
Stereoselective synthesis of a 9-O-sulfo Neu5Gc-capped O-linked oligosaccharide found on the sea urchin egg receptor†
Anindya
Das
a,
Pei-Jhen
Li
a,
Avijit K.
Adak
a,
Hsin-Ru
Wu
a,
Mohammad Tarique
Anwar
a,
Pei-Yun
Chiang
a,
Chung-Ming
Sun
b,
Jih-Ru
Hwu
a and
Chun-Cheng
Lin
 *a
*a
aDepartment of Chemistry, National Tsing Hua University, Hsinchu-30013, Taiwan. E-mail: cclin66@mx.nthu.edu.tw
bDepartment of Applied Chemistry, National Chiao Tung University, Hsinchu-30013, Taiwan
First published on 9th November 2018
Abstract
A straightforward synthesis of the α(2→5)-linked 9-O-sulfated Neu5Gc-capped O-linked tetrasaccharide (1) identified in the egg cell surface glycoproteins of the sea urchin Strongylocentrotus purpuratus receptor for sperm is reported. The construction of the α(2→5)Neu5Gc trimer by the formation of an amide linkage rather than a glycosidic bond avoids the requirement for α-stereoselective glycosylation. To highlight this amide bond formation strategy as a relatively facile method for synthesizing oligo-Neu5Gc containing O-linked glycans, its versatility is demonstrated by application to the coupling of SO3H-9Neu5Gcα(2→5)Neu5Gc glycolic acid and a sialyl-Tn-derived amine for achieving the target tetrasaccharide. This synthetic strategy may be implemented in the generation of other structurally similar O-sulfo Neu5Gc-capped α2→5-Oglycolyl-linked oligo-Neu5Gc chains, enabling additional biological and chemical investigations.
Introduction
Sialic acids (Sias) are nine-carbon core-structure acidic capping monosaccharides on glycan chains attached to cell surfaces and secreted glycoconjugates in animals.1 The ubiquitous terminal position and net negative charge at physiological pH of Sias underscore their importance in mediating numerous cellular and extracellular binding events and their essentialness for early embryogenesis.2 The three major forms of the Sia family are derivatives of N-acetylneuraminic acid (Neu5Ac), N-glycolylneuraminic acid (Neu5Gc), and deaminated neuraminic acid (KDN). Although Neu5Ac is commonly found in nature and in most mammals, the corresponding Sia Neu5Gc is rare in vertebrates.3 However, Neu5Gc-rich polysaccharides have been ubiquitously found in the egg jelly coats of various sea urchin species.4 These Sia-rich glycoproteins consist of unique α2→5-Oglycolyl-linked oligo/poly-Neu5Gc chains linked to core oligosaccharides with an average degree of polymerization (DP) of approximately 20. In these structures, each Neu5Gc residue is attached to the hydroxyl group of the glycolyl moiety of the nonreducing Neu5Gc terminus. The presence of α2→5-Oglycolyl-linked poly-Neu5Gc structures in egg jelly coat glycoproteins is common to two different species of sea urchin, Hemicentrotus pulcherrimus and Strongylocentrotus purpuratus, and has attracted significant interest because of their critical roles in increasing the intracellular pH and inducing the sperm acrosome reaction.5,6In pursuit of the identification of a novel (→5-OglycolylNeu5Gcα2→)n-linked poly-Sia chain, Kitazume and co-workers7 first isolated a (HSO3)9Neu5Gc-capped (→5-OglycolylNeu5Gcα2→)n oligosaccharide from the glycopeptide fraction of the egg cell surface complex of the sea urchin H. pulcherrimus in 1996. A subsequent study of the egg vitelline layer glycopeptide fractions of the S. purpuratus egg receptor8 for sperm led to the validation that this linear 9-O-sulfated α2→5-Oglycolyl-linked oligo-Neu5Gc chain is α-O-glycosidically linked through the C6-OH of the core N-acetylgalactosamine (GalNAc) moiety to the hydroxyl group of serine (Ser) and/or threonine (Thr) residues of the peptide backbone (1, Fig. 1). The oligo/poly-Neu5Gc units of the glycoprotein complex on the egg cell surface, however, are relatively short: the average DP was determined to be approximately 3 but may vary among sea urchin species.7,8 Because of their additional negatively charged sulfate moiety, both 9-O-sulfo Neu5Gcα2(→5-Oglycolyl-Neu5Gcα2→)1–2 epitopes show complete resistance to exosialidase treatment, which might serve as a protective mechanism and a stop signal for the elongation of the poly-Neu5Gc structure in sea urchins.5 In addition, the sulfated (HSO3)9Neu5Gcα2(→5-Oglycolyl Neu5Gcα2→)2 glycan motif of 1 displays significantly higher inhibitory activity on fertilization than the nonsulfated form of this oligo-Neu5Gc chain at a concentration of 600 μM.8 However, the α2→5-Oglycolyl-linked oligo-Neu5Gc structures isolated from natural sources could be difficult to obtain in large quantities, and their biological profiling may be hampered, partially due to the lack of highly homogeneous (HSO3)Neu5Gc-capped α2→5-Oglycolyl-linked glyco-peptide-containing samples. Thus, the development of efficient synthetic strategies for (HSO3)9Neu5Gc-capped O-linked tetra-saccharide and its nonsulfated analogs would be of great value.
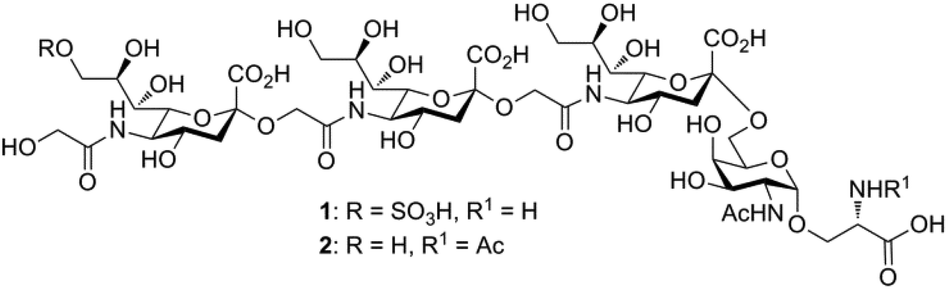 | ||
| Fig. 1 Structure of the 9-O-sulfo Neu5Gc-containing O-linked tetrasaccharide attached to the Ser residue (1) found as a component of the sea urchin egg receptor for sperm. The corresponding nonsulfated structural variant glycan 2 is also shown. | ||
Methods toward the synthesis of these privileged (→5-OglycolylNeu5Gcα2→) oligo-Sia glycan structures preferentially utilize the amide condensation between two readily accessible building blocks: a C-5 amino α-O-glycolyl sialoside and a glycolic acid-derived Neu5Ac.9 Although the peptide coupling yield was higher, O-sialylation using thiosialoside donors with a lower-molecular-weight alcohol-based benzyl glycolate acceptor often suffers from the formation of an α-/β-anomeric mixture, which is mainly due to the low stereoselectivity of sialyl donors. In addition, the use of an N-acetyl protecting group at the C-5 position of the nonreducing end of the Sia residue makes elongation of the Nu5Gc chain difficult because of the tedious deprotection step. In this regard, a 2,2,2-trichloroethoxy-carbonyl (Troc)-protected α-2-benzyl glycolate sialoside was reported, which allows sialylation and amide-coupling reactions from both the reducing end to the nonreducing end and vice versa during the synthesis of α(2→5)Neu5Gc-linked oligomers.10 Alternatively, an α(2→5)-linked Neu5Gc dimer was obtained via a regioselective ring opening of the minimally protected Sia derivative of a spirocyclic intermediate, which eliminated some of the aforementioned limitations.11
We previously established a facile method for the stereoselective construction of α(2→5)-linked di-Sia using an α-2-allyloxy sialoside that served as a common substrate for both Sia intermediates: the donor (acid) and acceptor (amine).12 The synthetic strategy was based on a highly α-selective sialylation between an allyl alcohol and an electrophilic β-sialyl chloride. For Sia chain extension, amide bond formation was employed instead of sialylation reactions to build the α(2→5)-linked allyloxy disialoside, an intermediate poised for conversion to the Neu5Gc oligomer. To avoid the notoriously low α-selectivity in sialylation, several research groups have favored amide bond formation to construct the α(2→5)-linkage in Neu5Gc-containing echinodermatous gangliosides.13–16 However, no synthetic efforts toward the naturally occurring (HSO3)9Neu5Gc-containing O-galactosaminyl-Ser 1 or its nonsulfated structural variant glycan 2 have been reported. This synthetic strategy could afford important chemical biology tools to investigate proteins that utilize these glycans and to enable a greater diversity to clarify the function of the egg and sperm recognition event during fertilization in nonmammalian sea urchins. Herein, we present the first chemical synthesis of the biologically active (HSO3)9Neu5Gcα2(→5OglycolylNeu5Gcα2→)2Neu5Gcα(2→6)GalNAc-Ser 1, a tetrasaccharide constituent of the cell surface glycoprotein found in the sea urchin egg receptor for sperm.
Results and discussion
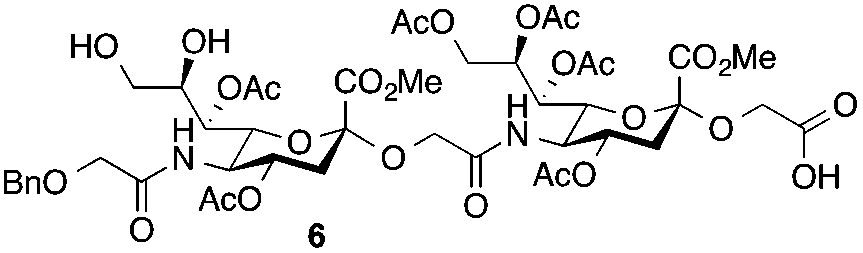
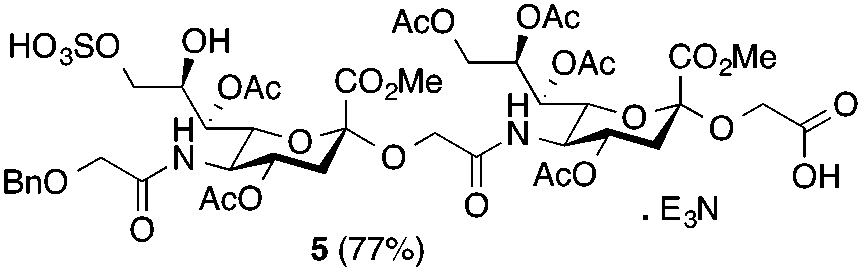
Glycan 1 features a terminal 9-O-sulfo Neu5Gc motif with three contiguous α(2→5)-linkages in conjunction with an α-linked O-galactosaminyl-Ser group at the reducing end. Tetrasaccharide glycan 2 shares similar structural features but lacks the 9-SO4− group at the nonreducing end Neu5Gc residue. Our retrosynthetic plan for the construction of Neu5Gc-capped O-galactosaminyl-Ser tetrasaccharide glycans 1 and 2 is outlined in Fig. 2. The target glycans could be achieved by a global deprotection of fully protected tetrasaccharide 3 and its di-O-isopropylidene protected analog 4. At the very outset, access to the reducing-end internal α(2→5) linkage in 3 was envisioned through the amide bond formation between 9-O-sulfo Neu5Gcα(2→5)Neu5Gc-glycolic acid (5) and a Fmoc-sialyl-Tn (sTn)-derived amine (9) obtained from 10. It was anticipated that regioselective 9′-O-sulfation could be performed on an 8,9-diol intermediate (6) generated from 8,9-O-isopropylidene Neu5Gcα(2→5)Neu5Gc-linked disialoside 7 to obtain the characteristic structure of modified sialic acid SO3H-9Neu5Gcα(2→5)Neu5Gc 5. The fully protected tetrasaccharide (4) could in turn similarly be assembled by a [2 + 2] coupling from two disaccharide fragments: Neu5Gcα(2→5)Neu5Gc-glycolic acid (8) and a Fmoc-sTn-derived amine (11 or 12), as outlined in Fig. 2. The di-Neu5Gc building blocks (7 and 8) could be easily derived from readily accessible simple sialosides using a [1 + 1] coupling between acid 15 (or 16) and amine 17. The sTn derivatives (9–12) were synthesized either by an enzymatic approach using 13 and N-carboxybenzyl carbamate (Cbz)-protected mannosamine or chemical sialylation between Tn-derived glycosyl acceptor 14 and the Troc-protected thiosialyl donors (18 or 19).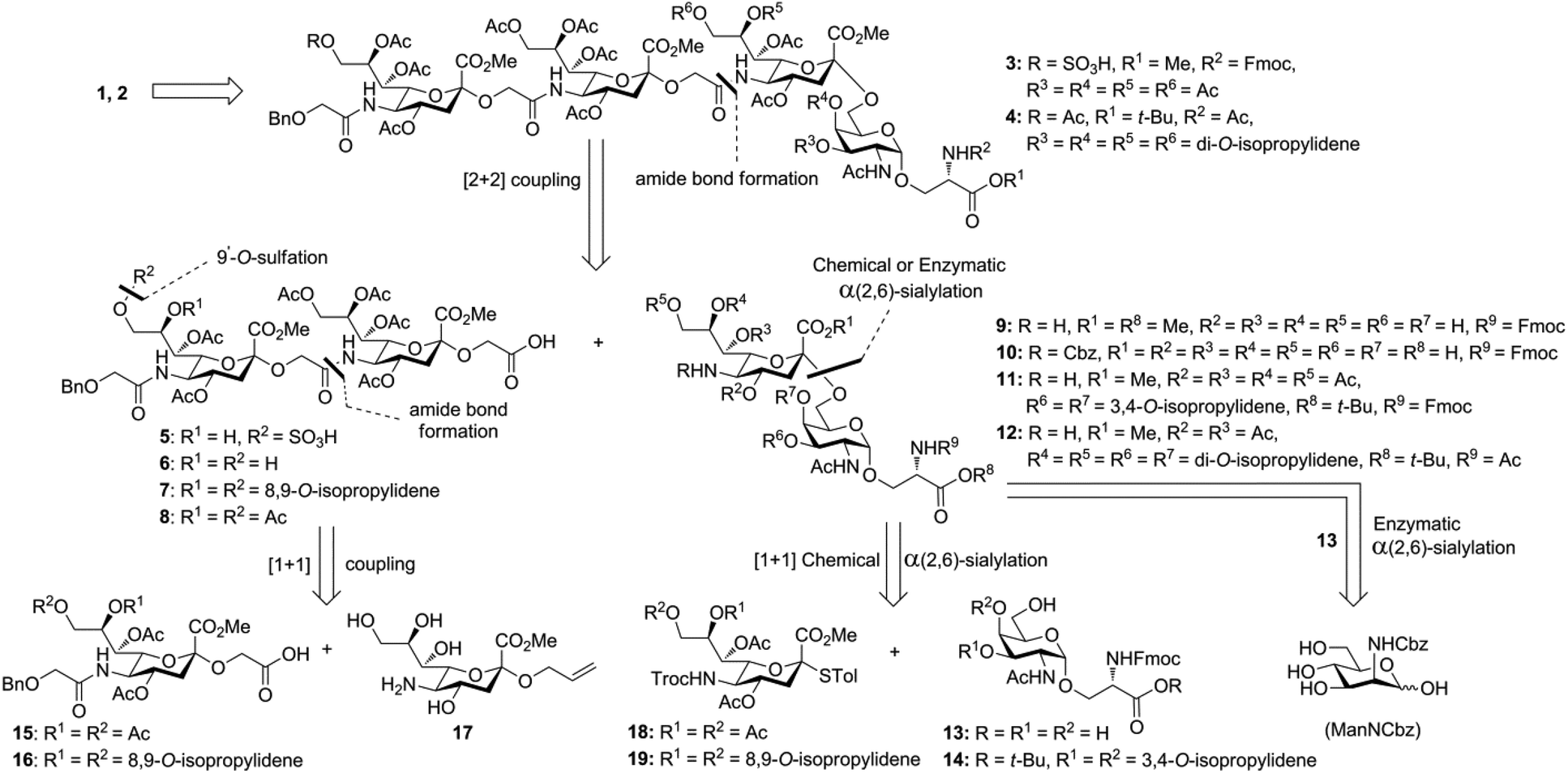 | ||
| Fig. 2 Retrosynthetic analysis of compounds 1 and 2. | ||
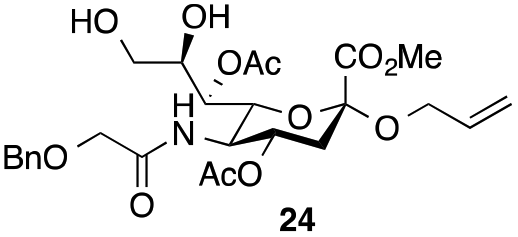
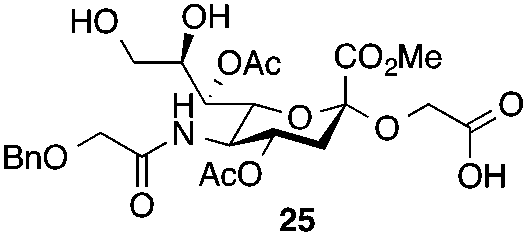
The synthesis of the target oligo-Neu5Gc compounds was started by the construction of di-Neu5Gc. As illustrated in Scheme 1, the synthesis of the C5 N-glycolyl-protected Neu5Gc acids (15 and 16) and their conversion to the corresponding disaccharide building blocks Neu5Gcα(2→5)Neu5Gc glycolic acids (6–8) were conducted by modifying our previously developed method.12 Accordingly, to install the glycolyl moiety at the nonreducing end, the known sialoside 1712 was coupled with benzyloxyacetic acid using PyBOP as a coupling reagent in the presence of Et3N in DMF at 0 °C to afford benzyloxyacetyl-protected Neu5Gc 20 in an isolated yield of 68%. The hydroxyl groups in 20 were either acetylated to give 21 or transformed into a fully protected cyclic 8,9-O-isopropylidene Neu5Gc derivative through a two-step regioselective acetonidation–acetylation procedure to afford 23 in good yield. The RuCl3–NaIO4-mediated oxidative cleavage of the terminal olefin at the reducing end in 21 and 23 generated the corresponding acids 15 (73%) and 16 (71%), respectively. These acids (15 and 16) were coupled with amine 17 to furnish the α(2→5)-linked disialosides 26 and 27 in yields of 54% and 52%, respectively. The DMAP-catalyzed O-acetylation of 26 and 27 produced fully protected Neu5Gc dimers 28 and 29, respectively. The α-stereochemistry at the anomeric centers of 27 and 28 was determined by selective proton-decoupled 13C NMR spectroscopy,17,18 using the coupling constants (3JC-1,H-3ax) of the axial Neu5Gc proton H3 (H-3ax) and the C-1 carbonyl carbon, which were 6.0 Hz at 169.53 ppm and 4.5 Hz at 167.51 ppm, respectively, for 27 and 5.8 Hz at 168.39 ppm and 5.5 Hz at 167.53 ppm, respectively, for 28. Subsequent oxidation of the double bond in 28 (RuCl3 and NaIO4) allows access to protected Neu5Gcα(2→5)Neu5Gc glycolic acid building block 8 in a satisfactory yield (63%). Notably, prolonged treatment with oxidants should be avoided, as it causes oxidative cleavage of the glycosidic bond. Similarly, 8,9-O-isopropylidene-protected Neu5Gcα(2→5)Neu5Gc glycolic acid building block 7 was prepared from α-2-allyloxy disialoside 29 in a 65% yield. Neu5Gc intermediates 23, 16, and 7 were further subjected to hydrolysis of the isopropylidene acetal by treatment with 80% aq. AcOH, generating 8,9-diol-containing sialosides 24 (86%) and 25 (75%) and Neu5Gcα(2→5)Neu5Gc glycolic acid 6 (74%), respectively, which are suitable substrates for regioselective 9-O-sulfation.
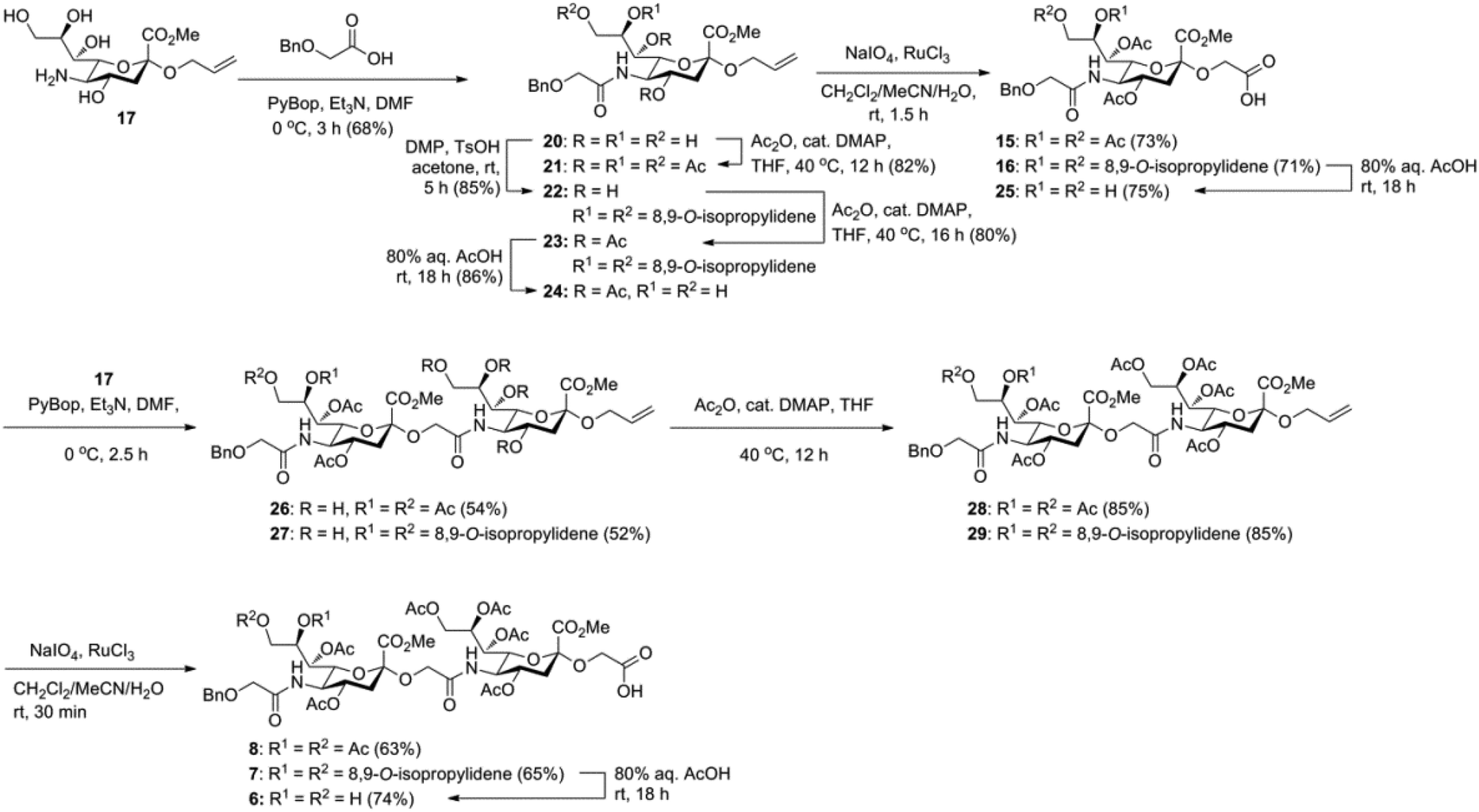 | ||
| Scheme 1 Synthesis of Neu5Gcα(2→5)Neu5Gc glycolic acid building blocks 6–8. | ||
Due to the presence of the destabilizing electron-withdrawing carboxyl group at the anomeric center and the lack of a participating auxiliary at the C-3 position of the sialyl donor,19,20 the major challenge encountered during the incorporation of Sia into the sugar chain was the realization of sialylation in high yield and with complete α-selectivity. The use of a stereodirecting nitrile solvent in combination with a diverse range of sialyl donors is commonly employed to promote α-stereoselectivity in the preparation of sTn derivatives.21–30 However, in most cases, these sialylation reactions do not provide the desired α-anomer exclusively. In the past decade, modification of the N-acetyl group at the C-5 position of Neu5Ac has been attempted to improve the reactivity of the sialyl donor.19,31 These C-5-modified donors generally provided dramatic increases in the reactivity and α-anomeric selectivity toward sialylation. Furthermore, a cyclic N-unsubstituted carbamate group at the C4 and C5 positions of the sialic acid was found to be an excellent sialyl donor for forming the α-glycosidic bond32–34 and has recently been applied for the synthesis of an α-aminooxy sTn derivative.34 We envisaged that an N-Troc-protection35 in the sialic acid donor would be suitable for the synthesis of sTn because the selective Troc deprotection can be accomplished under mild acidic conditions to produce a Neu5Acα(2–6)-Gal intermediate bearing a free amino group for further derivatization.
As outlined in Scheme 2, the synthesis of tetrasaccharide 30 was initiated by the chemical sialylation of Fmoc-protected O-(2-acetamido-2-deoxy-3,4-O-isopropylidene-α-D-galactopyranosyl)-Ser t-butyl ester 14.18 Subjecting 14 to N-iodosuccinimide/triflic acid (NIS-TfOH)-promoted sialylation with Troc-protected 4-thiotolyl sialyl donor 1836 in a mixture of CH2Cl2 and CH3CN (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)33 at −40 °C afforded Troc-protected sTn 31 in a 74% yield as an α-anomer. Importantly, no β-anomer was detected by TLC analysis. The anomeric configuration of the newly formed α-sialyl linkage was determined from the coupling constant between C1 and H-3ax, which was 6.0 Hz at 169.83 ppm. Reductive cleavage of the Troc group in 31 using Zn dust and 20% AcOH in acetonitrile afforded the requisite amine 11 in only a low 46% yield accompanied by the formation of byproduct 32 (37%) arising from C-8 to C-5 O→N acetyl migration,10 which could be separated from the desired product. We next tried to directly conjugate Fmoc-sTn amine 11 with Neu5Gcα(2→5)Neu5Gc glycolic acid 8 using PyBOP-mediated coupling conditions (DMF, Et3N, 0 °C). Unfortunately, none of the desired tetrasaccharide (30) was obtained. Analysis of the reaction mixture showed that most of the amine acceptor 11 was recovered as C-5 N-acetyl sTn 32.
:1)33 at −40 °C afforded Troc-protected sTn 31 in a 74% yield as an α-anomer. Importantly, no β-anomer was detected by TLC analysis. The anomeric configuration of the newly formed α-sialyl linkage was determined from the coupling constant between C1 and H-3ax, which was 6.0 Hz at 169.83 ppm. Reductive cleavage of the Troc group in 31 using Zn dust and 20% AcOH in acetonitrile afforded the requisite amine 11 in only a low 46% yield accompanied by the formation of byproduct 32 (37%) arising from C-8 to C-5 O→N acetyl migration,10 which could be separated from the desired product. We next tried to directly conjugate Fmoc-sTn amine 11 with Neu5Gcα(2→5)Neu5Gc glycolic acid 8 using PyBOP-mediated coupling conditions (DMF, Et3N, 0 °C). Unfortunately, none of the desired tetrasaccharide (30) was obtained. Analysis of the reaction mixture showed that most of the amine acceptor 11 was recovered as C-5 N-acetyl sTn 32.
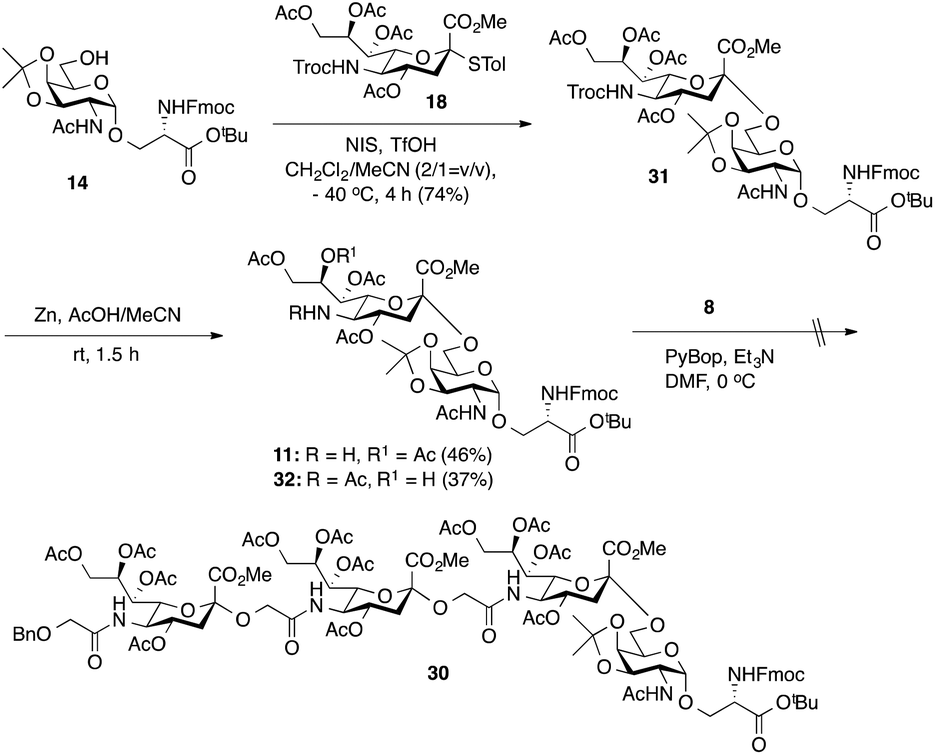 | ||
| Scheme 2 Chemical synthesis of Fmoc-sTn amine building block 11 and attempted synthesis of tetrasaccharide 30. | ||
Considering the low yield from the synthesis of key C-5 amino sTn building block 11 arising from O-acetyl migration of and the difficulty with the assembly of the target tetrasaccharide, we decided to switch to an alternative and possibly more effective synthetic pathway. As shown in Scheme 3, instead of 18, 8,9-O-isopropylidene-protected sialyl donor 1918 was used for the sialylation reaction because of its stereoselectivity in glycosylation and its isopropylidene group, which can be selectively removed under mild conditions. Most importantly, the use of 19 can avoid the competitive late stage C-8→C-5 acetyl migration on the sTn disaccharide. Gratifyingly, glycosylation of the Fmoc-Tn acceptor (14) with a stoichiometric amount of 19 under NIS-TfOH promotion conditions yielded α(2→6)-linked disaccharide 33 in a stereoselective manner (68% yield, α-only). The α-stereochemistry of the sTn disaccharide (33) was confirmed by the 3JC1–H3ax coupling constant of 6.3 Hz at 168.51 ppm. The Fmoc group on the Ser residue in 33 was then removed and exchanged to a more stable acetate group in 34 (92% for two steps) prior to NHTroc-deprotection to avoid undesired Fmoc cleavage in the following hydrogenolysis step. Finally, the cleavage of the Troc protecting group (activated zinc powder, 20% AcOH in CH3CN) in 34 proceeded smoothly, forming di-O-isopropylidene-protected C-5 amino sTn disaccharide 12 in 62% yield as the sole isolated product without affecting the acetonide or t-butyl ester groups. In this route, intramolecular C-8 to C-5 acetyl migration was avoided. The exposed amine in 12 is poised for coupling with the glycolic group of Neu5Gcα(2→5)Neu5Gc 8.
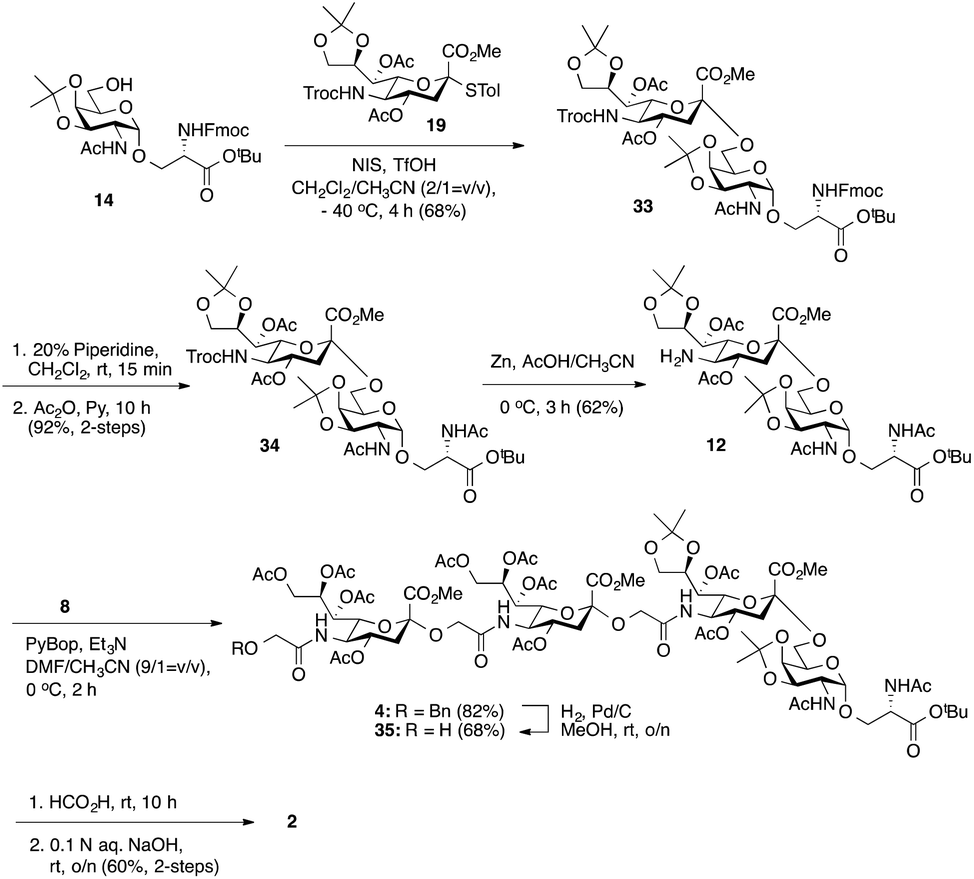 | ||
| Scheme 3 Improved synthesis of a C′-5 amino sTn disaccharide 12 and its exploitation for the construction of non-sulfated tetrasaccharide 2 following a [2 + 2] coupling strategy. | ||
As shown in Scheme 3, coupling between the amine in 12 and carboxylic acid in α(2→5)-linked Neu5Gc dimer 8 was performed under slightly modified conditions from those used previously. PyBOP, Et3N, and DMF/CH3CN (9/1 = v/v) were utilized to give fully protected tetrasaccharide 4 in good yield (82%). Importantly, the presence of CH3CN can suppress the migration of the acetyl group.14 However, the desired product was mixed with traces of the coupling reagent (PyBop) after purification. Thus, without further purification, the total synthesis of the nonsulfated tetrasaccharide 2 was accomplished by the global deprotection of 4. Initially, we tried a three-step deprotection process to afford the desired compound 2: (1) removal of the benzyl group under hydrogenolysis conditions, (2) hydrolysis of the acetonide and t-butyl ester groups using 40% trifluoroacetic acid (TFA) in wet CH2Cl2, and (3) saponification. Although ESI mass spectrometry confirmed the production of the desired product, we were unable to obtain the pure compound, presumably due to the strongly acidic nature of TFA, which might have degraded the glycoside structures.7,37 Alternatively, benzyl ethers were removed first by hydrogenolysis (Pd/C, H2) in methanol to give 35 (68%). Fortunately, PyBOP can be removed at this stage. Subsequently, the deprotection of the acetonide and t-butyl ester groups was performed by using formic acid followed by saponification (0.1 N aq. NaOH, rt) to afford nonsulfated tetrasaccharide 2 (60% yield for two steps).
Having prepared Neu5Gc-capped O-linked tetrasaccharide 4 and validated critical elements of our synthetic design, such as the [2 + 2] coupling strategy, we turned our attention to the synthesis of the 9-O-sulfated tetrasaccharide. However, it was first necessary to incorporate a Neu5Gc residue adorned with an 8,9-O-isopropylidene functional group handle that could deliver the 8,9-diol required for the proposed 9-O-sulfation at the nonreducing end of the underlying glycan chain. This goal could be achieved by using a [2 + 2] coupling of 8,9-O-isopropylidene-protected Neu5Gcα(2→5)Neu5Gc glycolic acid donor 7 (Scheme 1) with a suitable C-5 NH2 sTn disaccharide, as shown in Scheme 4.
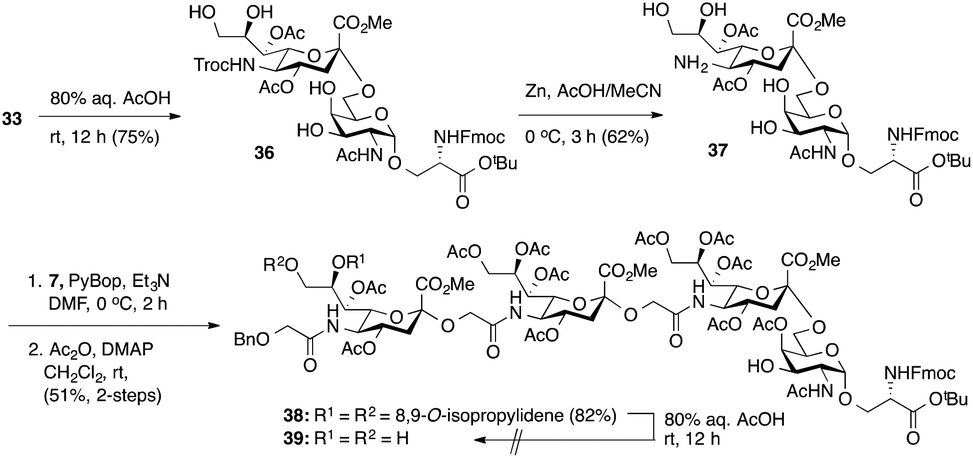 | ||
| Scheme 4 Attempted synthesis of a partially protected presulfated tetrasaccharide 39. | ||
Thus, following cleavage (80% aq. AcOH, rt) of the di-O-isopropylidene protecting group in sTn disaccharide 33, the resulting polyol 36 was converted to C-5 amine 37 by treatment with Zn/AcOH in 62% yield. Subsequent PyBOP-mediated coupling of 37 with Neu5Gcα(2→5)Neu5Gc glycolic acid (7) followed by O-acetylation afforded the desired tetrasaccharide 38 bearing an 8,9-O-isopropylidene protecting group in 51% yield in two steps. Surprisingly, de-O-isopropylidenation of the nonreducing Neu5Gc residue of 38 under acidic conditions (80% aq. AcOH, rt) failed to provide the desired tetrasaccharide 39, even though the conditions were previously successfully applied to hydrolyze the acetonide group in several Neu5Gc-containing mono- and disialosides (vide supra). Only unreacted compound 38 was recovered from the reaction mixture. Other conditions tested, including p-toluenesulfonic acid (pTsOH) in MeOH, proved ineffective. Notably, stronger acids such as TFA were not employed due to the acid sensitive glycosidic bond of the Neu5Gc-containing glycoside structure, which might have a detrimental effect (vide supra). These unsuccessful attempts suggest that it is difficult to unmask the 8,9-O-isopropylidene protecting group in tetrasaccharide 38 to form the 8,9-diol at the nonreducing end Neu5Gc unit.
Since the generation of the 8,9-diol in Neu5Gc at the nonreducing terminus of the underlying glycan chain (38) proved to be challenging, a viable alternative was to first construct the C9′-SO4− α(2→5)-Neu5Gc disaccharide and then couple it with an sTn disaccharide. To develop a convergent route for the 9-O-sulfo Neu5Gcα2(→5-OglycolylNeu5Gcα2→)-containing oligo-saccharide, we opted for the highly regio- and stereospecific enzymatic α(2,6)-sialylation reaction using a αGalNAc-linked glycoside for synthesizing a C′-5 NH2 sTn disaccharide. For the synthesis of 9-O-sulfated Neu5Gc oligomers, the SO3H-9Neu5Gcα(2→5)Neu5Gc disaccharide with a latent glycolic acid can serve as an ideal substrate to form α(2→5)-glycosidic linkages. However, to date, there is only one report on the 9-O-sulfation of a fully protected Neu5Gc derivative with a non-functionalizable coumarin aglycone.38
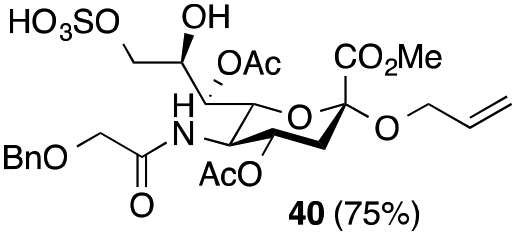
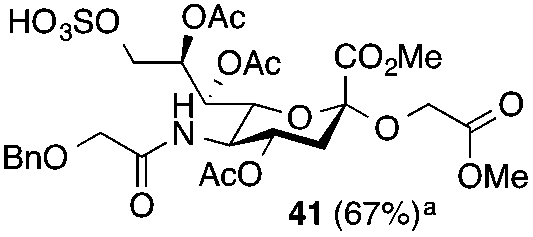
Thus, initial attempts were made to introduce a sulfate group at the nonreducing end of sialic acid by using Neu5Gc-based monosialosides (24 and 25) as model studies (Table 1). The treatment of α-2-allyloxy sialoside 24 with the SO3–Et3N complex (10 eq.) in DMF39 at 50 °C for 6 h exclusively produced 9-O-sulfate derivative 40 in 75% yield (Table 1, entry 1). Encouraged by this initial success, we next used 25 carrying a free glycolic acid at the reducing end to achieve regioselectivity in 9-O-sulfation. Importantly, after acetylation (Ac2O, Py), only 9-O-sulfo-Neu5Gc derivative 41 was isolated in 67% overall yield as the glycolic acid methyl ester, which was obtained because of the use of methanol to quench the excess sulfonation reagent (Table 1, entry 2). The chemical shifts of the C-9 protons in 41 clearly demonstrated the presence of a C-9 sulfate ester: δ 4.23 (dd, 1H, J8′,9′a = 3.3 Hz, J9′a,9′b = 11.1 Hz, H-9′a) and 4.01 (dd, 1H, J8′,9′b = 5.9 Hz, J9′a,9′b = 11.1 Hz, H-9′b), which are characteristic downfield-shifted signals of protons at the sulfated carbon according to published reports.7,38 In addition, in the 13C NMR spectrum, the C-9 carbon peak moved from 64.2 ppm to 62.8 ppm upon sulfation of the C-9 hydroxyl group. Notably, sulfation of the relatively less reactive secondary hydroxyl group at C-8 was not detected even in the presence of a large amount of the SO3–Et3N reagent. To our knowledge, there are no reports of the selective sulfation of the C-9 OH group of Neu5Gc derivatives in the presence of unprotected C-8 OH.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Time (h) | Product (%) |
| a Yield represented after acetylation for two steps. | |||
| 1 |
|
5 |
|
| 2 |
|
6 |
|
| 3 |
|
6 |
|
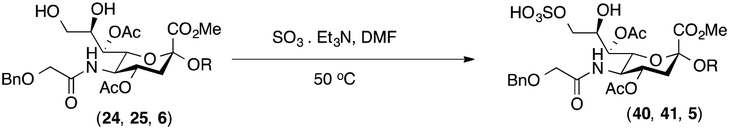
The synthesis of sulfated compound 5 was accomplished under the aforementioned conditions using Neu5Gcα(2→5)-Neu5Gc dimer 6 (Table 1, entry 3). The regioselective sulfation at the C9′-position of the terminal Neu5Gc residue afforded the desired SO3H-9Neu5Gcα(2→5)Neu5Gc 5 possessing a glycolic acid moiety at the reducing end in 77% yield. This transformation permits the installation of the terminal sulfate group present in the target molecule and simultaneously provides an advanced disaccharide building block suitable for subsequent amide bond formation (see below).
Bacterial sialyltransferases (STs) show a wide scope in the structures of their glycan acceptors and are capable of transferring modified sialic acid derivatives to produce sialylglycoconjugates.40,41 It was anticipated that a C-5′ Cbz-protected 2,6-sialyl-Tn derivative (10, Scheme 5) would be readily accessible by chemoenzymatic synthesis using ManN-Cbz as the sialic acid biosynthetic precursor. The N-Cbz group could subsequently be selectively removed after sialylation to generate 9 bearing a free amino group at the C5′-position. To simplify the procedure, a one-pot three-step cascade reaction was developed by using a sialic acid aldolase (from Pasteurella multocida),42 a CMP–sialic acid synthetase (NmCSS from Neisseria meningitidis)43 and an α-2,6-sialyltransferase (Pd2,6ST from Photobacterium damselae)44,45 (Scheme 5). Although the optimal pH values for these three enzymes are not the same, they all retain acceptable reactivity at pH 7.5. Thus, the CMP–sialic acid derivative was synthesized from ManNCbz by the actions of aldolase and NmCSS in a mixture containing Mg2+, pyruvate, and CTP in Tris-HCl buffer at pH 7.5 for 2 h at 37 °C. The Pd2,6ST-catalyzed α(2,6)-sialylation reaction was carried out in the same pot by adding ST and Tn glycoside 13.44 The desired product was purified using a C-18 reversed-phase column to obtain C-5′ N-Cbz-protected sTn disaccharide 10 in 54% yield. Notably, the Fmoc group was deprotected when the enzymatic reactions were performed at pH 8.0.
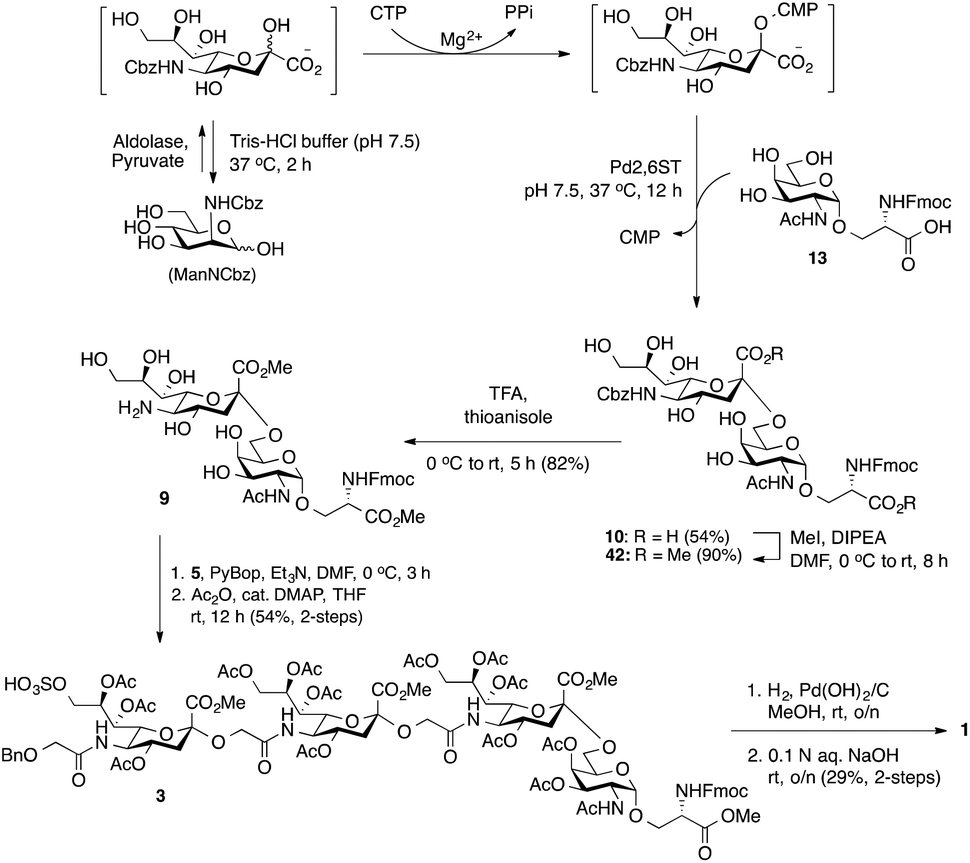 | ||
| Scheme 5 Convergent synthesis of 9-O-sulfated tetrasaccharide glycan 1. Enzymatic α(2,6)-sialylation to prepare a C5′ N-Cbz-protected sTn disaccharide 10, final coupling and global deprotection to deliver 1. | ||
As shown in Scheme 5, esterification of the two carboxylic acids in 10 using iodomethane in the presence of Hünig's base in DMF provided 42 in good yield (90%). The α-sialyl linkage in 42 was established from the chemical shift of the sialic acid H-3′eq proton (2.66 ppm, dd, J = 3.8, and 12.8 Hz) and H-3′ax/C-1 coupling constant (3JC-1,H-3′ax = 5.9 Hz).18 The cleavage of the Cbz-protecting group was effected by TFA in the presence of thioanisole,46 producing C5′-NH2 sTn building block 9 in satisfactory yield (82%). PyBOP-mediated coupling of 9-O-sulfo Neu5Gcα(2→5)Neu5Gc glycolic acid 5 and sTn-derived amine 9 followed by O-acetylation (Ac2O, cat. DMAP, THF) of the free hydroxyl groups (for ease of characterization) afforded protected tetrasaccharide 3 (54% for two steps). The successful preparation of tetrasaccharide 3 demonstrated the generality of the [2 + 2] route. Global deprotection of 3via hydrogenolytic removal of the benzyl ether over Pd(OH)2 on carbon in methanol with concomitant cleavage of the Fmoc group (confirmed by ESI MS) was followed by alkaline hydrolysis of the methyl esters and acetates with aqueous NaOH to furnish 9-O-sulfo Neu5Gc-containing tetrasaccharide 1 (29% for two steps). The target molecule was satisfactorily characterized by 1H, 13C NMR, and HRMS analyses.
Conclusions
In conclusion, the total synthesis of 9-O-sulfo O-linked α(2→5)Neu5Gc-containing tetrasaccharide 1 from the sea urchin species S. purpuratus has been achieved for the first time. Highlights of the synthesis include peptide coupling of Neu5Gc-containing latent glycolic acid units, including a SO3H-9Neu5Gcα(2→5)Neu5Gc dimer, and an sTn disaccharide, which reinforces the utility of the amide bond formation for constructing α2→5-Oglycolyl-linkages stereoselectively in Neu5Gc-containing oligosialic acids. We anticipate that the strategy outlined in this work will allow the concise preparation of structurally defined homogeneous 2→5-Oglycolyl-linked sialic acids, which are extensively found among heterogeneous natural isolates, to study their structure–activity relationship.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Ministry of Education of Taiwan, Ministry of Science and Technology of Taiwan, National Tsing Hua University, Academia Sinica, and Frontier Research Center on Fundamental and Applied Sciences of Matters.Notes and references
- R. L. Schnaar, R. Gerardy-Schahn and H. Hildebrandt, Physiol. Rev., 2014, 94, 461–518 CrossRef.
- A. Varki and P. Gagneux, Ann. Acad. Sci., 2012, 1253, 16–36 CrossRef CAS.
- L. R. L. Davies and A. Varki, Top. Curr. Chem., 2015, 366, 31–54 CrossRef CAS.
- S. Kitazume, K. Kitajima, S. Inoue, F. A. Troy, J.-W. Cho, W. J. Lennarz and Y. Inoue, J. Biol. Chem., 1994, 269, 22712–22716 CAS.
- H. V. Pomin, Glycoconjugate J., 2015, 32, 9–15 CrossRef.
- S. Miyata, C. Sato and K. Kitajima, Trends Glycosci. Glycotechnol., 2007, 19, 85–98 CrossRef CAS.
- S. Kitazume, K. Kitajima, S. Inoue, S. M. Haslam, H. R. Morris, A. Dell, W. J. Lennarz and Y. Inoue, J. Biol. Chem., 1996, 22, 6694–6701 CrossRef.
- S. K. Kitazume, S. Inoue, Y. Inoue and W. J. Lennarz, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3650–3655 CrossRef.
- C.-T. Ren, C.-S. Chen and S.-H. Wu, J. Org. Chem., 2002, 67, 1376–1379 CrossRef CAS.
- C.-T. Ren, C.-S. Chen, Y.-P. Yu, Y.-F. Tsai, P.-Y. Lin, Y.-J. Chen, W. Zou and S.-H. Wu, Chem. – Eur. J., 2003, 9, 1085–1095 CrossRef CAS.
- J. C. McAuliffe, D. Rabuka and O. Hindsgaul, Org. Lett., 2002, 4, 3067–3069 CrossRef CAS.
- G.-T. Fan, C.-C. Lee, J.-M. Fang and C.-C. Lin, J. Org. Chem., 2002, 67, 7565–7568 CrossRef CAS.
- S. Hanashima, D. Ishikawa, S. Akai and K.-I. Sato, Carbohydr. Res., 2009, 344, 747–752 CrossRef CAS.
- H. Tamai, H. Ando, H.-N. Tanaka, T. Hosoda-Yabe, T. Yabe, H. Ishida and M. Kiso, Angew. Chem., Int. Ed., 2011, 50, 2330–2333 CrossRef CAS.
- J. R. Rich and S. G. Withers, Angew. Chem., Int. Ed., 2012, 51, 8640–8643 CrossRef CAS.
- Y. Iwayama, H. Ando, H.-N. Tanaka, H. Ishida and M. Kiso, Chem. Commun., 2011, 47, 9726–9728 RSC.
- H. Hori, T. Nakajima, Y. Nishida, H. Ohrui and H. Meguro, Tetrahedron Lett., 1988, 29, 6317–6320 CrossRef CAS.
- See the ESI.†.
- A. K. Adak, C.-C. Yu, C.-F. Liang and C.-C. Lin, Curr. Opin. Chem. Biol., 2013, 17, 1030–1038 CrossRef CAS.
- C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872–11923 CrossRef CAS.
- G. A. Winterfeld and R. R. Schmidt, Angew. Chem., Int. Ed., 2001, 40, 2654–2657 CrossRef CAS.
- J. Wu and Z. Guo, Bioconjugate Chem., 2006, 17, 1537–1544 CrossRef CAS.
- M. Wagner, S. Dziadek and H. Kunz, Chem. – Eur. J., 2003, 9, 6018–6030 CrossRef CAS.
- R. Okamoto, S. Souma and Y. Kajihara, J. Org. Chem., 2008, 73, 3460–3466 CrossRef CAS.
- S. Sahabuddin, T.-C. Chang, C.-C. Lin, F.-D. Jan, H.-Y. Hsiao, K.-T. Huang, J.-H. Chen, J.-C. Horng, J. A. Ho and C.-C. Lin, Tetrahedron, 2010, 66, 7510–7519 CrossRef CAS.
- F. Yang, X. J. Zheng, C. X. Huo, Y. Wang, Y. Zhang and X. S. Ye, ACS Chem. Biol., 2011, 6, 252–259 CrossRef CAS.
- Q. Wang and Z. Guo, ACS Med. Chem. Lett., 2011, 2, 373–378 CrossRef CAS.
- P. Thompson, V. Lakshminarayanan, N. T. Supekar, J. M. Bradley, P. A. Cohen, M. A. Wolfert, S. J. Gendler and G. J. Boons, Chem. Commun., 2015, 51, 10214–10217 RSC.
- X.-G. Yin, X.-F. Gao, J.-J. Du, X.-K. Zhang, X.-Z. Chen, J. Wang, L.-M. Xin, Z. Lei, Z. Liu and J. Guo, Org. Lett., 2016, 18, 5796–5799 CrossRef CAS.
- N. Takeda, T. Takei, Y. Asahina and H. Hojo, Chem. – Eur. J., 2018, 24, 2593–2597 CrossRef CAS.
- C. De Meo and U. Priyadarshani, Carbohydr. Res., 2008, 343, 1540–1552 CrossRef CAS PubMed.
- H. Tanaka, Y. Nishiura and T. Takahashi, J. Am. Chem. Soc., 2006, 128, 7124–7125 CrossRef CAS.
- D. Crich and W. Li, J. Org. Chem., 2007, 72, 7794–7797 CrossRef CAS.
- M. Shi, K. A. Kleski, K. R. Trabbic, J.-P. Bourgault and P. R. Andreana, J. Am. Chem. Soc., 2016, 138, 14264–14272 CrossRef CAS.
- H. Tanaka, M. Adachi and T. Takahashi, Chem. – Eur. J., 2005, 11, 849–862 CrossRef CAS PubMed.
- G. Akcay, J. Y. Ramphal, M. d'Alarcao and K. Kumar, Tetrahedron Lett., 2015, 56, 109–114 CrossRef CAS.
- M. Murakami, R. Okamoto, M. Izumi and Y. Kajihara, Angew. Chem., Int. Ed., 2012, 51, 3567–3572 CrossRef CAS.
- N. Yamakawa, C. Sato, S. Miyata, E. Maehashi, M. Toriyama, N. Sato, K. Furuhata and K. Kitajima, Biochimie, 2007, 89, 1396–1408 CrossRef CAS.
- A.-T. Wu, Y.-P. Yu, C.-T. Ren, W. Zou and S.-H. Wu, Carbohydr. Res., 2005, 340, 1219–1223 CrossRef CAS.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS.
- C.-C. Yu and S. G. Withers, Adv. Synth. Catal., 2015, 357, 1633–1654 CrossRef CAS.
- Y. Li, H. Yu, H. Cao, K. Lau, S. Muthana, V. K. Tiwari, B. Son and X. Chen, Appl. Microbiol. Biotechnol., 2008, 79, 963–970 CrossRef CAS.
- Y. Li, H. Yu, H. Cao, S. Muthana and X. Chen, Appl. Microbiol. Biotechnol., 2012, 93, 2411–2423 CrossRef CAS.
- C.-F. Teo, T.-S. Hwang, P.-H. Chen, C.-H. Hung, H.-S. Gao, L.-S. Chang and C.-H. Lin, Adv. Synth. Catal., 2005, 347, 967–972 CrossRef CAS.
- L. Ding, H. Yu, K. Lau, Y. Li, S. Muthana, J. Wang and X. Chen, Chem. Commun., 2011, 47, 8691–8693 RSC.
- Y. Kiso, K. Ukawa and T. Akita, J. Chem. Soc., Chem. Commun., 1980, 101–102 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and characterization of all new compounds (1H NMR, 13C NMR, and HR-MS). See DOI: 10.1039/c8qo00996a |
| This journal is © the Partner Organisations 2019 |