
Anion–π and anion–π-radical interactions in bis(triphenylphosphonium)-naphthalene diimide salts†
Chee Koon
Ng‡
 a,
Teck Lip Dexter
Tam‡
*a,
Fengxia
Wei
a,
Xuefeng
Lu
b and
Jishan
Wu
b
a,
Teck Lip Dexter
Tam‡
*a,
Fengxia
Wei
a,
Xuefeng
Lu
b and
Jishan
Wu
b
aInstitute of Materials Research and Engineering (IMRE), Agency of Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis, #08-03, Singapore 138634, Singapore. E-mail: dexter-tam@imre.a-star.edu.sg
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore, 117543, Singapore
First published on 21st November 2018
Abstract
We have conducted a systematic study of anion–π and anion–π-radical interactions in bis(triphenylphosphonium)-naphthalene diimide (BTPP-NDI) salts which was made possible with the synthesis of different anionic analogues. DFT calculations and experimental evidence show anion–π and anion–π-radical interactions and that the cationic π-radical can induce magnetic spin polarization of the anion.
Introduction
Anion–π interactions have been widely studied due to their potential applications in anion recognition,1 ion transport,2 catalysis3 and anion doping.4 Recently, organic π-radicals have also attracted academic interest due to their connection with polarons/solitons in the electrical and magnetic properties of doped conjugated polymers.5 However, the nature of the interactions between the dopant/counter-ion and doped conjugated polymers is not well understood. In many cases, the doped conjugated polymers involve oxidized cationic or reduced anionic radical species that are chemically unstable towards air and/or water,6 making the study of such interactions challenging. The recent advances in molecular design has enabled the syntheses of stable organic π-radicals.7 Theoretical calculations on the influence of anion–π interactions on the magnetic spin polarization of the anion have also been reported.8 Here we leverage on a stable bis(triphenylphosphonium)-naphthalene diimide (BTPP-NDI) cationic radical reported by Mukhopadhyay's group7a to study the effect of anion–π-radicals by systematic variation of the anion. We found that weakly Lewis basic anions are able to induce subtle red shifts in the solution UV-Vis absorption that is dominated by an optical transition from the cationic π-radical in the 1+ salts. Very weakly Lewis basic π-anions like NO3− interact strongly with the NDI π system via π–π interactions in the 2+ salt and behaves as an electron acceptor in the 1+ salt that can undergo photoinduced electron transfer (PET) from the radical cation to NO3−. The radical cation is also able to induce magnetic spin polarization of the anions as observed from NMR. Our results provide evidence for elusive anion–π-radical interactions and thus allow a better understanding of organic ferromagnets and p-doped conductors.Results and discussion
Synthetic procedure
The syntheses of the BTPP-NDI compounds are shown in Scheme 1. 1-Br was synthesized using reported procedures,7a with the solvent replaced by 1,4-dioxane. 1-Br was oxidized using bromine to give 1-2Br. 1-PF6, 1-2PF6, 1-Cl and 1-2Cl were obtained via anion metathesis. 1-2NO3 was obtained via reacting 1-Br with silver nitrate, and subsequent reduction using excess triethylamine yielded 1-NO3. Br2-NDI was reacted with sodium iodide to give I2-NDI, which can undergo a similar reaction to give 1-I. Subsequent oxidation of 1-I with I2 did not yield the 2+ diiodide. Attempts to obtain 1-2I using anion metathesis with sodium iodide and 1-2Br resulted in the formation of 1-I instead. The neutral compound 1 can be isolated via reduction with hydrazine.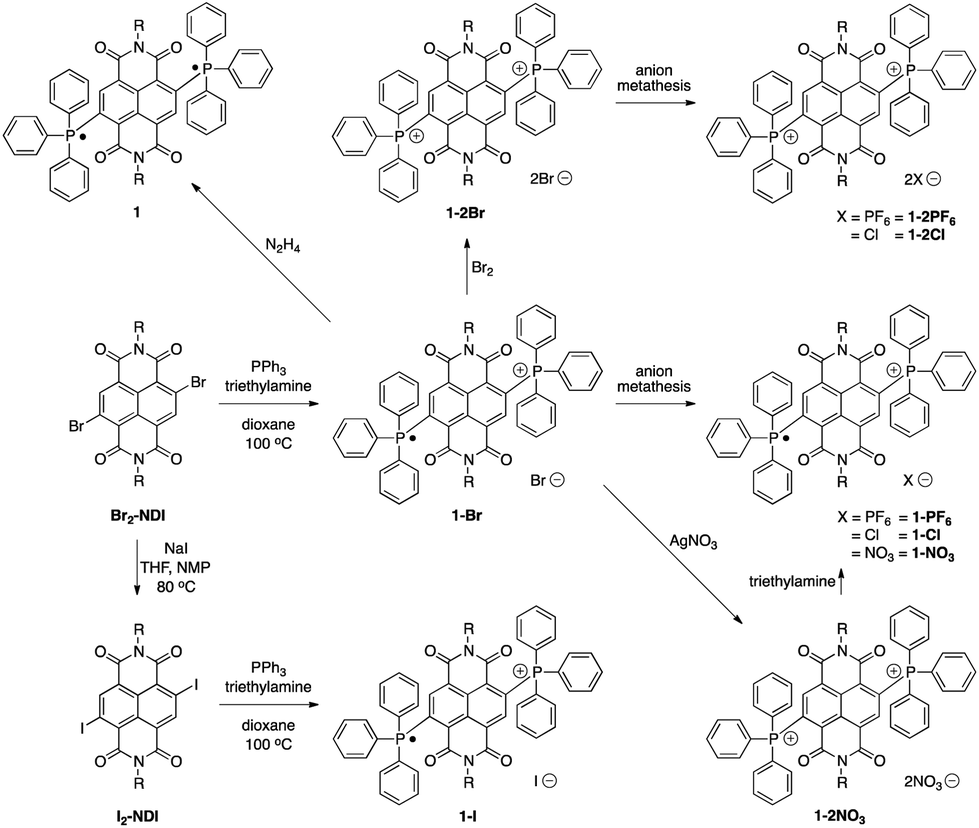 | ||
| Scheme 1 Syntheses of the BTPP-NDI compounds. | ||
X-ray crystallographic analysis
Single crystal XRD structures of all the compounds except 1-2Cl and 1-NO3 are shown in Fig. 1 and Fig. S1–S8, ESI.† We were unable to obtain the single crystal XRD structures of 1-2Cl and 1-NO3 due to their PET nature (vide infra). The anion⋯π distances were found to be 2.91 Å for 1-PF6, 3.15 Å for 1-Cl, 3.32 Å for 1-Br, 3.76 Å for 1-I, 2.81 Å for 1-2PF6, 3.19 Å for 1-2Br and 2.83 Å for 1-2NO3. All except 1-I have anion⋯π distances shorter than the sum of van der Waals distances between carbon and their respective anion element (F for PF6−, N and O for NO3−). In general, the 1+ salts displayed longer anion⋯π distances than their respective 2+ salts due to their weaker anion–π interactions, and the anion⋯π distances seemed to increase down the group for the halides for both 1+ and 2+ salts. For the multi-nuclear anions, both PF6− and NO3− showed very short anion⋯π distances, suggesting the importance of multiple short-contacts for very weakly Lewis basic anions.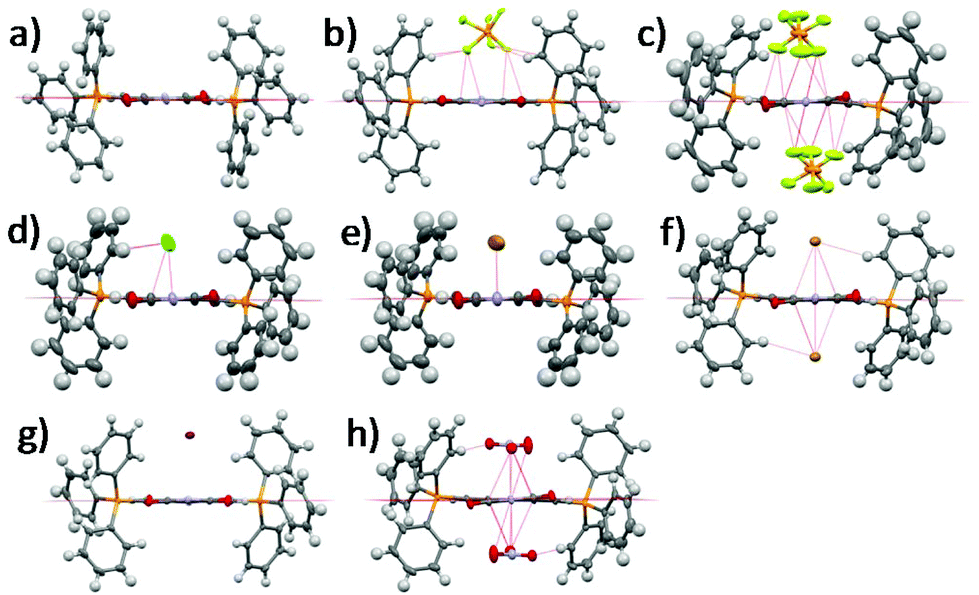 | ||
| Fig. 1 Single crystal XRD structures of (a) 1, (b) 1-PF6, (c) 1-2PF6, (d) 1-Cl, (e) 1-Br, (f) 1-2Br, (g) 1-I and (h) 1-2NO3. Solvent molecules and alkyl groups are omitted for clarity. | ||
X-ray photoelectron spectroscopy (XPS) analysis
The XPS spectra of the BTPP-NDI salts are shown in Fig. S9, ESI.† Peak shifts toward higher binding energies were observed for F 1s (PF6−), Cl 2p3/2 (Cl−), Br 3d5/2 (Br−) and N 1s (NO3−) in 2+ salts as compared to 1+ salts. Higher binding energy species were observed for 1-Cl, 1-2Cl, 1-Br, 1-2Br and 1-I, as well as 1-2PF6. To the best of our knowledge, such an additional peak for the binding energy of F 1s in PF6− is unheard of. The XPS results corroborate the short contacts observed in the single crystal XRD structures, and hence provide strong evidence of anion–π interactions in the solid state for the BTPP-NDI salts.UV-Vis absorption
The solution UV-Vis absorption spectra of the BTPP-NDI salts in acetonitrile and chloroform are shown in Fig. 2. In polar aprotic solvents like acetonitrile where anion–π interactions are diminished, the anions do not show any observable impact on the λmax for both the 1+ and 2+ salts, except Cl− in 1-2Cl that shows absorption peaks corresponding to the 1+ salts, indicating that some form of electron transfer is involved. Saha and co-workers9 have proposed that very Lewis basic anions like F− can undergo direct thermal electron transfer to the NDI while Gabbaï et al.10 suggested that F− deprotonates the solvent (acidified by NDI) and the deprotonated solvent acts as the reducing agent for NDI. Regardless of the mechanism of reduction, the HOMO of Cl− (−5.54 eV)11 is not high enough to reduce BTPP-NDI2+ (LUMO = −4.75 eV, determined via the onset of first reduction in cyclic voltammetry in Fig. S10, ESI†) and Cl− is not Lewis basic enough (pKa = −6.3) to deprotonate either acetonitrile (pKa = 25) or chloroform (pKa = 15.5). Hence, photoinduced electron transfer (PET) is required to reduce the BTPP-NDI core from 2+ to 1+. PET is known to occur from Cl− (but not Br− and I−) to strongly Lewis π-acidic NDIs due to the stronger Lewis basicity of Cl−.9,12 The paramagnetic species arising from PET can also be observed in the broadening of 1H NMR signals of 1-2Cl (Fig. S21, ESI†). We also noted that there is a difference in the UV-Vis absorption of 1-NO3 as compared to that of the other 1+ salts from 320 to 430 nm.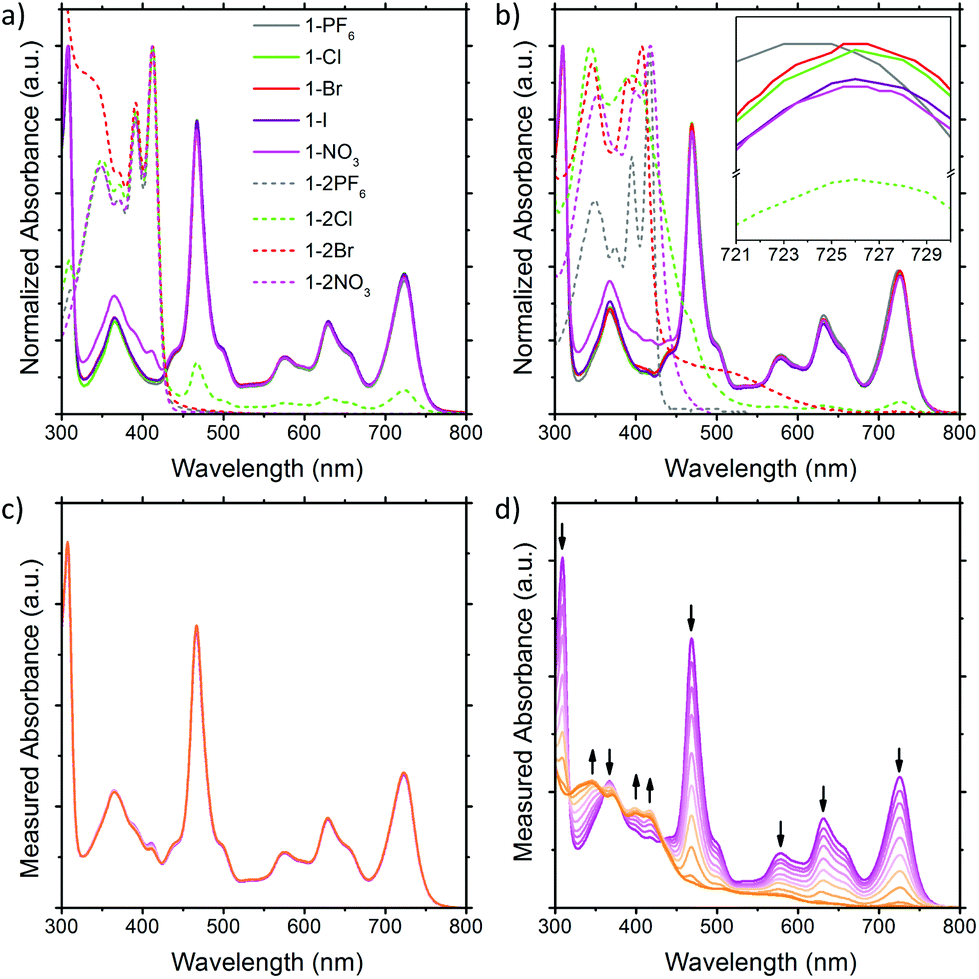 | ||
| Fig. 2 Solution UV-Vis of the BTPP-NDI salts in (a) acetonitrile and (b) chloroform. Solution UV-Vis of 1-NO3 in (c) acetonitrile and (d) chloroform upon illumination. | ||
In nonpolar solvents where anion–π-radical interactions are expected to be stronger, the λmax values of the 1+ halide and nitrate salts were red-shifted from 723 nm in acetonitrile to about 726 nm in chloroform. In contrast, the λmax of 1-PF6 was only red-shifted from 723 nm in acetonitrile to 724 nm in chloroform. This miniscule shift for halide and nitrate salts as compared to that of 1-PF6 is probably due to electronic interactions between the frontier orbitals of the anions and the SUMO of the radical cation arising from their similar energy levels (Fig. S44, ESI†) when the cation and anion are in close proximity, which is also supported by TDDFT calculations (Fig. S13 and Table S5, ESI†). The difference in the UV-Vis absorption of 1-NO3 as compared to the other 1+ salts from 320 to 430 nm remained observable in chloroform. For the 2+ salts, their absorption profiles in chloroform are largely different from those in acetonitrile, with the exception of 1-2PF6 and 1-2NO3 due to their electrochemical inactivity towards the 2+ cation for the latter. For 1-2Cl, the absorption peaks corresponding to the 1+ salts remain due to PET (vide supra). For 1-2Br, a charge transfer band at around 520 nm, arising from the stronger anion–π interaction in chloroform, was observed.
Based on time-dependent density functional theory (TDDFT) calculations, the lowest energy D0 → D1 transition for the 1+ salts is mainly contributed by SOMOα → LUMOα (Table S5, ESI†). With the SUMO value estimated to be −4.75 eV from CV measurements and an optical bandgap of 1.65 eV for the 1+ salts, the LUMO value of the 1+ salts is estimated to be −3.10 eV. Based on the reduction potential of NO3− (NO3− + 2H+ + 2e− → NO2− + 2H2O, E° = +0.42 V;13 Fc/Fc+vs. SHE = +0.62 V;14 HOMO of Fc = 4.8 eV; LUMO = −4.60 eV) we postulate that 1-NO3 will undergo “reverse” PET, i.e., π → anion PET cf. anion → π “normal” PET reported by Saha and co-workers (see the energy diagram in Fig. S44, ESI†).9 Upon illumination, the UV-Vis absorption of 1-NO3 remained similar in acetonitrile but degraded rapidly in chloroform, giving rise to two new peaks at ∼400 nm corresponding to the 2+ cation and a new peak at 350 nm corresponding to the 2+ cation and NO2− (Fig. 2c & d).15 Based on these observations, we reasoned that the pKa values of the solvents are the likely cause for the distinct stability of 1-NO3. As stated in the redox equation, the reduction of NO3− requires the presence of H+, which is promoted by the much higher acidity of chloroform as compared to acetonitrile.
Nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR)
To further characterize the BTPP-NDI salts, their halogen NMR spectra were collected (Fig. S27–S38, ESI†) and their chemical shifts (δ) and linewidths at the half peak height (LW1/2) were tabulated (Table S3, ESI†). Unfortunately, we were not able to perform 14N NMR due to the unavailability of the required NMR probe. Respective tetrabutylammonium salts (TBA-X) were used as the control. Restricted motion (19F) and boundedness (35Cl, 81Br and 127I) often result in a broader LW1/2 for their NMR signals.16 In polar aprotic solvents like acetonitrile-d3, the anions in TBA.X salts behave like free ions and hence sharp halogen NMR signals were observed. The 19F NMR signals for 1-2PF6 showed a very narrow LW1/2 (1.5 Hz) and were essentially the same as the control while 1-PF6 showed a slightly broader LW1/2 (1.9 Hz). For 1-Cl, the LW1/2 of the 35Cl NMR signal was 63 Hz while 1-2Cl did not show any observable signal. Similarly, the LW1/2 of the 81Br NMR signal for 1-Br was 915 Hz while 1-2Br did not show any observable signal. The broadening of the 35Cl and 81Br NMR signals in 1-Cl and 1-Br as compared to their TBA.X salts suggests that Cl− and Br− are somewhat bound to the BTPP-NDI core. Similarly, the absence of the 35Cl and 81Br NMR signals in 1-2Cl and 1-2Br suggests that Cl− and Br− are strongly bound, or due to the occurrence of PET for the former, resulting in the NMR signals being too broad to be observed. No 127I NMR signal could be observed for 1-I due to the large nuclear quadrupole moment (I > 5/2). Although the LW1/2 of halogen NMR can be used to estimate the restricted motion/boundedness of the halides, the presence of paramagnetic radical species further complicates matters. This is because apart from the omnipresence of diamagnetic relaxation, paramagnetic compounds also exhibit paramagnetic relaxation which involves several contributions that make estimation difficult.17Based on the solution UV-Vis absorption, acetonitrile is able to provide effective electrostatic screening in the 1+ salts via its high dielectric constant of 37.5. However, it has a very small magnetic susceptibility of −2.8 × 10−5 cm3 mol−1; hence it is unable to provide effective magnetostatic screening. Thus, deviations from the halogen δ of TBA.X are most likely caused by magnetic spin polarization induced by the π-radical. The Δδ with respect to TBA.X are 18.8 Hz for 1-PF6, 114.4 Hz for 1-Cl and 389.8 Hz for 1-Br. Thus, the degree of the anion–π-radical interaction for the 1+ salts is 1-Br > 1-Cl > 1-PF6. In nonpolar solvents like chloroform-d, the anions should be more strongly bound and hence we expect broader halogen NMR signals. The LW1/2 of the 19F NMR signal for 1-PF6 (162.5 Hz) is more than two orders of magnitude larger than the control (1.5 Hz) as shown in Fig. 3. The LW1/2 of the 19F NMR signal for 1-2PF6 (4.4 Hz) is only about three times larger than the control. It is thus clear that the very broad LW1/2 for 1-PF6 in chloroform-d was caused by stronger magnetic spin polarization arising from the stronger anion–π-radical interaction between PF6− and the π-radical. Similar 19F NMR signal broadening was previously reported, but in a selenium-centered radical SbF6− salt.18 To our knowledge, there is no report on such 19F NMR signal broadening in solution with π-radicals. The observed LW1/2 of the 35Cl NMR signal for 1-Cl was 415 Hz while no 81Br NMR signal for 1-Br could be observed. Similarly, no 35Cl and 81Br NMR signals could be observed for 1-2Cl and 1-2Br. The LW1/2 trend of the halogen NMR signals is in agreement with the polarizabilities of the anions and the polarizing power of the cations.
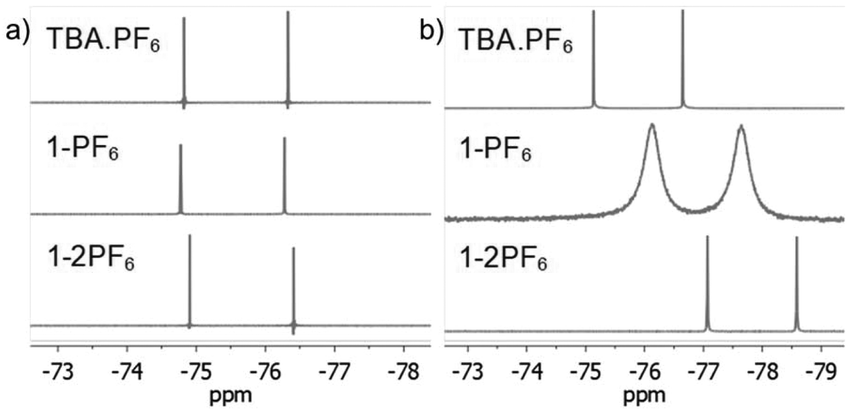 | ||
| Fig. 3 19F NMR of the PF6− salts in (a) acetonitrile-d3 and (b) chloroform-d. | ||
EPR of the 1+ salts was measured in dry dichloromethane (Fig. S10, ESI†). The 1+ halides did not show any noticeable difference while 1-NO3 showed a broader signal, indicating some exchange interaction between the BTPP-NDI radical cation and NO3−. We believe that this difference in the EPR signal for 1-NO3, together with the difference in its UV-Vis absorption, is caused by strong anion–π-radical interactions, as shown by the unexpected short anion⋯π distance calculated using DFT (2.93 Å, Table S4, ESI,†vide infra). The difference in the EPR signal and UV-Vis absorption for 1-NO3 is also supported by the Lewis basicity of the anions, based on the pKa of their respective acids: HPF6 < HI < HBr < HCl < HNO3.
Electrospray ionization-mass spectrometry (ESI-MS)
The presence of anion–π/anion–π radical interactions in BTPP-NDI salts was also supported by electrospray ionization mass spectrometry (ESI-MS) data. Peaks corresponding to m/z values of 1157.41 [1 + PF6]+ were found for 1-PF6 and 1-2PF6; 1047.42 [1 + Cl]+ for 1-Cl and 1-2Cl; 1091.37 [1 + Br]+ for 1-Br and 1-2Br; and 1074.21 [1 + NO3]+ for 1-NO3 and 1-2NO3 (Fig. S39–S42, ESI†). However, [1 + I]+ could not be observed in 1-I as most of the I− was oxidized to I3− in the ESI-MS, which does not have any significant anion–π/anion–π radical interactions with BTPP-NDI, consistent with our previous findings from the pyridinium–NDI system11 (Fig. S43, ESI†).Density functional theory (DFT) calculations
To provide molecular interpretation for the halogen NMR, we performed density functional theory (DFT) calculations by employing long-range corrected hybrid functional CAMB3LYP19 with Pople-type basis 6-311G+(d,p) for atoms in the cation and LANL2DZdp20 for atoms in the anion. The optimized structures agree very well (<5% deviation) with the obtained single crystal structures (refer to Table S4 in the ESI† for the experimental and calculated bond lengths), with exceptions to the larger calculated anion⋯π distances (>5% deviation) for the 1+ halides. This systematic overestimation of the anion⋯π distances for the 1+ halides is likely the consequence of electron self-interactions, leading to over-stabilization of the coulombic terms relative to the exchange–correlation terms, a situation similar to a two-center three-electron system.21 All anion⋯π distances calculated for 1+ salts were larger than those of the corresponding 2+ salts except for 1-NO3 (2.933 Å) which showed a smaller value than that of 1-2NO3 (2.964 Å), with the former in an edge-on configuration and the latter in a face-on configuration agreeing with the single crystal structure. We believe that the shortest anion⋯π distance for 1-NO3 among the 1+ salts is the likely explanation for its different UV-Vis absorption and EPR as compared to the other 1+ salts. The edge-on configuration for 1-NO3 is in agreement with the absorbed nitrate on various metal electrodes during reduction.22 In 1-2NO3, the interaction between the highly π-acidic BTPP-NDI core and the π-rich NO3− in the face-on configuration is very favorable. In 1-NO3, the BTPP-NDI core is relatively π-rich and hence to minimize electronic repulsion, the edge-on configuration is favored. The different configurations of NO3− on the π-acidic BTPP-NDI core support the concepts used in anion–π catalysis, e.g., stabilization of nitronate intermediates in enamine chemistry.3,23 The 1+ salts show non-zero spin densities on the anions with Br− > Cl− > PF6− > I− > NO3− (Fig. 4a). This is in accordance with the trend of anion–π-radical interactions based on the Δδ in the halogen NMR (vide supra). Even though PF6− is very weakly polarizable, there is a significant amount of delocalized spin density that accounts for the severe broadening of the 19F NMR signal for 1-PF6. The significant amount of the delocalized spin density on PF6− arises from the fact that PF6− has short anion⋯π distances and the strength of the magnetostatic field (from the π-radical) obeys an inverse-cube law with respect to the distance. Hence, even the highly polarizable I− showed the least spin density due to its large anion⋯π distance.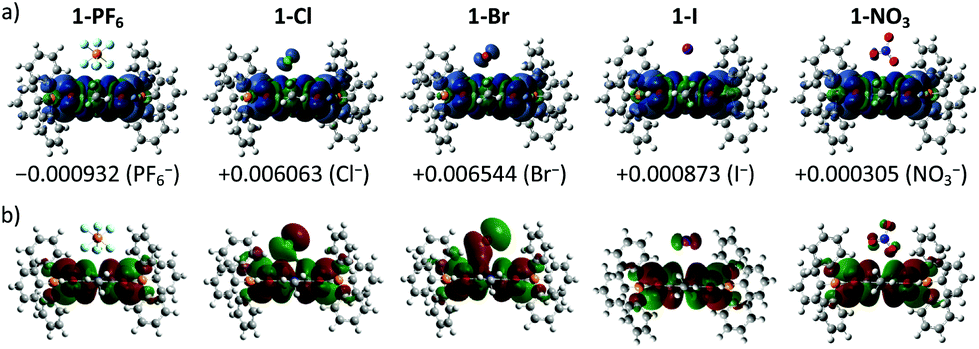 | ||
| Fig. 4 (a) Spin density (isovalue = 0.0001 e−/au3) and (b) SOMO (isovalue = 0.01 e−/au3) of the geometry optimized 1+ salts in the gas phase using DFT UCAMB3LYP 6-311G+(d,p) for atoms in the cation and LANL2DZ with polarization and diffuse functions for atoms in the anion. Numerical values denote the sum of Mulliken atomic spin densities on anions. | ||
It is also worth noting that the spin densities on the 1+ radical and the halides/NO3− are of the same spin type while being of different spin types for PF6−. Even when considering a Ci symmetry point group with an additional anion, i.e. 1-X + X−, the trend of the spin density remains the same. The same spin type in m-phenylene-based bis(aminoxyl) diradical–anion complexes was believed to enhance the intramolecular ferromagnetic exchange interaction between the diradicals.8a In the case of our BTPP-NDI system, the same spin type may facilitate intermolecular ferromagnetic exchange interactions in single crystals for the halide salts while antiferromagnetic exchange interactions may be observed in the single crystal for 1-PF6 due to its different spin types. In the solid state, the intermolecular antiferro/ferromagnetic exchange interactions are expected to be stronger due to the stronger anion–π radical interactions as shown in the XPS data (Fig. S10†). Magnetic measurements of the single crystals are currently underway.
The strength of the electrostatic field (arising from the coulombic attraction between the cation and anion) obeys an inverse-square law with respect to the distance. This means that even though the magnetic polarization of I− is weak in 1-I, the electric polarization is stronger. This is true for all anions and is reflected in the SOMO in Fig. 4b. The polarizable halides and NO3− show significant contribution to the SOMO while the very weakly polarizable PF6− shows negligible contribution. Furthermore, the trend of the calculated λmax calculated via TDDFT (Fig. S13 and Table S5, ESI†) is in accordance with the experimentally observed UV-Vis absorption. This explains the red-shifted λmax for 1-Cl, 1-Br, 1-I and 1-NO3 as compared to 1-PF6 in chloroform.
Conclusions
In conclusion, we extend the concept of anion–π interactions to anion–π-radical interactions. We note that even in solution, anions are able to induce changes in the photophysical and magnetic properties of the cationic π-radical. Apart from the “normal” PET from the anion to the Lewis π-acidic 2+ cation for 1-2Cl, we were able to observe a “reverse” PET from the radical cation to the anion for 1-NO3. The radical cation is also able to induce magnetic spin polarization of the anions in NMR. These effects should be much stronger in the solid state without the electrostatic screening from the solvent molecules. We believe that our results will provide better understanding of organic ferromagnets and p-doped conductors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We like to thank Ms Tan Geok Kheng (X-ray Diffraction Laboratory in National University of Singapore) and Ms Debbie Seng (Structural Materials Department in IMRE) for their efforts to solve the single crystal structures and collect the XPS data.Notes and references
- (a) D.-X. Wang and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 892–897 CrossRef CAS; (b) M. M. Watt, L. N. Zakharov, M. M. Haley and D. W. Johnson, Angew. Chem., Int. Ed., 2013, 52, 10275–10280 CrossRef CAS.
- (a) L. Adriaenssens, C. Estarellas, A. Vargas Jentzsch, M. Martinez Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324–8330 CrossRef CAS; (b) M. Giese, M. Albrecht and K. Rissanen, Chem. Commun., 2016, 52, 1778–1795 RSC.
- (a) Y. Zhao, C. Beuchat, Y. Domoto, J. Gajewy, A. Wilson, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101–2111 CrossRef CAS; (b) Y. Zhao, Y. Cotelle, L. Liu, J. López-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS.
- (a) C.-Z. Li, C.-C. Chueh, F. Ding, H.-L. Yip, P.-W. Liang, X. Li and A. K.-Y. Jen, Adv. Mater., 2013, 25, 4425–4430 CrossRef CAS; (b) B. Lüssem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef; (c) X. Zhao, D. Madan, Y. Cheng, J. Zhou, H. Li, S. M. Thon, A. E. Bragg, M. E. DeCoster, P. E. Hopkins and H. E. Katz, Adv. Mater., 2017, 29, 1606928 CrossRef; (d) Y. Xu, J. Yuan, J. Sun, Y. Zhang, X. Ling, H. Wu, G. Zhang, J. Chen, Y. Wang and W. Ma, ACS Appl. Mater. Interfaces, 2018, 10, 2776–2784 CrossRef CAS.
- (a) J.-L. Brédas and G. B. Street, Acc. Chem. Res., 1985, 1985, 309–315 CrossRef; (b) H. Fukutome, A. Takahashi and M.-a. Ozaki, Chem. Phys. Lett., 1987, 133, 34–38 CrossRef CAS; (c) P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus and A. Pron, Chem. Soc. Rev., 2013, 42, 8895–8999 RSC.
- D. M. d. Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef.
- (a) S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, J. Am. Chem. Soc., 2014, 136, 12004–12010 CrossRef CAS; (b) Q. Song, F. Li, Z. Wang and X. Zhang, Chem. Sci., 2015, 6, 3342–3346 RSC; (c) S. K. Keshri, S. Kumar, K. Mandal and P. Mukhopadhyay, Chem. – Eur. J., 2017, 23, 11802–11809 CrossRef CAS; (d) S. Debnath, C. J. Boyle, D. Zhou, B. Wong, K. R. Kittilstved and D. Venkataraman, RSC Adv., 2018, 8, 14760–14764 RSC; (e) A. J. Greenlee, C. K. Ofosu, Q. Xiao, M. M. Modan, D. E. Janzen and D. D. Cao, ACS Omega, 2018, 3, 240–245 CrossRef CAS; (f) Z. Bin, H. Guo, Z. Liu, F. Li and L. Duan, ACS Appl. Mater. Interfaces, 2018, 10, 4882–4886 CrossRef CAS.
- (a) M. E. Ali and P. M. Oppeneer, J. Phys. Chem. Lett., 2011, 2, 939–943 CrossRef CAS; (b) D. Bhattacharya, S. Shil, A. Misra, L. Bytautas and D. J. Klein, J. Phys. Chem. A, 2016, 120, 9117–9130 CrossRef CAS.
- S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679–13691 CrossRef CAS.
- G. Bélanger-Chabot, A. Ali and F. P. Gabbaï, Angew. Chem., Int. Ed., 2017, 56, 9958–9961 CrossRef.
- T. L. D. Tam, C. K. Ng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 7374–7377 RSC.
- S. Saha, Acc. Chem. Res., 2018, 51, 2225–2236 CrossRef CAS.
- B. C. Berks, S. J. Ferguson, J. W. B. Moir and D. J. Richardson, Biochim. Biophys. Acta, Bioenerg., 1995, 1232, 97–173 CrossRef.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- J. Mack and J. R. Bolton, J. Photochem. Photobiol., A, 1999, 128, 1–13 CrossRef CAS.
- (a) C. Hall, Q. Rev., Chem. Soc., 1971, 25, 87–109 RSC; (b) J.-X. Yu, R. R. Hallac, S. Chiguru and R. P. Mason, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 70, 25–49 CrossRef CAS.
- J. Koehler and J. Meiler, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 360–389 CrossRef CAS.
- S. Zhang, X. Wang, Y. Su, Y. Qiu, Z. Zhang and X. Wang, Nat. Commun., 2014, 5, 4127, DOI:4110.1038/ncomms5127.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- C. E. Check, T. O. Faust, J. M. Bailey, B. J. Wright, T. M. Gilbert and L. S. Sunderlin, J. Phys. Chem. A, 2001, 105, 8111–8116 CrossRef CAS.
- B. Braïda, P. C. Hiberty and A. Savin, J. Phys. Chem. A, 1998, 102, 7872–7877 CrossRef.
- (a) M. C. P. M. da Cunha, M. Weber and F. C. Nart, J. Electroanal. Chem., 1996, 414, 163–170 CrossRef; (b) S.-E. Bae, K. L. Stewart and A. A. Gewirth, J. Am. Chem. Soc., 2007, 129, 10171–10180 CrossRef CAS; (c) K. Nakata, Y. Kayama, K. Shimazu, A. Yamakata, S. Ye and M. Osawa, Langmuir, 2008, 24, 4358–4363 CrossRef CAS PubMed.
- (a) Y. Zhao, Y. Domoto, E. Orentas, C. Beuchat, D. Emery, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2013, 52, 9940–9943 CrossRef CAS; (b) Y. Zhao, S. Benz, N. Sakai and S. Matile, Chem. Sci., 2015, 6, 6219–6223 RSC; (c) J. López-Andarias, A. Frontera and S. Matile, J. Am. Chem. Soc., 2017, 139, 13296–13299 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis procedure, characterization data, including X-ray data and NMR spectra. CCDC 1871419–1871426. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo01122b |
| ‡ Both authors contributed equally to the manuscript. |
| This journal is © the Partner Organisations 2019 |