
Identification of a common recognition center for a photoactive non-steroidal antiinflammatory drug in serum albumins of different species†
Oscar
Molins-Molina
 a,
Emilio
Lence
b,
Daniel
Limones-Herrero
a,
Concepción
González-Bello
b,
Miguel A.
Miranda
*a and
M. Consuelo
Jiménez
*a
a,
Emilio
Lence
b,
Daniel
Limones-Herrero
a,
Concepción
González-Bello
b,
Miguel A.
Miranda
*a and
M. Consuelo
Jiménez
*a
aDepartamento de Química/Instituto de Tecnología Química UPV-CSIC, Universitat Politècnica de València, Camino de Vera s/n, 46071 Valencia, Spain. E-mail: mcjimene@qim.upv.es
bCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, Jenaro de la Fuente s/n, 15782 Santiago de Compostela, Spain
First published on 19th November 2018
Abstract
The non-steroidal anti-inflammatory drug (S)-carprofen (CPF) has been used as a photoactive probe to investigate the possible existence of a common recognition center in serum albumins (SAs) of different species. The methodology involves irradiation of the CPF/SA complexes, coupled with gel filtration chromatography or proteomic analysis of the photolysates, docking and molecular dynamics simulations. Photolysis of CPF/SA complexes at λ = 320 nm, and gel filtration chromatography, revealed that the protein fraction still contained the drug fluorophore, in agreement with covalent attachment of the photogenerated radical intermediate CBZ˙ to SAs. After trypsin digestion and ESI-MS/MS, the incorporation of CBZ˙ was detected at several positions in the different albumins. Remarkably, modifications at the IB/IIIA interface were observed in all cases (Tyr452 in HSA, RbSA and RtSA and Tyr451 in BSA, PSA and SSA). The molecular basis of this common recognition, studied by docking and molecular dynamics simulation studies on the corresponding non-covalent complexes, corroborated the experimentally observed covalent modifications. Our computational studies also revealed that the previously reported displacement of CPF by (S)-ibuprofen, a site II specific drug, would be due to an allosteric effect in site II, rather than a direct molecular displacement, as expected.
Introduction
Covalent photobinding of a ligand to a protein has recently been used as a tool for mapping the protein recognition center. This has been proven for a model system (carprofen methyl ester/bovine α1 acid glycoprotein) using a systematic approach that combines photophysics, reactivity, proteomics and molecular dynamics simulation studies.1The parent drug carprofen [2-(6-chloro-9H-carbazol-2-yl)propanoic acid] is a non-steroidal antiinflammatory drug (NSAID) of the 2-arylpropionic acid family employed for the treatment of pain and inflammation for almost a decade in the 90's. Then, it was removed from the market for human use, and currently tablets or injections are only used for veterinary purposes. It possesses a chiral center and the (S)-enantiomer is the pharmacologically active form (CPF, Chart 1).2 Rather than glycoproteins, the major transport proteins for CPF are serum albumins (SAs), which are very abundant in mammals and play a crucial role in bio-distribution, metabolism and elimination of exogenous compounds.3
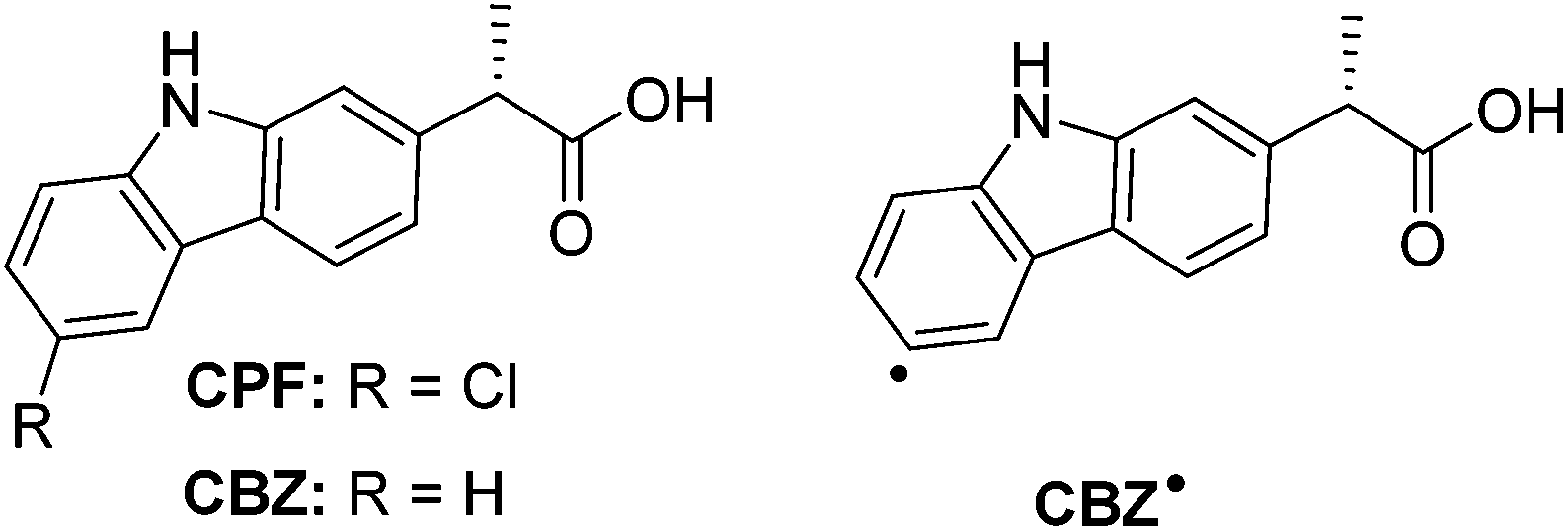 | ||
| Chart 1 Chemical structures of (S)-carprofen (CPF), its photoreaction product (CBZ) and radical intermediate (CBZ˙). | ||
Human serum albumin (HSA) consists of a single polypeptide chain with a heart shaped three dimensional structure and three homologous domains I–III, each of them containing two sub-domains (A and B).4 Small organic ligands bind to HSA in regions located within the hydrophobic cavities of sub-domains IIA and IIIA.5 While neutral, bulky heterocyclic compounds mostly bind to subdomain IIA by means of strong hydrophobic interactions, acidic molecules bind preferentially to subdomain IIIA, through electrostatic and/or H-bonding interactions.6 The binding properties of other albumins, such as bovine, rabbit and rat serum albumins (BSA, RbSA and RtSA, respectively), have been explored using different techniques, such as high performance displacement chromatography, fluorescence or equilibrium dialysis.7
The in vitro supramolecular dark binding of CPF to HSA has been investigated by several methods, including photophysical techniques such as fluorescence and transient absorption spectroscopy.8,9 The results show that although subdomain IIIA is the major binding site for CPF within HSA, subdomain IIA is also populated to some extent.
In contrast to the reversible, non-covalent complexation, photobinding corresponds to the formation of a covalent photoadduct upon irradiation of a ligand in the presence of a protein. Photobinding is relevant to photoallergy, which is generally attributed to covalent conjugation of proteins with photosensitizing agents, photochemical intermediates or photoproducts (usually denoted as haptens); the resulting modified proteins can act as antigens, thus provoking the immune system response.10 In this context, transport proteins, especially serum albumins, have been employed as models to investigate the photobinding process.11
In connection with the photoallergic properties of CPF,12 its photobinding to HSA has been reported; the process is thought to involve the formation of aryl radicals (CBZ˙) resulting from C–Cl photocleavage and can be followed by incorporation of the fluorophore into the biomacromolecule.8b,13 The process can be monitored by following the changes in the fluorescence band at increasing irradiation times, since the fluorescence quantum yield (ϕF) of the resulting CBZ is much higher than that of parent CPF.
The analogies and differences between species in drug response are due to pharmacokinetic or pharmacodynamic aspects. Species differences are often unpredictable and generalizations are not easy to make. In the case of NSAIDs, some pharmacokinetic aspects (such as protein binding and distribution volumes) do not display significant variations between species, while others (i.e. clearance and elimination half-life) do. Concerning the pharmacodynamic aspects, significant differences exist among animal species for this family of drugs, such as the relative potencies for the inhibition of the COX-1 and COX-2 isoforms.14
In this context, the aim of the present work is to elucidate if there is a common recognition center for NSAIDs in SAs of different species, using the CPF as a photoactive probe for different SAs [human (HSA), bovine (BSA), porcine (PSA), rabbit (RbSA), rat (RtSA) and sheep (SSA)]. As a matter of fact, the combined information obtained from photoreactivity, proteomic and molecular dynamics simulation studies unambiguously demonstrated the existence of a common recognition center for CPF in SAs of the different species, in the interface between subdomains IB and IIIA.
Results and discussion
The obtained results are presented below, arranged under separate headings dealing with dark binding interactions, covalent photobinding and theoretical calculations.Dark binding interactions in CPF/SA complexes
Non-covalent binding of CPF to SAs resulted in very subtle changes in the intensity and relative position of the CPF characteristic bands in the absorption spectra. Thus, shifts in the order of 3–5 nm were noticed in the spectra of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes (CPF/SA) with respect to those obtained by the addition of independent drug and protein contributions (CPF + SA) (Table 1). The set of spectra for all the SAs is presented in Fig. S1 of the ESI.†
:1 complexes (CPF/SA) with respect to those obtained by the addition of independent drug and protein contributions (CPF + SA) (Table 1). The set of spectra for all the SAs is presented in Fig. S1 of the ESI.†
| Complex | Δλ1, Δλ2a |
τ
Tb |
I
Fc |
|---|---|---|---|
| a Shifts of the position of characteristic absorption bands in CPF/SA with respect to CPF + SA, in nm; Δλ1 corresponds to the band at ca. 270 nm and Δλ2 to the band at ca. 305 nm. b Average triplet lifetime, at λexc = 355 nm, in air, in microseconds. The value in the absence of protein is lower than 1 μs. c Relative fluorescence intensity (λexc = 320 nm) at the maxima for irradiated CPF/SAs, after Sephadex filtration, taking CPF/HSA as a reference. | |||
| CPF/HSA | 3, 4 | 9.3 | 1.0 |
| CPF/BSA | 3, 5 | 15.7 | 0.8 |
| CPF/PSA | 3, 3 | 12.4 | 1.4 |
| CPF/RbSA | 3, 5 | 6.4 | 2.0 |
| CPF/RtSA | 4, 5 | 10.6 | 2.2 |
| CPF/SSA | 3, 4 | 16.1 | 1.0 |
Laser flash photolysis was performed at λexc = 355 nm for CPF/SAs. Complexation was evidenced by lengthening (by one order of magnitude) of triplet excited state decays (3CPF*) monitored at λ = 450 nm in the presence of SAs (Table 1 and Fig. 1).
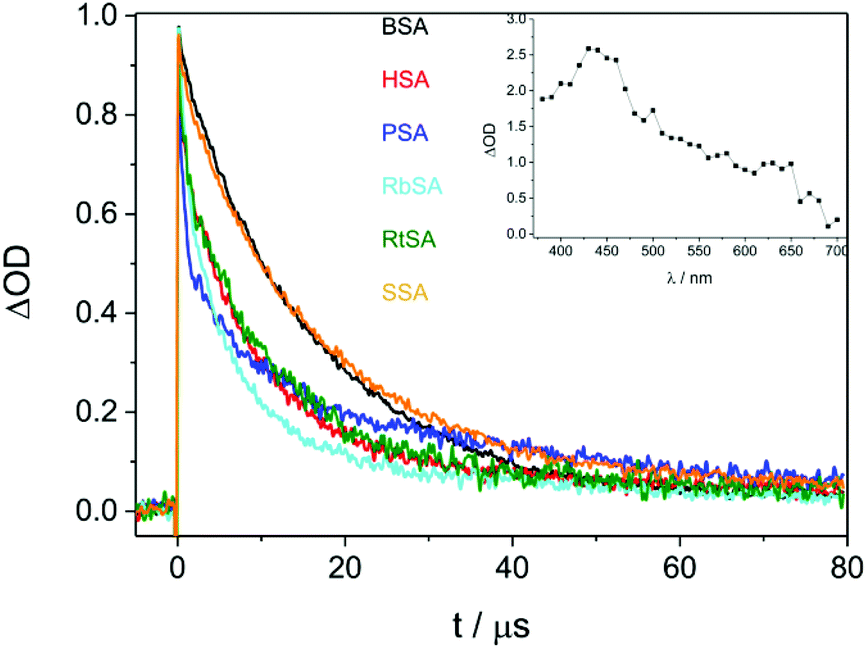 | ||
| Fig. 1 Laser flash photolysis of CPF ([CPF] = 10−4 M, λexc = 355 nm, PBS, air) in the presence of different SAs at a 1:1 drug/protein molar ratio. Decays monitored at λ = 450 nm. Inset: Transient absorption spectra of CPF/HSA 1.4 μs after the laser shot. | ||
Covalent drug–protein photobinding
Solutions of CPF/SA (1:1 molar ratio) were irradiated at λ = 320 nm. The spectra recorded before (black traces) and after 3 minutes of irradiation (red traces) are shown in Fig. 2. In general, the emission bands of the irradiated samples were more intense. This is in agreement with the occurrence of photodehalogenation, leading to products with a ϕF much higher than that of CPF.15
 | ||
| Fig. 2 Fluorescence spectra of CPF/SA mixtures at 1:1 drug/protein molar ratio: before irradiation (black trace), after 3 min of irradiation at λexc = 320 nm (red trace) and after Sephadex filtration of the photolyzates (blue trace). | ||
To check whether covalent modification of the proteins had occurred, the photolysates were treated with 6 M guanidinium chloride and filtered through Sephadex (see experimental details in the ESI†), a process that allows the removal of the species of low molecular weight. The obtained spectra are also shown in Fig. 2 (blue traces). In general, filtration through Sephadex was associated with a remarkable decrease of the emission intensity (compare red and blue traces), indicating that an important fraction of the generated photoproducts was not bound to the protein. Nonetheless, emission was still clearly observed, demonstrating that CPF-derived species are covalently attached to the SAs. In parallel, non-irradiated samples of CPF/SAs were subjected to the same work up, as control experiments; as expected from the lack of covalently bound adducts, no emission was observed in these cases. The extent of photobinding relative to the CPF/HSA system is given in Table 1.
Having demonstrated the photobinding of CPF to the SAs by gel filtration chromatography coupled with fluorescence measurements, the formation of covalent photoadducts was investigated in more detail by proteomic analysis. These studies were expected to provide precise information on which amino acid(s) are covalently modified. Thus, the photoreactivity of CPF with the six SAs was addressed. The irradiated CPF/SA systems were filtered to remove excess of ligand; then, trypsin or endoproteinase Glu-C digestions were followed by HPLC-MS/MS. This was intended to obtain information on the modified peptide sequence and to characterize the adduct. Full scan and fragmentation data files were analyzed by using the Mascot® database search engine (Matrix Science, Boston, MA, USA) and by entering variable modifications that take into account the main possible residues (FWY) able to react with the carbazolyl radical CBZ˙ obtained after dehalogenation of CPF.
For the human protein, the main results confirmed the identification of CBZ-HSA derived peptide adducts at Phe134, Trp214 and Tyr452 (Fig. 3 and Table 2), according to the ESI-MS/MS spectra and fragmentation pattern of the modified peptides, that agreed well with the “y” and “b” ion series (see pages S3–S5 in the ESI†).
 | ||
| Fig. 3 Amino acid sequence of HSA, with the amino acids modified by covalent binding of CBZ indicated in red. | ||
The procedure was performed for the whole set of investigated proteins. The observed modified amino acids are indicated in Table 3; the protein sequence with the modified amino acids highlighted together with the ESI-MS/MS spectra and fragmentation pattern of all the peptide sequences are presented in pages S6–S28 of the ESI.† Remarkably, for the six homologous proteins, covalent modification of the tyrosine residue located at the interface between sub-domains IB and IIIA was obtained (Tyr452 for HSA).
| Binding pocket | HSA | BSA | PSA | RbSA | RtSA | SSA |
|---|---|---|---|---|---|---|
| a Only non-external modifications are considered. b The modifications are organized according to the binding pocket. c The indicated numbering is the one used in the available crystallographic structures where the position of the first 24 amino acids is not resolved. | ||||||
| IB | Phe134 | Tyr137 | Phe134 | Phe134 | Tyr137 | |
| Tyr138 | Tyr138 | |||||
| Phe149 | Phe149 | |||||
| Tyr161 | ||||||
| IIA | Trp214 | Tyr291 | ||||
| IIB | Phe325 | Phe330 | Phe325 | |||
| Phe329 | Phe329 | |||||
| IB/IIIA interface | Tyr452 | Tyr451 | Tyr451 | Tyr452 | Tyr452 | Tyr451 |
| IIIB | Phe508 | Phe550 | Phe509 | |||
A plausible mechanism for the photomodification of SAs is depicted in Scheme 1. It involves the reaction of 3CPF* with Trp to afford the corresponding radical ion pairs; the CBZ radical formed after the loss of Cl− would react with Phe, Tyr or Trp resulting in covalent binding to these residues.
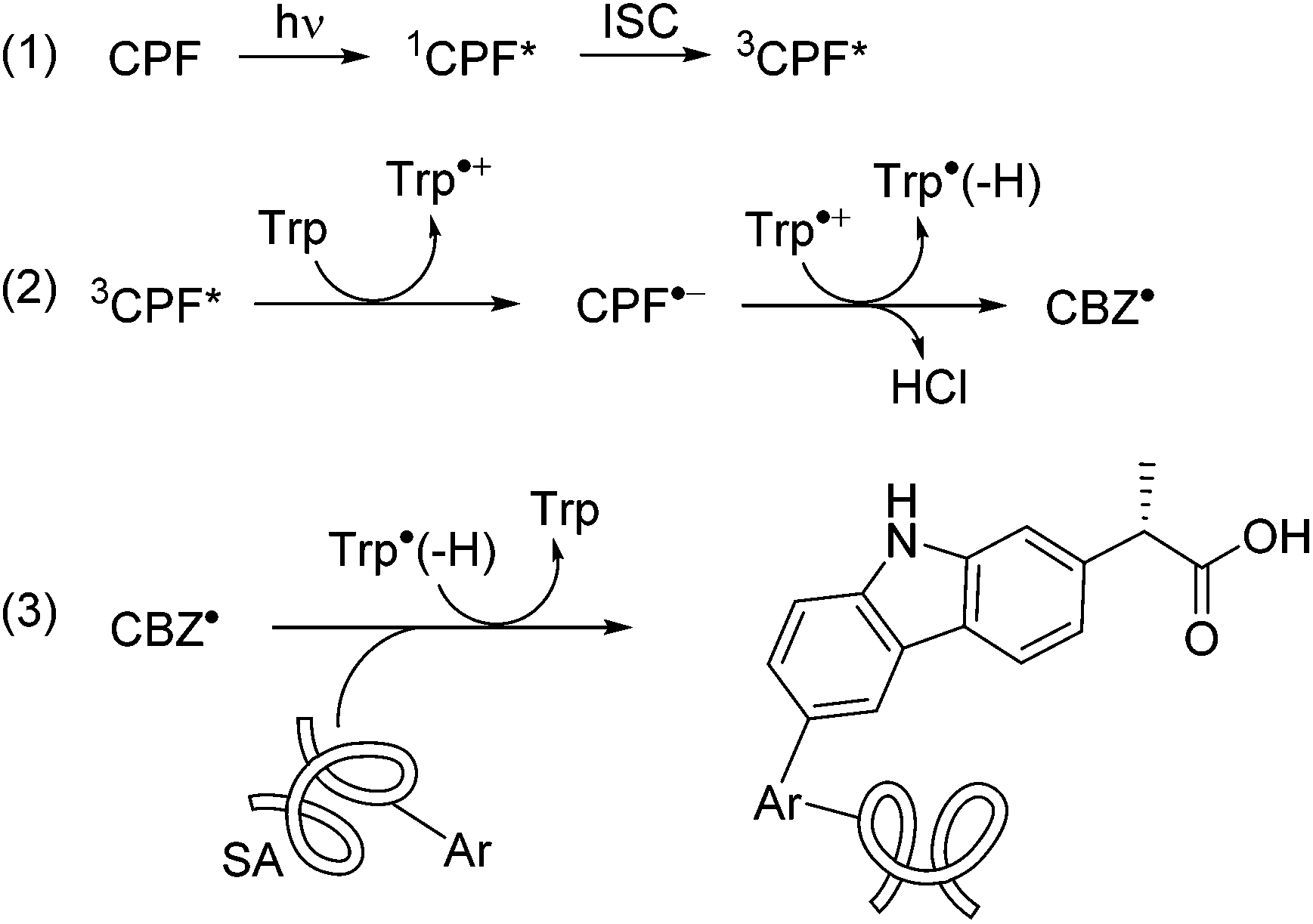 | ||
| Scheme 1 Proposed covalent modification mechanism. | ||
Molecular modelling studies
For the six homologous proteins the covalent modification of the tyrosine residue located in helix h4-III16 of sub-domain IIIA was obtained (Tyr452 for HSA). This suggested that the main binding pocket of CPF is the interface region of sub-domains IIIA and IB, in which the tyrosine residue is located (Fig. 4A). The latter residue is placed on the cleft of the serum albumin “V” structure and about 10 Å away from Trp214 (in HSA). Another relevant binding pocket of CPF to serum albumins, which was identified in four of the six proteins, is the most accessible part of sub-domain IB involving helixes h7-I, h8-I, h9-I and h10-I (Fig. 4B). Moreover, with the exception of HSA and PorSA, binding to the pocket composed of helixes h7-II, h8-II and h9-II in sub-domain IIB was identified (Fig. 4B). The proteomic results also showed that for HSA, as well as the RbSA protein, binding to site I (sub-domain IIA) takes place. | ||
| Fig. 4 Determined binding pockets of carprofen to serum albumin proteins. (A) Main recognition pocket that is located in the cleft of the serum albumin “V” structure (green) involving the interface region between sub-domains IIIA and IB. The experimentally observed modified residue is highlighted in yellow. The structure corresponds to wild-type HSA (PDB 1E7B). (B) Other relevant binding pockets observed in two of the proteins studied. The crystal structures of HSA (PDB 1E7B, grey) and BSA (PDB 3 V03, blue) are compared showing the determined pockets and the modified residues as spheres (orange for HSA and purple for BSA). The sub-domains of serum albumins are also labeled. | ||
In order to get a better understanding of the binding interactions responsible for the affinity of CPF to SA proteins, the binding mode of this compound in atomic detail was explored. These studies were focused on HSA and on the main binding pockets identified by the proteomic studies: (i) “V” cleft between domains I and II; and (ii) sub-domain IB (Fig. 4). To this end, molecular docking using the GOLD 5.2.217 program and the protein coordinates found in the crystal structure of HSA in complex with palmitic acid (PDB code 4BKE,18 2.35 Å) was first used. This structure was chosen because it contains ligand molecules (palmitic acid) in some of the regions studied in this work and has higher resolution than other HSA structures. The proposed binding mode was further analysed by Molecular Dynamics (MD) simulation studies in order to assess the stability and therefore the reliability of the postulated binding. The results of these studies in the three pockets are discussed below.
As Fig. 5 shows, CPF would be anchored to the “V” cleft pocket through its carboxylate group through strong electrostatic interactions with the guanidinium group of Arg114 and the ε-amino group of Lys190 and a strong hydrogen bonding between the NH group of its aromatic moiety and the side chain carbonyl group of Asn429. In addition, the aromatic ring is stabilized by numerous lipophilic interactions with the apolar residues that surround it involving residues Ala191, Ala194, Val433, Val455, Val456, Val426 and Gln459. Under this arrangement, the C4 carbon atom attached to the chlorine atom in CPF would be located pointing towards the side chain of Tyr452, which is the only aromatic residue in the vicinity of the ligand. The proposed binding mode would explain therefore the experimentally observed covalent modification of Tyr452.
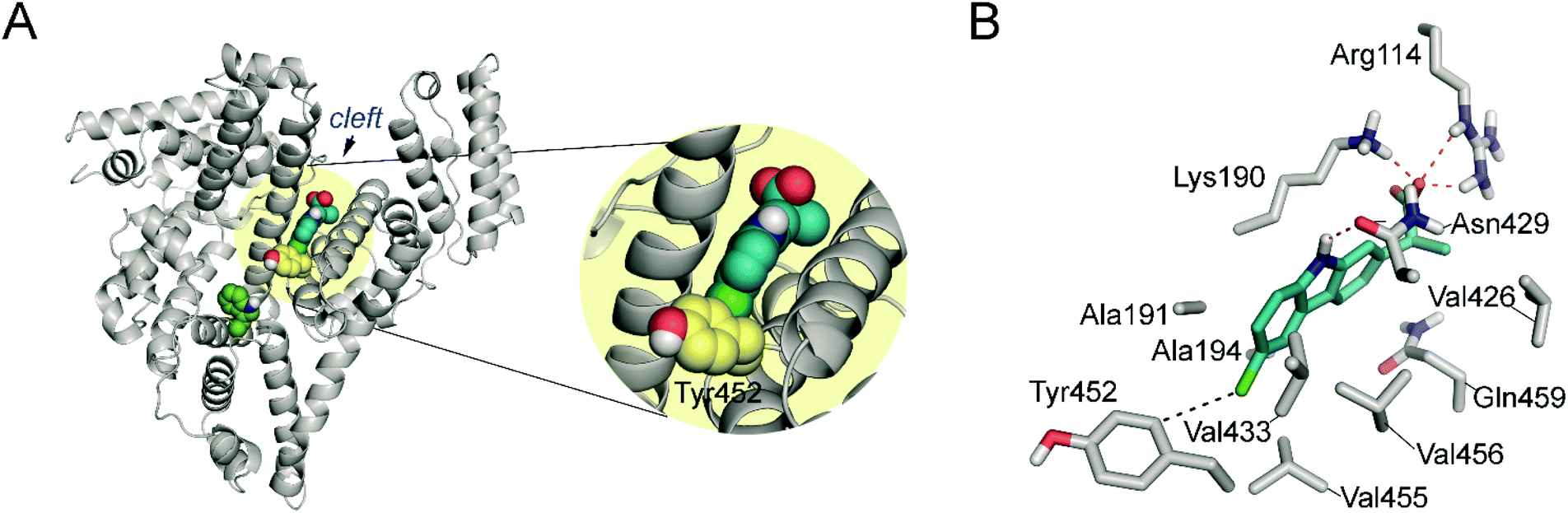 | ||
| Fig. 5 (A) Overall view of the proposed binary CPF/HSA-V cleft complex obtained by docking and MD simulation studies. A snapshot after 150 ns is shown. (B) Detailed view of the CPF/HSA-V cleft complex. Relevant side chain residues are shown and labelled. Polar (red) contacts and distances (black) to Tyr452 are shown as dashed lines. | ||
It was previously suggested that CPF binds with high affinity to site II and with weaker affinity to site I on HSA.20 This was corroborated with diverse competition studies with site specific drugs such as (S)-ibuprofen (IBU, site II) and warfarin (site I). As will be discussed below, the proteomic and molecular modelling studies reported here also corroborate site I as a secondary binding site of CPF. However, our results clearly showed that the main binding pocket of CPF is the cleft of the serum albumin “V” structure and not site II, as no covalent modifications of the aromatic residues of this pocket were observed in any of the six proteins studied. Reasoning that: (1) as observed in the crystal structure of HSA in complex with two molecules of ibuprofen (PDB code 2BXG,21 2.7 Å), this cleft is located nearby to site II; and (2) HSA can undergo significant conformational changes upon binding ligands, specifically in domains I and III, as previously reported by Curry et al.22 and also by us,19 we considered that the experimentally observed displacement of CPF by ibuprofen, which suggests site II as the main binding pocket, is also in agreement with the herein proposed binding. Thus, we believe that the higher affinity of ibuprofen than CPF to HSA and the expected conformational changes resulting from ibuprofen binding, in particular in the proximity of site II, might disfavor the binding of CPF to the “V” cleft of the protein. As a consequence, CPF would be displaced from its main binding pocket by an allosteric effect in site II, rather than a molecular displacement. In order to corroborate this allosteric modulation hypothesis, the conformational changes that the CPF/HSA complex undergoes upon ibuprofen binding were studied by MD simulation studies. To this end, CPF was manually docked into the “V” cleft of the tertiary IBU/HSA complex (PDB code 2BXG) with the arrangement observed in the binary CPF/HSA complex (Fig. 5B) and the resulting quaternary complex was subjected to 100 ns of MD simulation. The opposite case was also explored, i.e. the addition of two molecules of ibuprofen to the binary CPF/HSA complex, providing similar results. Whereas no significant changes were observed in the binding mode of the two ibuprofen molecules (S-isomer), which remain stable during the whole simulation (Fig. 6A and B), CPF underwent a large displacement from its original binding pocket (up to 15 Å) as well as significant variations in its binding arrangement (rotation) as a consequence of the loss of its main anchoring interactions (Fig. 6C). This is clearly visualized by comparison of its position in the binary CPF/HSA complex and after 100 ns of dynamics simulation in the quaternary IBU + IBU + CPF/HSA complex (Fig. 6D). The binding free energy calculated for each ligand in their corresponding pockets of the complex also highlighted the lower binding affinity of CPF (Table 4, entries 3 vs. 1). This was calculated using the MM/PBSA23 approach in implicit water (Generalised Born, GB) as implemented in Amber. Thus, whereas the affinity of ibuprofen to both pockets is retained or even increased, a large decrease in the calculated binding free energy of CPF is predicted (∼6.8 kcal).
 | ||
| Fig. 6 (A–C) Comparison of the quaternary IBU + IBU + CPF HSA/complex after minimisation and prior to simulation (grey) and after 100 ns of dynamics simulation (green). A close view of IBU (A and B) and CPF (C) binding sites is provided. (D) Comparison of the position of CPF in the binary CPF/HSA/complex (see Fig. 5B) and in the quaternary one (A–C). Note that for IBU no significant changes are observed during the simulation, whereas CPF is displaced from its binding pocket even prior to simulation. | ||
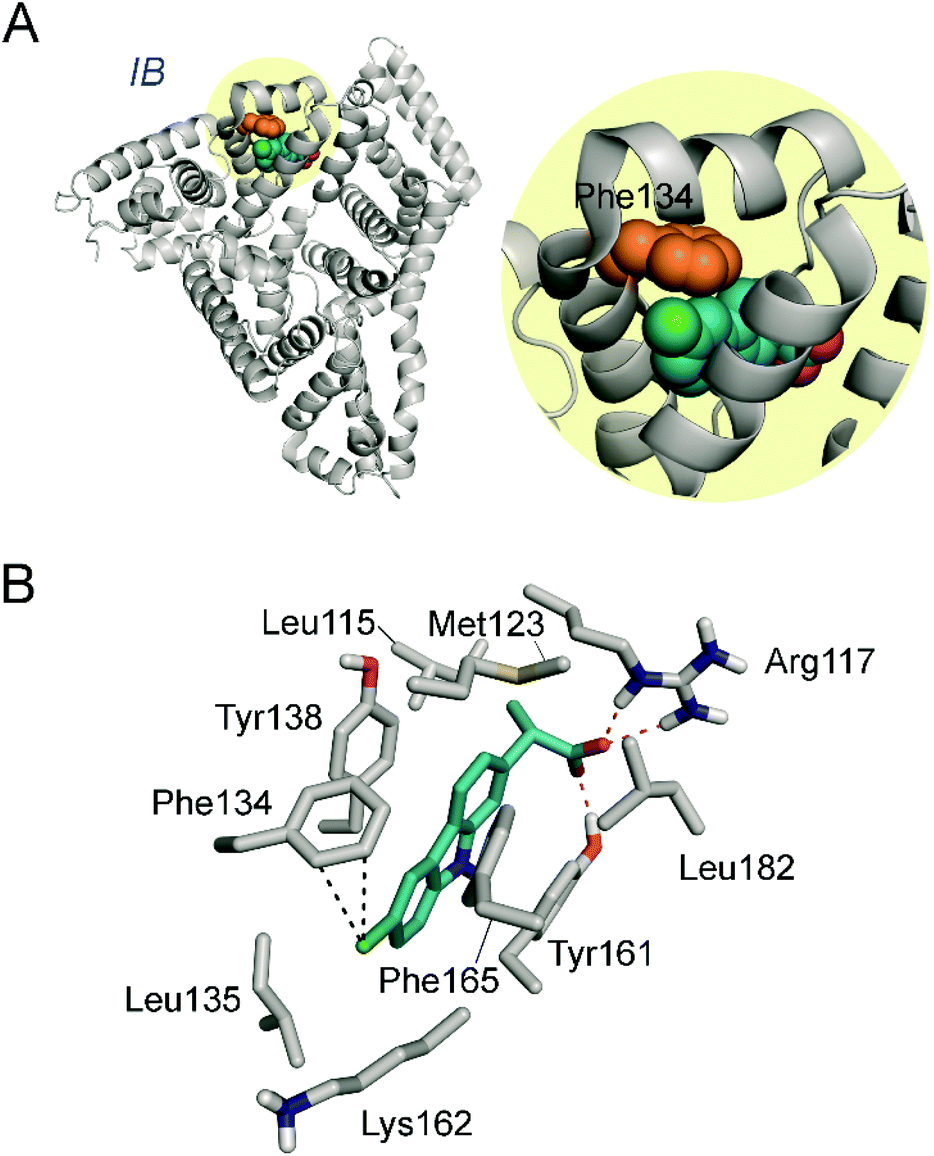 | ||
| Fig. 7 (A) Overall view of the binary CPF/HSA-IB complex obtained by docking and MD simulation studies. The ligand and residue modified are shown as spheres. (B) The viewpoint showed corresponds to the snapshot after 50 ns of MD simulation. Relevant side chain residues are shown and labelled. Polar (red) contacts and distances (black) to Phe134 are shown as dashed lines. | ||
Conclusions
In the dark, CPF binds to serum albumins of different species, giving rise to non-covalent complexes. These complexes show characteristic features in their photophysical properties, being especially noteworthy in the lengthening of their triplet excited state lifetimes by at least one order of magnitude. Irradiation of the complexes by selective excitation of the CPF chromophore leads to irreversible covalent binding, as demonstrated by gel filtration chromatography, where incorporation of the fluorophore into the protein fraction is clearly observed. Digestion of the irradiated CPF/SA complexes and subsequent proteomic analysis reveal the attachment of the photodehalogenated drug to selected amino acid residues. Remarkably in all the investigated proteins, covalent modification of Tyr452 (in HSA, RbSA, RtSA) or Tyr451 (in BSA, PSA, SSA) is consistently observed. This indicates the presence of a common recognition center in SAs, irrespective of the involved species, at the interface between sub-domains IB and IIIA (“V” cleft pocket). Another relevant binding pocket of CPF to serum albumins, which was identified in four of the six proteins, proved to be the most accessible part of sub-domain IB.The molecular basis of this common recognition was studied by molecular dynamics simulation studies of the corresponding non-covalent complexes. The results revealed that these complexes are very stable and the carbon atom of CPF in which the radical is generated, would be in close contact with the identified aromatic residues. Moreover, diverse previously reported competition studies with site specific drugs such as (S)-ibuprofen (site II) and warfarin (site I) revealed that CPF binds with high affinity to site II and with weaker affinity to site I on HSA. The proteomic and molecular modelling studies reported here also corroborate this idea. Our studies revealed that CPF would be displaced by (S)-ibuprofen from its main binding pocket by an allosteric effect in site II, rather than an expected direct molecular displacement. The former would be due to: (i) the large conformational changes that HSA can undergo upon binding ligands, and (ii) the close proximity of this cleft to site II. The reported integrated strategy that involves irradiation of CPF/SA complexes, coupled with fluorescence, identification of the photoinduced modified amino acid residues by proteomic analysis, docking and MD simulation studies could, in principle, be extended to a variety of protein/ligand complexes if an active chromophore is present in the ligand.
Experimental
General
Racemic carprofen, HSA, BSA, PSA, RbSA, RtSA and SSA were commercially available. Spectrophotometric, HPLC or reagent grade solvents were used without further purification. Solutions of phosphate-buffered saline (PBS) (0.01 M, pH 7.4) were prepared by dissolving phosphate-buffered saline tablets in Milli-Q water. Steady state absorption spectra were recorded in a JASCO V-630 spectrophotometer. Analytical HPLC analysis was performed by means of a Waters HPLC system connected to a PDA Waters 2996 detector. HPLC isolation was carried out on a JASCO HPLC equipment, composed of a DG-2080-54 degasification system, an LG-2080-04 mixer and a PU-2080 pump connected to a UV-1575 detector. The (S)-enantiomer of carprofen was separated from a 1.8 M racemic mixture in methyl tert-butyl ether by HPLC (Technocroma Kromasil 100-TBB column, mobile phase hexane/methyl tert-butyl ether/acetic acid (45:55:0.1, v/v/v), flow 2.2 mL min−1).
Fluorescence experiments
Spectra were recorded on a JASCO FP-8500 spectrofluorometer system, provided with a monochromator in the wavelength range of 200–850 nm, at 22 °C. Experiments were performed in solutions of CPF (2.5 × 10−5 M) in the presence of SAs (at 1:1 CPF/SA), employing 10 × 10 mm2 quartz cells with 4 mL capacity. The excitation wavelength was 320 nm.
Laser flash photolysis experiments
A Q-switched Nd:YAG laser (Quantel Brilliant, 355 nm, 15 mJ per pulse, 5 ns fwhm) coupled to a mLFP-111 Luzchem miniaturized equipment was employed. This transient absorption spectrometer includes a ceramic xenon light source, 125 mm monochromator, Tektronix 9-bit digitizer TDS-3000 series with 300 MHz bandwidth, compact photomultiplier, power supply, cell holder and fiber optic connectors, fiber optic sensor for laser-sensing pretrigger signal, computer interfaces, and a software package developed in the LabVIEW environment from National Instruments. The LFP equipment supplies 5 V trigger pulses with a programmable frequency and delay. The rise time of the detector/digitizer is ∼3 ns up to 300 MHz (2.5 GHz sampling). The monitoring beam is provided by a ceramic xenon lamp and delivered through fiber optic cables. The laser pulse is probed by a fiber that synchronizes the LFP system with the digitizer operating in the pretrigger mode. Transient spectra and kinetic traces were recorded employing 10 × 10 mm2 quartz cells with 4 mL capacity. The concentration of CPF was 1.0 × 10−4 M, and the CPF/SA molar ratio was 1:1. All the experiments were carried out at room temperature. The <τT> values of CPF were determined by fitting the decay traces at λmax = 450 nm by means of a monoexponential function.
Steady-state photolysis experiments
Steady-state photolysis of CPF (2.5 × 10−5 M) was performed by using a 150 W Xe lamp coupled to a monochromator at lamp output (λexc = 320 nm) in PBS in air and in the presence of protein (CPF/SA 1:1 molar ratio), through Pyrex. The course of the reaction was followed by monitoring the changes in the fluorescence spectra of the reaction mixtures at increasing times.
Treatment with guanidinium chloride and filtration through Sephadex
Guanidinium chloride (1.72 mL, 6 M) was added to 3 mL of CPF/SA in PBS, in order to cause protein denaturation. The mixture was then filtered through a Sephadex P-10 column. Firstly, 25 mL of pure PBS were eluted; then 2.5 mL of the CPF/SA mixture treated with GndCl were eluted. Subsequently, 3.5 mL of PBS were eluted again. The absorption and emission of the final sample were then measured. To take into account the dilution factor, a similar experiment was conducted directly on SA (in the absence of CPF). In this way, the ratio between the absorbance value before and after filtration was obtained, which was employed as a correction factor in the experiments.Protein digestion and LC-ESI-MS/MS analysis
After irradiation, the SAs were enzymatically digested into smaller peptides using trypsin or V8. Subsequently, these peptides were analyzed using nanoscale liquid chromatography coupled to tandem mass spectrometry (nano LC-MS/MS). Briefly, 20 μg of sample were taken (according to Qubit quantitation) and the volume was set to 20 μL. Digestion was achieved with sequencing grade trypsin (Promega) or Glu-C. For the case of trypsin, the steps are: (i) 2 mM DTT in 50 mM NH4HCO3, V = 25 μL, 20 min (60 °C); (ii) 5.5 mM IAM in 50 mM NH4HCO3, V = 30 μL, 30 min (dark); (iii) 10 mM DTT in 50 mM NH4HCO3, V = 60 μL, 30 min; (iv) trypsin (trypsin:protein ratio 1:20 w/w) V = 64 μL, overnight 37 °C. Digestion was stopped with 7 μL 10% TFA (cf. protein ca. 0.28 μg μL−1). Next, 5 μL of sample (except the main bands) were loaded onto a trap column (NanoLC column, 3 μ C18–CL, 350 μm × 0.5 mm; Eksigent) and desalted with 0.1% TFA at 3 μL min−1 for 5 min. The peptides were then loaded onto an analytical column (LC Column, 3 μ C18–CL, 75 μm × 12 cm, Nikkyo) equilibrated in 5% acetonitrile and 0.1% formic acid. Elution was carried out with a linear gradient of 5 to 45% B in A for 30 min (A: 0.1% formic acid; B: acetonitrile, 0.1% formic acid) at a flow rate of 300 μL min−1. Peptides were analyzed in a nanoESI qQTOF mass spectrometer (5600 TripleTOF, ABSCIEX). The TripleTOF was operated in information-dependent acquisition mode, in which a 0.25 s TOF MS scan from 350–1250 m/z was performed, followed by 0.05 s product ion scans from 100–1500 m/z on the 50 most intense 2–5 charged ions. ProteinPilot v4.5 (ABSciex) search engine default parameters were used to generate the peak list directly from 5600 TripleTOF wiff files. The obtained mgf was used for identification with MASCOT (v 4.0, Matrix-Science). Database search was performed on the SwissProt database. Searches were done with tryptic specificity allowing one missed cleavage and a tolerance on the mass measurement of 100 ppm in MS mode and 0.6 Da in MS/MS mode. Carbamidomethylation of Cys was used as a fixed modification and oxidation of Met and deamidation of Asn and Gln as variable modifications. A modification was defined in Phe, Tyr, and Trp for CPF.
Docking studies
They were carried out using the program GOLD 5.2.217 and the protein coordinates found in the crystal structure of HSA in complex with palmitic acid (PDB code 4BKE,18 2.35 Å). Ligand geometries were minimized using the AM1 Hamiltonian as implemented in the program Gaussian 0926 and used as MOL2 files. Each ligand was docked in 25 independent genetic algorithm (GA) runs, and for each of these a maximum number of 100000 GA operations were performed on a single population of 50 individuals. Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95, and 10, respectively), as well as the hydrogen bonding (4.0 Å) and van der Waals (2.5 Å) parameters. The position of the side chain of the experimentally observed modified residue was used to define the active-site and the radius was set to 8 Å. All crystallographic water molecules and the ligands were removed for docking. The “flip ring corners” flag was switched on, while all the other flags were off. The GOLD scoring function was used to rank the ligands in the order of fitness.
Building of the apo-serum albumin models
The Phyre2 homology modelling web server was used to model the three-dimensional structure of serum albumin from rat, rabbit, porcine and sheep.27 The coordinates of the crystallographically determined HSA (PDB code 1ETB,28 1.8 Å, chain A) were chosen as the main templates for building the model. The resulting protein structures had a 73%, 76%, 75% and 75% sequence identity and 95% of its sequence was modelled with 100% confidence by the template (Fig. S2†).Molecular dynamics simulations
000 TIP3P water molecules and neutralized by the addition of sodium ions. The system was minimized in five stages: (a) minimization of poorly unsolved residues 6, 41, 109, 170, 204, 205, 244, 276, 277, 317, 372, 396, 466, 519, 521, 538, 560, and 570 (1000 steps, first half using steepest descent and the rest using conjugate gradient); (b) minimization of the ligand (1000 steps, first half using steepest descent and the rest using conjugate gradient); (c) minimization of the solvent and ions (5000 steps, first half using steepest descent and the rest using conjugate gradient); (d) minimization of the side chain residues, water and ions (5000 steps, first half using steepest descent and the rest using conjugate gradient); (e) final minimization of the whole system (5000 steps, first half using steepest descent and the rest using conjugate gradient). A positional restraint force of 50 kcal mol−1 Å−2 was applied to not minimized residues of the protein during stages a–c and to α carbons during stage d, respectively.
MM/PBSA calculations
The binding free energies for CPF on binding sub-domains IIIA and IIIB were calculated by the MM/PBSA23 approach implemented in Amber Tools 1.5. The ante-MMPBSA.py module was used to create topology files for the complex, protein and ligands and binding free energies were calculated with the MMPBSA.py module.43 A single trajectory approach was used to calculate binding free energies considering only: (a) for 100 ns simulations, the last 80 ns (401 snapshots) of the whole MD trajectories; (b) for 150 or 200 ns simulations, the last 100 ns (401 snapshots) of the whole MD trajectories. The Poisson–Boltzmann (PB) and Generalized Born (GB) implicit solvation models were employed. The latter model provided relative free energy values more in agreement with the experimental results.Abbreviations
| SAs | Serum albumin(s) |
| HSA | Human serum albumin |
| BSA | Bovine serum albumin |
| PSA | Pig serum albumin |
| RbSA | Rabbit serum albumin |
| RtSA | Rat serum albumin |
| SSA | Sheep serum albumin |
| CPF | (S)-Carprofen |
| IBU | (S)-Ibuprofen |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Economy and Competiveness (CTQ2016-78875-P, SAF2016-75638-R and BES-2014-069404), Generalitat Valenciana (PROMETEO2017/075), Consellería de Cultura, Educación e Ordenación Universitaria (Centro singular de investigación de Galicia accreditation 2016-2019, ED431G/09) and the European Regional Development Fund (ERDF) is acknowledged. This work was also supported by Instituto de Salud Carlos III (ISCIII) co-funded by Fondo Europeo de Desarrollo Regional – FEDER for the Thematic Networks and Co-operative Research Centres: ARADyAL (RD16/0006/0030). EL thanks the Xunta de Galicia for his postdoctoral fellowship. We are also grateful to the Centro de Supercomputación de Galicia (CESGA) for use of the Finis Terrae II supercomputer. The proteomic analysis was performed in the proteomics facility of SCSIE University of Valencia that belongs to ProteoRed PRB2-ISCIII and is supported by grant PT13/0001, of the PE I+D+I 2013–2016, funded by ISCIII and FEDER.Notes and references
- D. Limones-Herrero, R. Pérez-Ruiz, E. Lence, C. González-Bello, M. A. Miranda and M. C. Jiménez, Chem. Sci., 2017, 8, 2621–2628 RSC.
- (a) W. M. O'Brien and G. F. Bagby, Pharmacotherapy, 1987, 7, 16–24 CrossRef; (b) S. L. Curry, S. M. Cogar and J. L. Cook, J. Am. Anim. Hosp. Assoc., 2005, 41, 298–309 CrossRef; (c) P. Lees, M. F. Landoni, J. Giraudel and P. L. Toutain, J. Vet. Pharmacol. Ther., 2004, 27, 479–490 CrossRef CAS.
- (a) T. J. Peter, All about albumin: biochemistry, genetics and medical applications, Academic press, California, 1996 Search PubMed; (b) U. Kragh-Hansen, Pharmacol. Rev., 1981, 33, 17–53 CAS.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS.
- (a) G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1975, 11, 824–832 CAS; (b) U. Kragh-Hansen, V. T. G. Chuang and M. Otagiri, Biol. Pharm. Bull., 2002, 25, 695–704 CrossRef CAS.
- (a) G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1252–1261 Search PubMed; (b) M. Fasano, S. Curry, E. Terreno, M. Galliano, G. Fanali, P. Narciso, S. Notari and P. Ascenzi, IUBMB Life, 2005, 57, 787–796 CrossRef CAS; (c) D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–203 CrossRef CAS.
- (a) T. Kosa, T. Maruyama and M. Otagiri, Pharm. Res., 1997, 14, 1607–1612 CrossRef CAS; (b) A. F. Aubry, N. Markoglou and A. McGann, Comp. Biochem. Physiol., Part C: Pharmacol., Toxicol. Endocrinol., 1995, 112, 257–266 CrossRef CAS; (c) M. H. Rahman, T. Maruyama, T. Okada, K. Yamasaki and M. Otagiri, Biochem. Pharmacol., 1993, 46, 1721–1731 CrossRef CAS; (d) H. Kohita, Y. Matsushita and I. Moriguchi, Chem. Pharm. Bull., 1994, 42, 937–940 CrossRef CAS.
- (a) I. Vayá, R. Pérez-Ruiz, V. Lhiaubet-Vallet, M. C. Jiménez and M. A. Miranda, Chem. Phys. Lett., 2010, 486, 147–153 CrossRef; (b) V. Lhiaubet-Vallet, Z. Sarabia, F. Boscá and M. A. Miranda, J. Am. Chem. Soc., 2004, 126, 9538–9539 CrossRef CAS; (c) V. Lhiaubet-Vallet, F. Bosca and M. A. Miranda, J. Phys. Chem. B, 2007, 111, 423–431 CrossRef CAS.
- (a) M. H. Rahman, T. Maruyama, T. Okada, K. Yamasaki and M. Otagiri, Biochem. Pharmacol., 1993, 46, 1721–1731 CrossRef CAS; (b) M. H. Rahman, T. Maruyama, T. Okada, T. Imai and M. Otagiri, Biochem. Pharmacol., 1993, 46, 1733–1740 CrossRef CAS.
- (a) M. Divkovic, C. K. Pease, G. F. Gerberick and D. A. Basketter, Contact Dermatitis, 2005, 53, 189–200 CrossRef CAS; (b) G. Johannesson, S. Rosqvist, C. H. Lindh, H. Welinder and B. A. G. Jönsson, Clin. Exp. Allergy, 2001, 31, 1021–1030 CrossRef CAS PubMed; (c) A. Lahoz, D. Hernández, M. A. Miranda, J. Pérez-Prieto, I. Morera and J. Castell, Chem. Res. Toxicol., 2001, 14, 1486–1491 Search PubMed.
- P. Jones, In vitro phototoxicity assays, in Principles and Practice of Skin Toxicology, ed. R. Chilcott and S. Price, John Wiley & Sons, 2008, p. 169 Search PubMed.
- (a) Y. Merot, M. Harms and J.-H. Saurat, Dermatologica, 1983, 166, 301–307 Search PubMed; (b) H. Kietzmann, T. H. Rüther and B. Scheuer, Akt. Dermatol., 1984, 10, 17–20 Search PubMed; (c) E. Hoting and K. H. Schultz, Dermatosen, 1984, 32, 215–217 CAS; (d) R. Roelandts, Dermatologica, 1986, 172, 64–65 CrossRef CAS; (e) F. Bosca, M. L. Marin and M. A. Miranda, Photochem. Photobiol., 2001, 74, 637–655 CrossRef CAS; (f) A. C. Kerr, F. Muller, J. Ferguson and R. S. Dawe, Br. J. Dermatol., 2008, 159, 1303–1308 CrossRef CAS.
- J. Moser, F. Boscá, W. W. Lovell, J. V. Castell, M. A. Miranda and A. Hye, J. Photochem. Photobiol., B, 2000, 58, 13–19 CrossRef CAS.
- P.-L. Toutain, A. Ferran and A. Bousquet-Mélou, Species Differences in Pharmacokinetics and Pharmacodynamics, in Handbook of Experimental Pharmacology, Vol. 199, Comparative and Veterinary Pharmacology, ed. F. Cunningan, J. Elliot and P. Lees, Springer-Verlag, Berlin, Heidelberg, 2010 Search PubMed.
- F. Bosca, S. Encinas, P. F. Heelis and M. A. Miranda, Chem. Res. Toxicol., 1997, 10, 820–827 Search PubMed.
- B. Sekula, A. Ciesielska, P. Rytczak, M. Koziołkiewicz and A. Bujacz, Biosci. Rep., 2016, 36, e00338, DOI:10.1042/BSR20160089.
- http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ .
- A. Sivertsen, J. Isaksson, H. S. Leiros, J. Svenson, J. Svendsen and B. O. Brandsdal, BMC Struct. Biol., 2014, 14, 4 CrossRef.
- R. Pérez-Ruiz, E. Lence, I. Andreu, D. Limones-Herrero, C. González-Bello, M. A. Miranda and M. C. Jiménez, Chem. – Eur. J., 2017, 23, 13986–13994 CrossRef.
- V. Lhiaubet-Vallet, F. Boscá and M. A. Miranda, J. Phys. Chem. B, 2007, 111, 423–431 CrossRef CAS.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS.
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Biol., 1998, 5, 827–835 CrossRef CAS.
- B. R. Miller III, T. D. McGee Jr., J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef.
- Z. M. Wang, J. X. Ho, J. R. Ruble, J. Rose, F. Rüker, M. Ellenburg, R. Murphy, J. Click, E. Soistman, L. Wilkerson and D. C. Carter, Biochim. Biophys. Acta, 2013, 1830, 5356–5374 CrossRef CAS.
- P. A. Zunszain, J. Ghuman, T. Komatsu, E. Tsuchida and S. Curry, BMC Struct. Biol., 2003, 3, 6–14 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.2, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS.
- J. A. Hamilton, L. K. Steinrauf, B. C. Braden, J. Liepnieks, M. D. Benson, G. Holmgren, O. Sandgren and L. Steen, J. Biol. Chem., 1993, 268, 2416–2424 CAS.
- (a) E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak, J. C. Delepine, P. Cieplak and F.-Y. Dupradeau, Nucleic Acids Res., 2011, 39, W511–W517 CrossRef CAS; (b) http://upjv.q4md-forcefieldtools.org/RED/ (accessed June 1, 2018). (c) F.-Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, Phys. Chem. Chem. Phys., 2010, 12, 7821–7839 RSC.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, O. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS.
- (a) J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS; (b) J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- (a) J. C. Gordon, J. B. Myers, T. Folta, V. Shoja, L. S. Heath and A. Onufriev, Nucleic Acids Res., 2005, 33(Web Server issue), W368 CrossRef CAS; (b) http://biophysics.cs.vt.edu/H++ .
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER 2015, University of California, San Francisco, 2015 Search PubMed.
- A. W. Goetz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef.
- R. Salomon-Ferrer, A. W. Goetz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS.
- S. Le Grand, A. W. Goetz and R. C. Walker, Comput. Phys. Commun., 2013, 184, 374–380 CrossRef CAS.
- T. A. Darden, D. York and L. G. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- W. L. DeLano, The PyMOL Molecular Graphics System, DeLano Scientific LLC, Palo Alto, CA, USA, 2008. http://www.pymol.org/ Search PubMed.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS.
- http://www.amber.utah.edu/AMBER-workshop/London-2015/pca/ .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qo01045e |
| This journal is © the Partner Organisations 2019 |