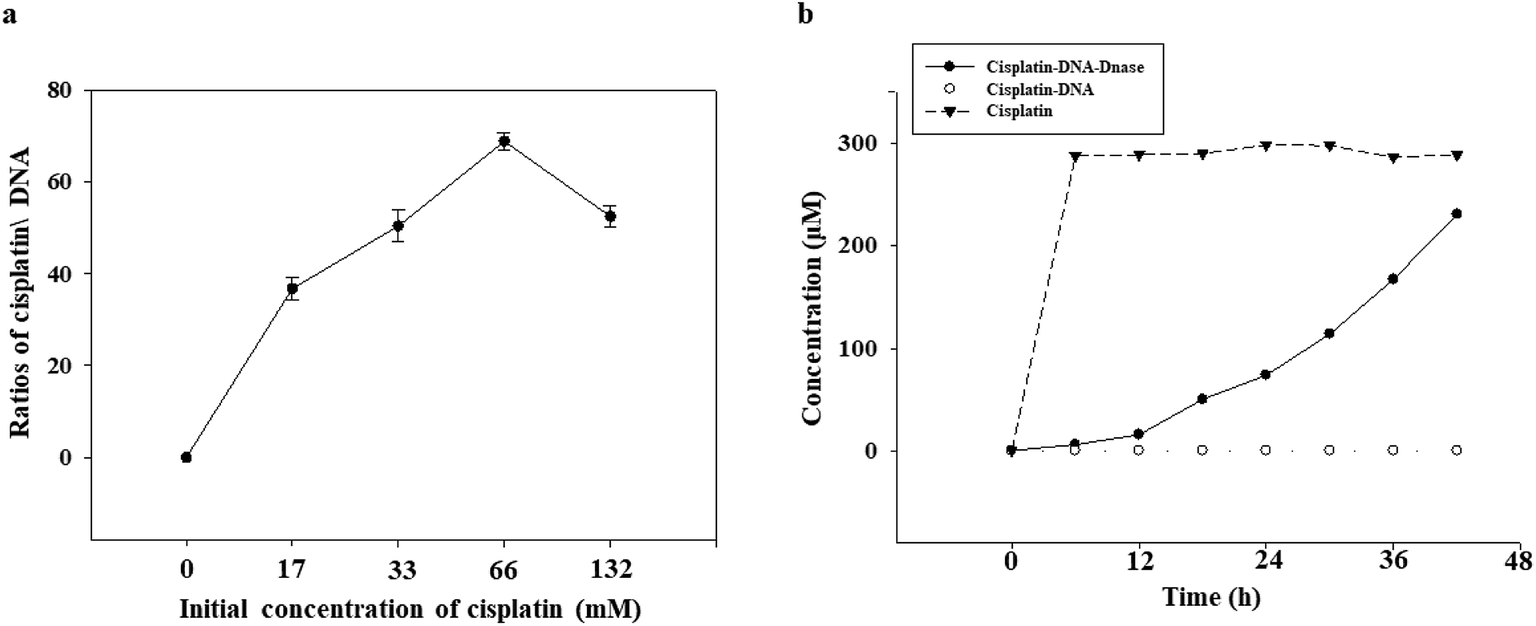 | ||
| Fig. 3 Binding and release assay of cisplatin. (a) Quantification of the cisplatin/DNA ratio in the cisplatin–DNA tetrahedron–affibody nanoparticle. Cisplatin–DNA tetrahedron–affibody nanoparticles were purified using a Sephadex G-25 column, and excess free cisplatin was subjected to derivatization by DDTC and analyzed with HPLC. The amount of cisplatin bound to the DNA tetrahedron–affibody was calculated in the detected sample. (b) Cisplatin release assay from the cisplatin–DNA tetrahedron–affibody nanoparticle. The concentration of cisplatin was measured by HPLC at designated time. | ||
The release of cisplatin from the DNA tetrahedron–affibody is another significant factor to affect its drug effect. The result was investigated and analysed. As shown in Fig. 3b, cisplatin was gradually released under the action of DNase I with time, and after 48 h, almost all cisplatin was released. These results indicated that the cisplatin was released rapidly and effectively from DNA tetrahedron–affibody and the drug activity performed. Moreover, compared with other nano-carriers like micelle, polymer and inorganic nano-particles, DNA tetrahedron–affibody carrier can be decomposed to deoxyribonucleic acid and amino acids, which will not be accumulated in vivo.
3.5 Assay for targeting of cisplatin–DNA tetrahedron–affibody
Affibody can mimic the antibody in its ability to specifically target the HER2 receptor. Therefore, a HER2 overexpressing cell line, BT474, and a HER2 low-expressing cell line, MCF-7, were used to evaluate the targeting of the cisplatin–DNA tetrahedron–affibody to the HER2 receptor and to compare it with cisplatin. The HER2 receptor was highly overexpressed in the BT474 breast cancer cell line, whereas it was expressed at a low level in the MCF-7 breast cancer cell line.27 Since cisplatin itself did not produce fluorescence, a fluorescent FAM label was added to the DNA tetrahedron. In the binding assay (Fig. 4), the cisplatin–DNA tetrahedron–affibody nanoparticle bound to the BT474 HER2-overexpressing cancer cells with greater affinity and higher selectivity compared to that for the MCF-7 HER2 low-expressing cell line. Specifically, its affinity for the BT474 HER2-overexpressing cells was three-fold higher than that for the MCF-7 HER2 low-expressing cells (Fig. S5†). These results manifested that the newly established nano-drugs had better target and a higher capacity to carry a certain amount of cisplatin than general antibody molecular drugs.26 Thus, this nano-drug if of potential for future application in preventing premature degradation of the cisplatin or interacting with healthy tissue and reducing systemic toxicity.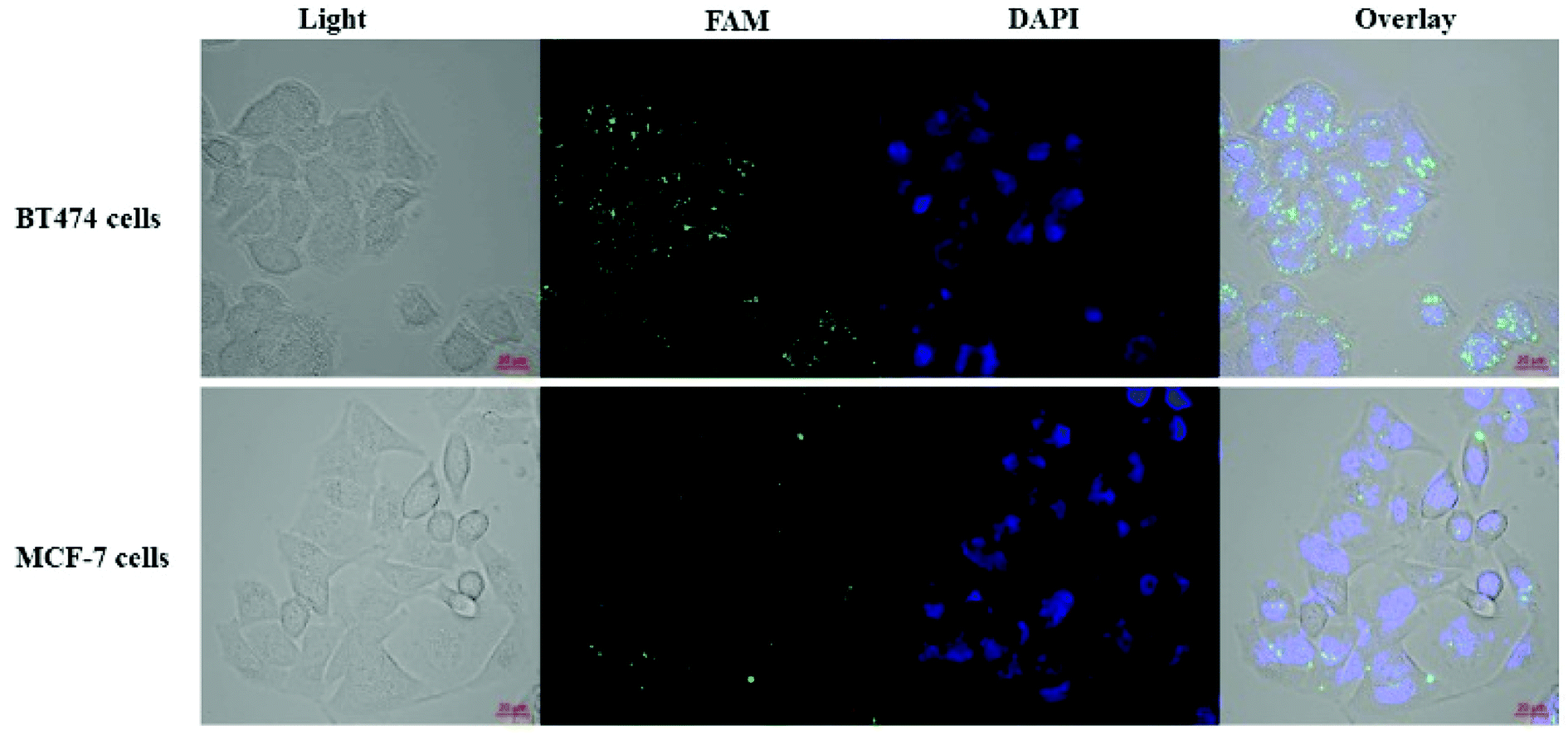 | ||
| Fig. 4 Binding assay using breast cancer cells. BT474 HER2-overexpressing cancer cells and MCF-7 cancer cells with low HER2 expression were incubated with the cisplatin–DNA tetrahedron–affibody nanoparticle for 1 h. The fluorescence images were obtained using a confocal laser scanning microscopy. | ||
3.6 Cytotoxicity evaluation
The inhibitory activity of the nanoparticle in HER2-overexpressing breast cancer cells was also examined using cisplatin as a reference. As shown in Fig. 5, 3.3 μM, 16.7 μM, and 33.3 μM cisplatin inhibited BT474 cell growth by 17%, 28%, and 68%, respectively, after 48 h. The nano-drug (1:68 cisplatin–DNA tetrahedron–affibody) inhibited the growth of BT474 cells by 27%, 39%, and 74% at 51 nM, 257 nM, and 512 nM, respectively, after 48 h. Cell growth inhibition by cisplatin and cisplatin–DNA tetrahedron–affibody increased after treatment for 72 h. The maximum rate of cell growth inhibition reached to 82.9% and 94.57% at a concentration of 33.3 μM cisplatin and 512 nM nano-drug, respectively. Comparatively, at a lower concentration (3.3 μM cisplatin vs. 51 nM nano-drug), both cisplatin and the nano-drug showed poor cell growth inhibition. However, at a higher concentration (33.3 μM cisplatin vs. 512 nM nano-drug), they exhibited excellent inhibition. The nano-drug (1:68 cisplatin–DNA tetrahedron–affibody) exhibited 1.6-fold higher growth inhibition of BT474 cells at a concentration of 512 nM (containing 33.3 μM cisplatin) after treatment for 72 h. Thus, these results indicated that cisplatin–DNA tetrahedron–affibody was more inhibitory in HER2 high-expressing cells than cisplatin.
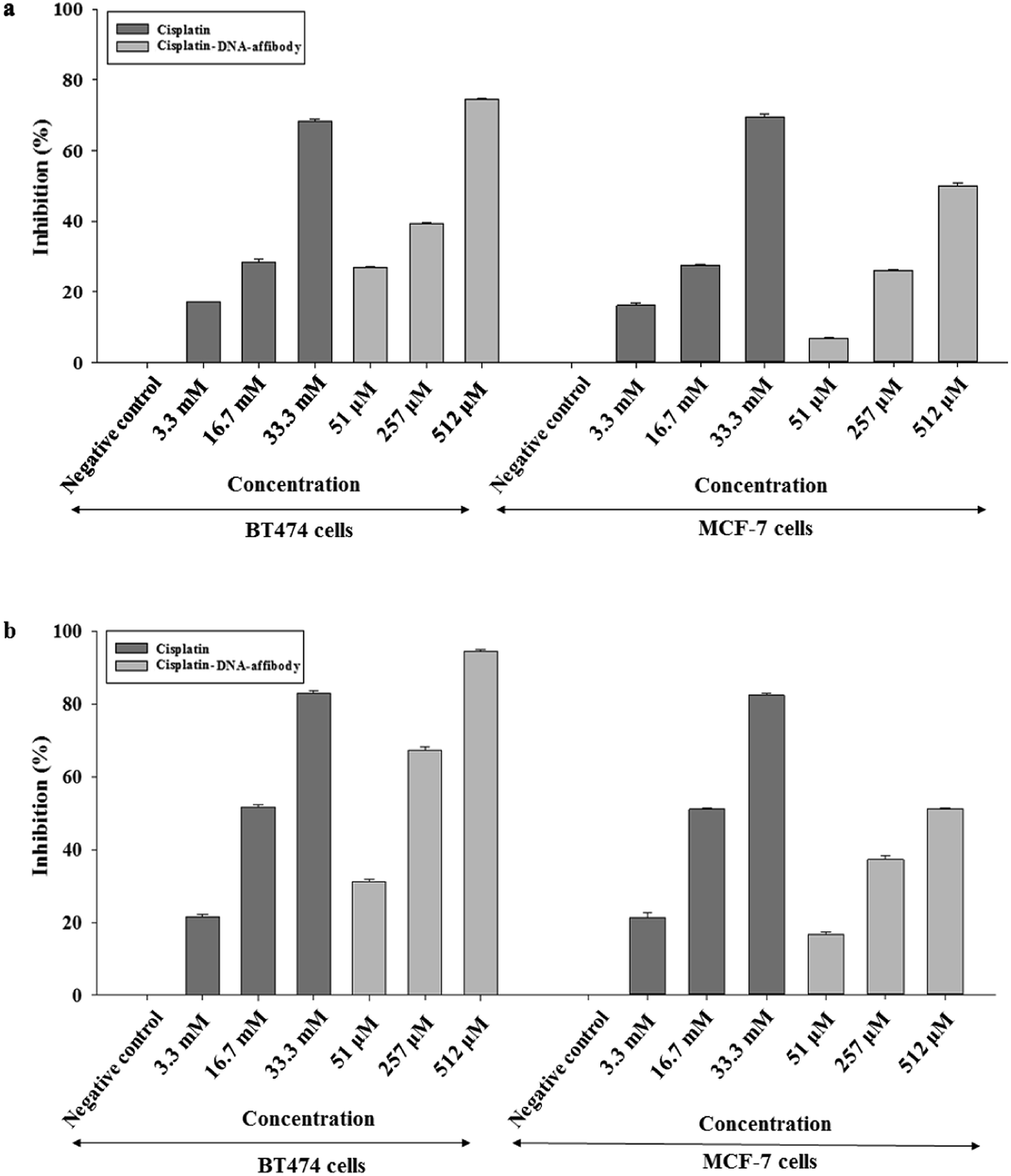 | ||
| Fig. 5 Inhibition of cell growth in BT474 and MCF-7 cancer cells by the cisplatin–DNA tetrahedron–affibody nanoparticle. The ratio of cisplatin–DNA tetrahedron–affibody and cisplatin was 1:68. Cell growth was measured using an MTT assay after (a) 48 h and (b) 72 h of treatment with cisplatin and cisplatin–DNA tetrahedron–affibody. The results are expressed as a percentage of the control as the mean ± standard deviation. | ||
However, when the nano-drug was used in HER2 low-expressing cell MCF-7, the cell growth inhibition trend was opposite to the nano-drug used in the HER2 overexpressing cell BT474. The average percent growth inhibition by cisplatin was 1.2–2.3 fold higher than nano-drug at 3.3 μM concentration; 1.0–1.3 fold higher than nano-drug at 16.7 μM; and 1.3–1.6 fold higher than nano-drug at 33.3 μM after 48 and 72 h treatments, respectively. The results of the MCF-7 cell assay also revealed that the cisplatin–DNA tetrahedron–affibody had much lower toxicity than cisplatin in HER2 low-expressing cells.
The possible route and mechanism of cisplatin–DNA tetrahedron–affibody for the treatment of HER2-overexpressing breast cancer was illustrated in Fig. 6. Affibody specifically targeted and bound to the HER2 receptor on the surface of breast cancer cells, thus enabling cisplatin–DNA tetrahedron–affibody to accumulate in cancer cells. Subsequently, cisplatin–DNA tetrahedron–affibody entered the cytoplasm by fusing with cell membranes or by endocytosis and then was digested by deoxyribonucleases and proteases to release cisplatin.28 Cisplatin interacted with DNA of cancer cells, causing DNA to fail to transcribe and translate properly, and finally to apoptosis. Another possible process was assumed to be direct interaction between cisplatin and protein, resulting in the dysfunction of protein and apoptosis of cells.29 It is inferred that loading and carrying of cisplatin with DNA tetrahedron–affibody greatly increased the uptake concentration of cisplatin in cytoplasm, which is different from the transport mechanism of cisplatin through copper transporter into cell membrane.30 We can get a conclusion from the experimental results that cisplatin–DNA tetrahedron–affibody is of targeting, high drug-loading and low toxicity. Further investigation is being carried out to optimize the drug loading system and to expand its application field.
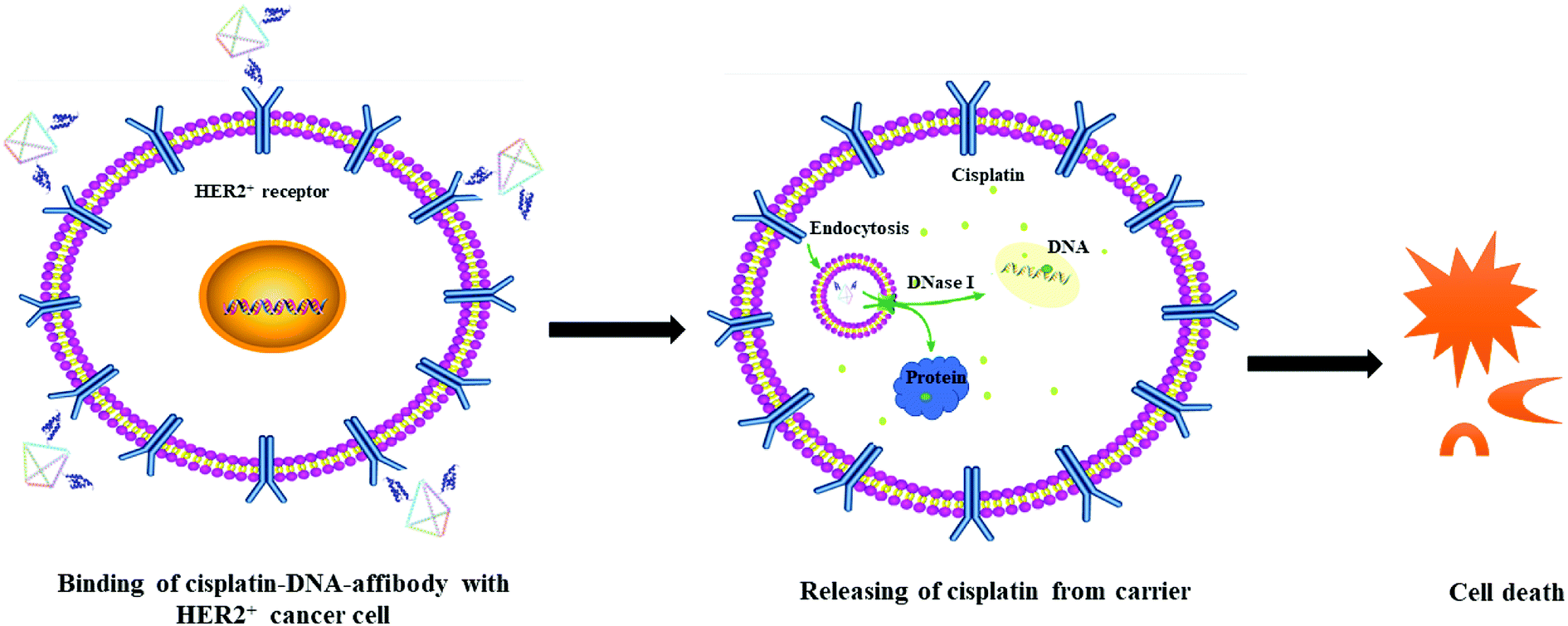 | ||
| Fig. 6 The schematic illustration of the mechanism of cisplatin–DNA tetrahedron–affibody in treatment of HER2-overexpressing cancer cells. | ||
4. Conclusions
A DNA tetrahedron–affibody nano-carrier was constructed and could deliver cisplatin effectively. This structure has a small size but exhibits greater drug-loading capacity. This novel nano-drug with high selectivity can specifically target HER2-overexpressing breast cancer cells. It also displays strong growth inhibition activity in HER2-overexpressing cancer cells. Comparatively, it has lower growth inhibitory activity against HER2 low-expressing cancer cells. This nanocarrier is a good candidate to be developed into a drug with high specificity, high efficacy, and low toxicity for the treatment of HER2-overexpressing breast cancers. In our future work, this cisplatin–DNA tetrahedron–affibody drug will be applied in a mouse model to verify the targeting effect and therapeutic outcome of the drug.Conflicts of interest
There is no competing financial interest in this work.Acknowledgements
This work was financially supported by the Natural Science Foundation of Hebei Province (grant number B2016201031), the Hebei Province Science Foundation for High-level Personnel (grant number GCC2014013), and the Hebei University Science Foundation (grant number 3333112).Notes and references
- J. Ma, X. Hu, J. Li, D. Wu, Q. Lan, Q. Wang, S. Tian and W. Dong, Oncotarget, 2017, 8, 85926–85939 Search PubMed.
- F. Liang, S. Zhang, H. Xue and Q. Chen, BMC Cancer, 2017, 17, 871 CrossRef PubMed.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- C. F. Harrington, R. C. Le Pla, G. D. Jones, A. L. Thomas and P. B. Farmer, Chem. Res. Toxicol., 2010, 23, 1313–1321 Search PubMed.
- R. J. Browning, P. J. T. Reardon, M. Parhizkar, R. B. Pedley, M. Edirisinghe, J. C. Knowles and E. Stride, ACS Nano, 2017, 11, 8560–8578 CrossRef CAS PubMed.
- S. Amptoulach and N. Tsavaris, Chemother. Res. Pract., 2011, 2011, 843019 Search PubMed.
- G. Cossa, L. Gatti, F. Zunino and P. Perego, Curr. Med. Chem., 2009, 16, 2355–2365 CrossRef CAS PubMed.
- P. Eisenberg, F. R. MacKintosh, P. Ritch, P. A. Cornett and A. Macciocchi, Ann. Oncol., 2004, 15, 330–337 CrossRef CAS PubMed.
- Z. Yue and Z. Cao, Curr. Cancer Drug Targets, 2016, 16, 480–488 CrossRef CAS.
- H. Liu, Y. Zhang, Y. Han, S. Zhao, L. Wang, Z. Zhang, J. Wang and J. Cheng, Colloids Surf., B, 2015, 131, 12–20 CrossRef CAS PubMed.
- H. Zhou, G. Wang, Y. Lu and Z. Pan, Biomater. Sci., 2016, 4, 1212–1218 RSC.
- P. J. Reardon, M. Parhizkar, A. H. Harker, R. J. Browning, V. Vassileva, E. Stride, R. B. Pedley, M. Edirisinghe and J. C. Knowles, Int. J. Nanomed., 2017, 12, 3913–3926 CrossRef CAS PubMed.
- S. Spreckelmeyer, N. Estrada-Ortiz, G. G. H. Prins, M. van der Zee, B. Gammelgaard, S. Stürup, I. A. M. de Graaf, G. M. M. Groothuis and A. Casini, Metallomics, 2017, 9, 1786–1795 RSC.
- J. Li, C. Fan, H. Pei, J. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- S. Surana, A. R. Shenoy and Y. Krishnan, Nat. Nanotechnol., 2015, 10, 741–747 CrossRef CAS PubMed.
- Y. J. Chen, B. Groves, R. A. Muscat and G. Seeling, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- V. Kumar, S. Bayda, M. Hadla, I. Caligiuri, C. Russo Spena, S. Palazzolo, S. Kempter, G. Corona, G. Toffoli and F. Rizzolio, J. Cell. Physiol., 2016, 231, 106–110 CrossRef CAS PubMed.
- V. J. Schüller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 5, 9696–9702 CrossRef PubMed.
- G. Zhu, J. Zheng, E. Song, M. Donovan, K. Zhang, C. Liu and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7998–8003 CrossRef CAS PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- Y. Zhang, S. Jiang, D. Zhang, X. Bai, S. M. Hecht and S. Chen, Chem. Commun., 2017, 53, 573–576 RSC.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef CAS PubMed.
- N. Berova, K. Nakanishi and R. W. Woody, Circular Dichroism: Principles and Applications, Wiley-VCH, New York, 2nd edn, 2000, pp. 703–718 Search PubMed.
- B. I. Kankia, V. Bukin and V. A. Bloomfiled, Nucleic Acids Res., 2001, 29, 2795–2801 CrossRef CAS PubMed.
- R. Raghavan, M. Burchett, D. Loffredo and J. A. Mulligan, Drug Dev. Ind. Pharm., 2000, 26, 429–440 CrossRef CAS PubMed.
- J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRef CAS PubMed.
- K. Subik, J. F. Lee, L. Baxter, T. Strzepek, D. Costello, P. Crowley, L. Xing, M. C. Hung, T. Bonfiglio, D. G. Hicks and P. Tang, Breast Cancer: Basic Clin. Res., 2010, 4, 35–41 Search PubMed.
- S. Yuan, X. Ding, Y. Cui, K. Wei, Y. Zheng and Y. Liu, Eur. J. Inorg. Chem., 2016, 2017, 1778–1784 CrossRef.
- G. P. Stathopoulos, D. Antoniou, J. Dimitroulis, P. Michalopoulou, A. Bastas, K. Marosis, J. Stathopoulos, A. Provata, P. Yiamboudakis, D. Veldekis, N. Lolis, N. Georgatou, M. Toubis, Ch. Pappas and G. Tsoukalas, Ann. Oncol., 2010, 21, 2227–2232 CrossRef CAS PubMed.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08735k |
| This journal is © The Royal Society of Chemistry 2019 |