
Thienylbenzotriazole promoted highly active gold nanoparticles supported on N-doped graphene as efficient catalysts in water and a mechanism exploration†
Dongdong
Ye
a,
Ronghui
Huang
a,
Haiyan
Zhu
a,
Liang-Hua
Zou
b and
Dawei
Wang
 *a
*a
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China. E-mail: wangdw@jiangnan.edu.cn
bSchool of Pharmaceutical Science, Jiangnan University, Wuxi 214122, China
First published on 12th November 2018
Abstract
An efficient and simple method for the in situ preparation of highly active gold nanoparticles on nitrogen-doped graphene was designed and developed with thienylbenzotriazole gold as the precursor and no additional surfactants or reductants. The developed gold nanoparticles were characterized through energy dispersive X-ray spectroscopy, scanning electron microscopy, transmission electron microscopy, X-ray powder diffraction and X-ray photoelectron spectroscopy studies. This kind of gold nanocomposite was proven to be effective for the chemoselective alkylation of ketones with alcohols, alcohols with alcohols, amines with alcohols, and particularly for amines with amines, under water-based and base-free conditions for the first time. Mechanism investigations were carried out to better discuss this catalyst system and these reactions.
Introduction
The construction of C–C and C–N bonds with alcohol as an alkylation reagent has been considered an atom-efficient, green method and has obtained considerable attention during the past several years.1 The alkylation process usually involves the activation of primary alcohols through the removal of hydrogen to form ketones or aldehydes, which react with ketones (amines or alcohols) to form C–C and C–N double bonds, followed by reduction using H2 generated in the upper dehydrogenation step to achieve the alkylated products.2,3 Water is usually produced as the sole byproduct, and the atom efficiency of this reaction is high.4 To date, relevant research has mainly focused on homogeneous catalytic systems based on iridium, ruthenium and palladium.5,6 Recently, we developed several homogeneous catalysts to realize this alkylation reaction and other borrowing hydrogen reactions.7 However, these catalysts usually included expensive iridium and couldn't be recovered, and this led to great waste in organic synthesis.8 These deficiencies reinforce the need for a reusable catalyst system, particularly under clean conditions such as those involving the use of water, for this alkylation reaction.Supported gold nanoparticles (NPs) are the most promising new catalytic nanocomposite for green reactions due to their unique structures and excellent properties.9,10 Recently, Cao and co-workers reported gold nanoparticles supported on titania as a heterogeneous catalyst for the N-alkylation of amines with alcohols through a borrowing hydrogen reaction.11 Later, they described that a Au/TiO2 catalyst was effective for the direct reductive N-alkylation of a range of aromatic nitro compounds and alcohols with excellent yields. Sabater et al. discovered that a secondary amine was formed through the N-monoalkylation of a primary amine with an alcohol, with Au/CeO2 as the catalyst.12 The Shi group reported a highly efficient heterogeneous Au/Ag–Mo nanorod catalyst for the tandem reaction of alcohols and nitrobenzene to generate N-alkyl amines and imines, using glycerol as the hydrogen source.13 Corma and Iborra reported the synthesis of quinoxalines via the oxidative coupling of vicinal diols with 1,2-phenylenediamine derivatives, with gold nanoparticles supported on nanoparticulated ceria (Au/CeO2) or hydrotalcite (Au/HT) as the catalyst.14 In 2013, Kobayashi et al. demonstrated the formation of imines from alcohols and amines using polymer incarcerated gold/palladium alloy nanoparticles under mild conditions, with high activity and selectivity.15 Cao and co-workers achieved a bimetal-catalysed effective C–C reaction of primary and secondary alcohols via a borrowing hydrogen reaction using Au–Pd/HT.11 Despite all the above-mentioned achievements, direct gold nanoparticle catalysed alkylation remains a difficult task, particularly with regards to: (1) the alkylation of amines with amines; (2) highly active catalysts for the alkylation reactions of ketones, alcohols and amines using alcohols and amines; and (3) reactions carried out in water.16 Herein, we report an efficient and simple method for the in situ preparation of gold nanoparticles on nitrogen-doped graphene, with thienylbenzotriazole gold as the precursor and no additional surfactants or reductants. This kind of gold nanocomposite demonstrated excellent catalytic activity for alkylation reactions in water under base-free conditions.
Results and discussion
The synthesis of the ligand thienylbenzotriazole and the heterogeneous catalyst TTA-Au-NG
Thienylbenzotriazole was synthesized using benzotriazole and thiophene as the raw materials, which were mixed with Cu(OAc)2, potassium carbonate and Selectfluor as additives and refluxed for 12 hours with nitromethane as the solvent (Scheme 1). With the ligand thienylbenzotriazole (TTA) prepared, the gold complex was synthesized with KAuCl4·2H2O and KOH in EtOH at room temperature overnight. Thermogravimetric analysis of the complex was carried out to examine its stability, and the results are available in the ESI.†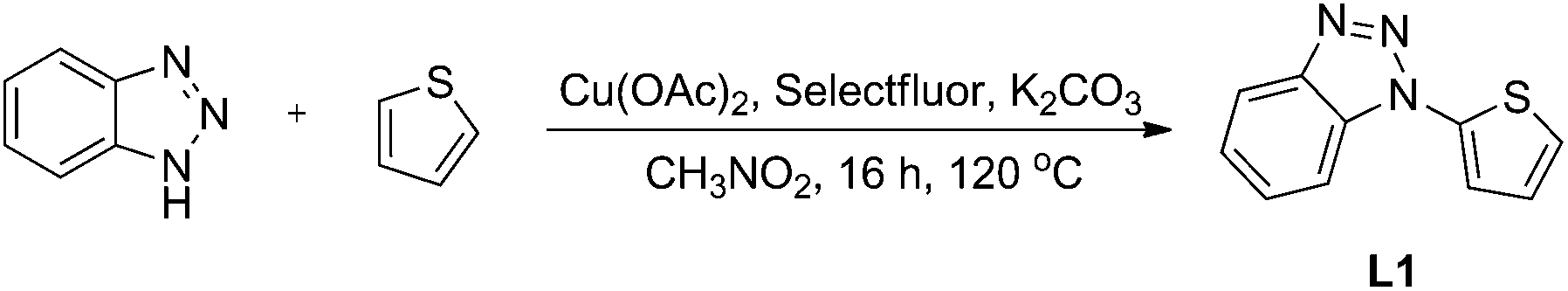 | ||
| Scheme 1 The synthesis of thienylbenzotriazole. | ||
Next, graphene oxide was obtained via a modified Hummers’ method (see the ESI†).17 Nitrogen-doped graphene was synthesized through the following method.18 Firstly, the prepared graphene oxide was uniformly dispersed in a methanol solution of urea and was then placed in a hydrothermal reactor at 150 °C for 12 hours. Next, the product was taken out and dried, placed in a tube furnace, subjected to nitrogen gas and calcined at 750 °C for 4 hours to obtain nitrogen-doped graphene (NG). The catalyst TTA-Au-NG (5% loading, w/w) was next synthesized easily with the nitrogen-doped graphene and gold complex in EtOH refluxed under ultrasonication overnight, without additional surfactants or reductants.
Characterization of the nanocomposite TTA-Au-NG
After synthesis, the supported gold nanocomposite (TTA-Au-NG) was characterized through transmission electron microscopy (TEM), energy dispersive X-ray spectroscopy (EDX), X-ray powder diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). The images in Fig. 1(a) and (b) show scanning electron microscopy images of nitrogen-doped graphene (NG). And the TEM images in Fig. 1(c) and (d) reveal that the gold nanoparticles were supported on nitrogen-doped graphene successfully. The structural characteristics and morphology of the prepared catalyst TTA-Au-NG were observed clearly and easily through transmission electron microscopy (TEM). | ||
| Fig. 1 SEM images of (a and b) NG, and TEM images of (c and d) TTA-Au-NG. | ||
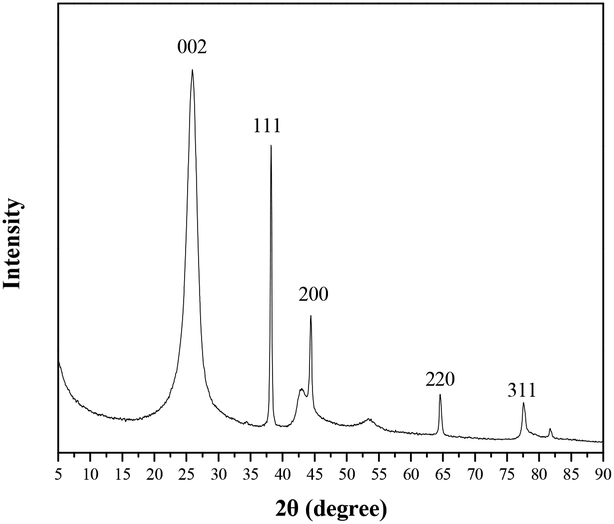
In addition, X-ray powder diffraction (XRD) studies were conducted to examine this catalyst, and the results revealed that gold nanoparticles were well loaded onto the nitrogen-doped graphene (NG) (Fig. 2). The peak at 2θ = 26.6° in the XRD pattern was a diffraction peak belonging to the graphite (002) plane, which proved that graphene oxide was reduced by urea. And the peaks at 2θ = 38.2°, 44.4°, 64.6° and 77.6° correspond to the (111), (200), (220) and (311) crystal planes of Au0, respectively, which reveals that the gold nanoparticles were well supported on the nitrogen-doped graphene. Meanwhile, the EDS pattern shows peaks at 2.1 and 9.7 keV, which are characteristic peaks of gold (Fig. 3).
 | ||
| Fig. 2 XRD pattern of TTA-Au-NG. | ||
 | ||
| Fig. 3 EDS pattern of TTA-Au-NG. | ||
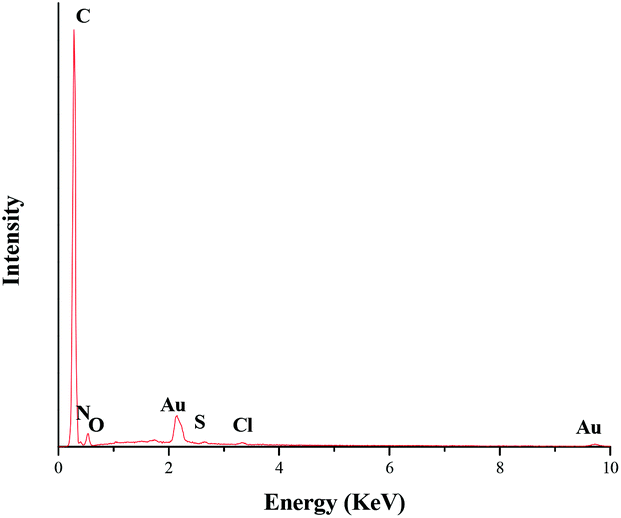
The chemical composition and surface chemical states of the TTA-Au-NG composite were also shown via X-ray photoelectron spectroscopy. The wide XPS spectra indicated the presence of C, N, O and Au elements, and the peaks at binding energies of 87.9 and 84.2 eV belonged to Au0 (Fig. 4).
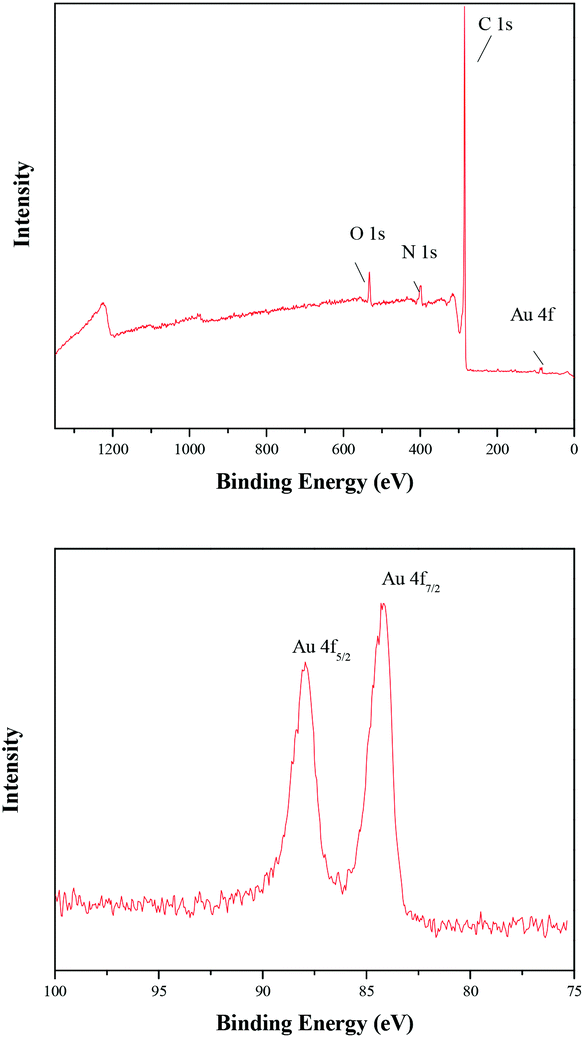 | ||
| Fig. 4 XPS spectra of TTA-Au-NG. | ||
Catalytic activity
Initially, the classical borrowing hydrogen reaction involving benzyl alcohol and acetophenone was studied to test the catalyst activity of TTA-Au-NG in water. After a series of experiments, it was revealed that base was not necessary for this transformation. Interestingly, we found that AgNTf2 was the best additive for this reaction (Table 1, entry 10). Next, the loading of additive was explored, and 5% additive could produce 88% yield of the desired product. The effect of catalyst loading was further studied. Increasing the catalyst loading to 2% produced 89% yield of 3a, however a lower yield was produced after increasing the catalyst loading to 5%. Meanwhile, there was only 56% yield of the product (3a) with 0.2% catalyst. Therefore, it was found the optimal conditions involved AgNTf2 as the additive, 1% catalyst and refluxing in water for 12 hours.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Time [h] | Yieldb [%] |
| a Reagents and conditions: 1a (1.0 mmol); 2a (1.1 mmol); catalyst (0.2–5 mol%, 5% loading, w/w); additive (0.05–1.0 mmol); H2O (3.0 mL); reflux. b Yield of isolated product. c TTA-Au-G was used. | ||||
| 1 | 1% | Na2CO3 (1.0 eq.) | 12 | 11 |
| 2 | 1% | K2CO3 (1.0 eq.) | 12 | 18 |
| 3 | 1% | Cs2CO3 (1.0 eq.) | 12 | 23 |
| 4 | 1% | K3PO4 (1.0 eq.) | 12 | <5 |
| 5 | 1% | NEt3 (1.0 eq.) | 12 | <5 |
| 6 | 1% | KOH (1.0 eq.) | 12 | 36 |
| 7 | 1% | AgOTf (1.0 eq.) | 12 | 82 |
| 8 | 1% | AgBF4 (1.0 eq.) | 12 | 37 |
| 9 | 1% | AgSbF6 (1.0 eq.) | 12 | 46 |
| 10 | 1% | AgNTf2 (1.0 eq.) | 12 | 93 |
| 11 | 1% | AgNTf2 (0.1 eq.) | 12 | 91 |
| 12 | 1% | AgNTf2 (0.05 eq.) | 12 | 88 |
| 13 | 2% | AgNTf2 (0.05 eq.) | 12 | 89 |
| 14 | 5% | AgNTf2 (0.05 eq.) | 12 | 81 |
| 15 | 1% | AgNTf2 (0.05 eq.) | 6 | 79 |
| 16 | 1% | AgNTf2 (0.05 eq.) | 24 | 87 |
| 17 | 0.2% | AgNTf2 (0.05 eq.) | 12 | 56 |
| 18 | — | AgNTf2 (0.05 eq.) | 12 | <5 |
| 19 | — | AgNO3 (0.05 eq.) | 12 | <5 |
| 20 | 1% | — | 24 | <5 |
| 21 | 1%c | AgNTf2 (0.05 eq.) | 12 | 47 |
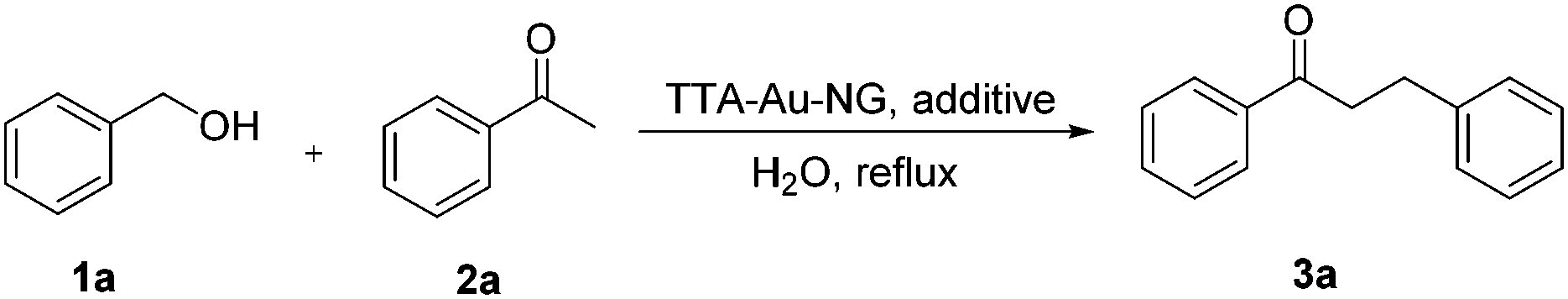
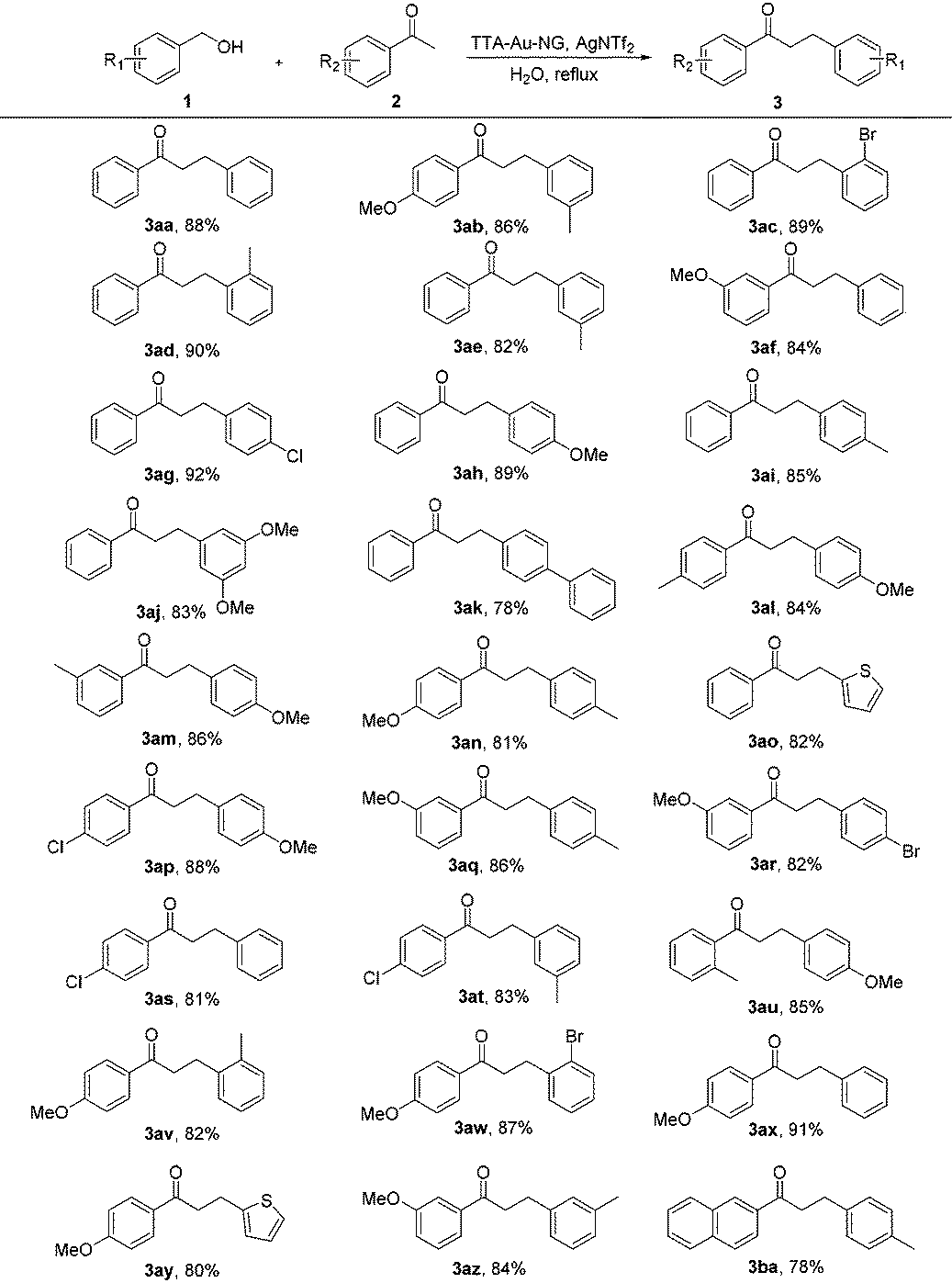
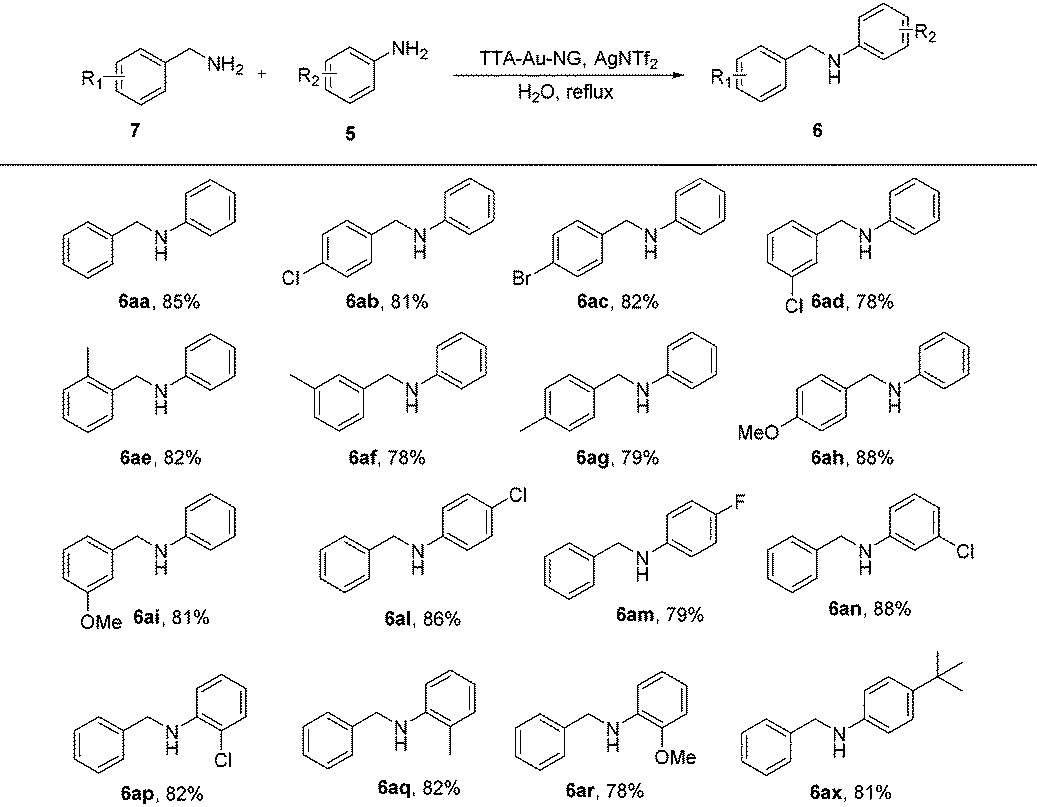
With the optimized reaction conditions in hand, substrate expansion of the borrowing hydrogen reaction was next examined (Table 2). We found that there were high yields involving substrates with both electron donating groups and electron withdrawing groups at different positions on the aryl group moieties. Interestingly, the naphthyl group could produce moderate yields (3ba), and the desired products 3ak and 3ao were obtained with good yields.
Borrowing hydrogen reactions involving alcohols and alcohols are much more promising and difficult. The reaction progression usually includes the successive dehydrogenation of alcohols to the corresponding carbonyl compounds. Highly active catalysts play a decisive role and determine the possible reaction pathway (Scheme 2).
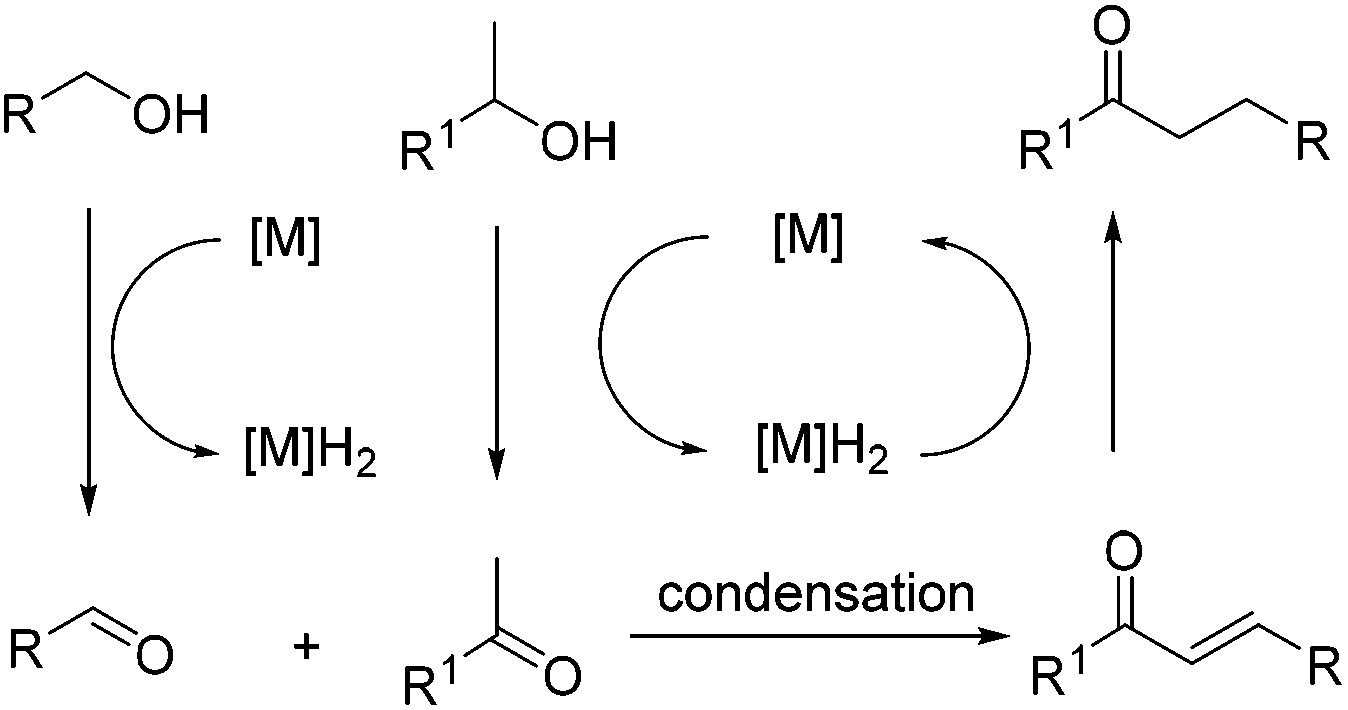 | ||
| Scheme 2 The borrowing hydrogen protocol for primary and secondary alcohols. | ||
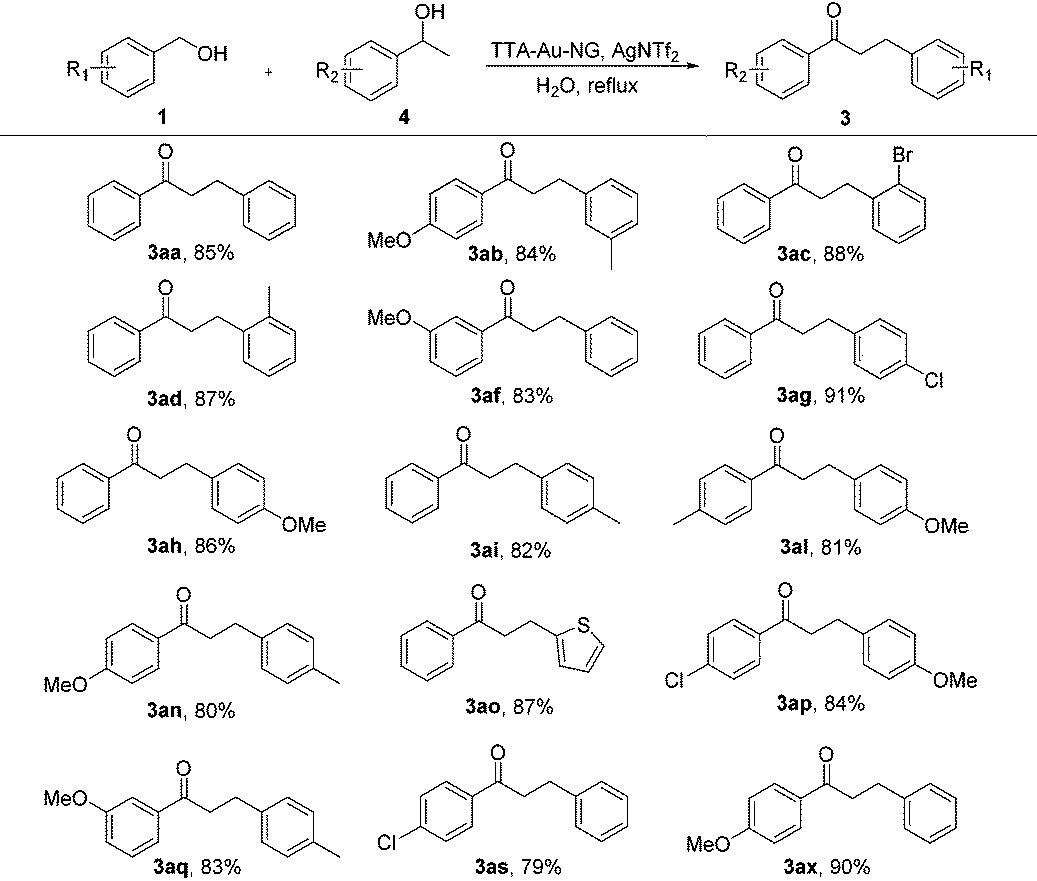
Encouraged by such promising results, we further employed the catalyst TTA-Au-NG in the reaction of primary and secondary alcohols, and the results are summarized in Table 3. The experiments revealed that most of the substrates could react smoothly and the corresponding functionalized ketones were gained in high to good yields through the use of the catalyst TTA-Au-NG. The highest yield was achieved based on a para-methoxy group in a secondary alcohol.
The much greener borrowing hydrogen reaction involving an amine and alcohol is still a very challenging transformation under clean conditions that has not been realized in water with gold nanoparticles as the catalyst until now. Here, we further attempted the reaction of amines with alcohols using this TTA-Au-NG catalyst in water (Table 4). After screening the reaction conditions, the product (6a) was achieved in water at 90 °C after 12 hours. It was observed that substrates with electron-donating and electron-withdrawing groups both had good functional group tolerance with high yields. It was interesting that substrates with cycloalkyl groups, such as cyclohexyl and cyclopropyl, could also produce high yields (6aj and 6ak). In addition, moderate yields were obtained with long chain alkyl groups, such as 6ay (78%) and 6az (74%). Notably, substrates such as 6ba also produced the corresponding secondary amines in moderate yields.
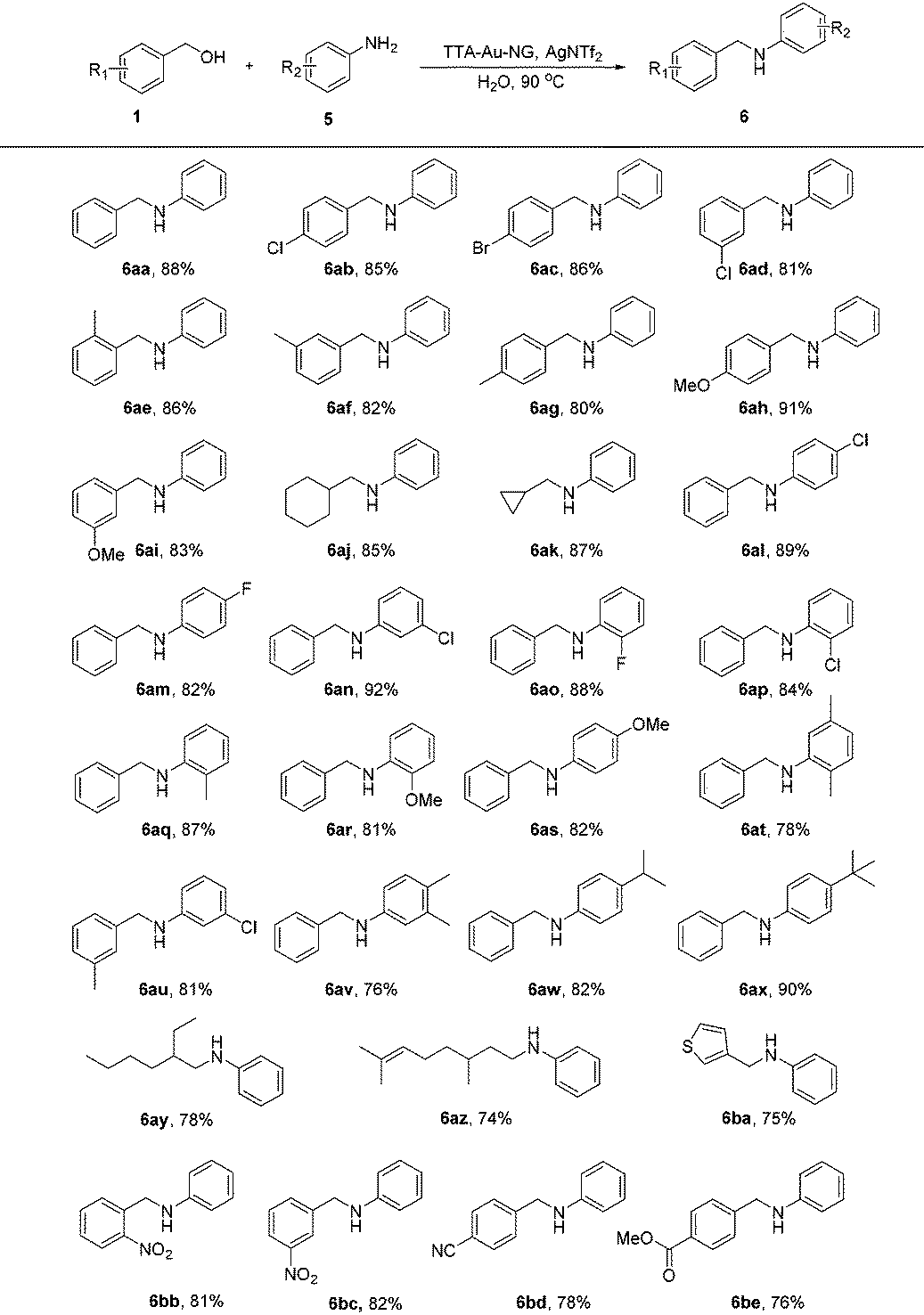
Compared to the borrowing hydrogen reactions of ketones with alcohols, alcohols with alcohols, and amines with alcohols, the borrowing hydrogen reaction of amines with amines is much more difficult and not well studied (Scheme 3). This transformation is an atom-efficient and green process; only ammonia and water are produced as byproducts.
 | ||
| Scheme 3 The borrowing hydrogen reaction of an amine with another amine. | ||
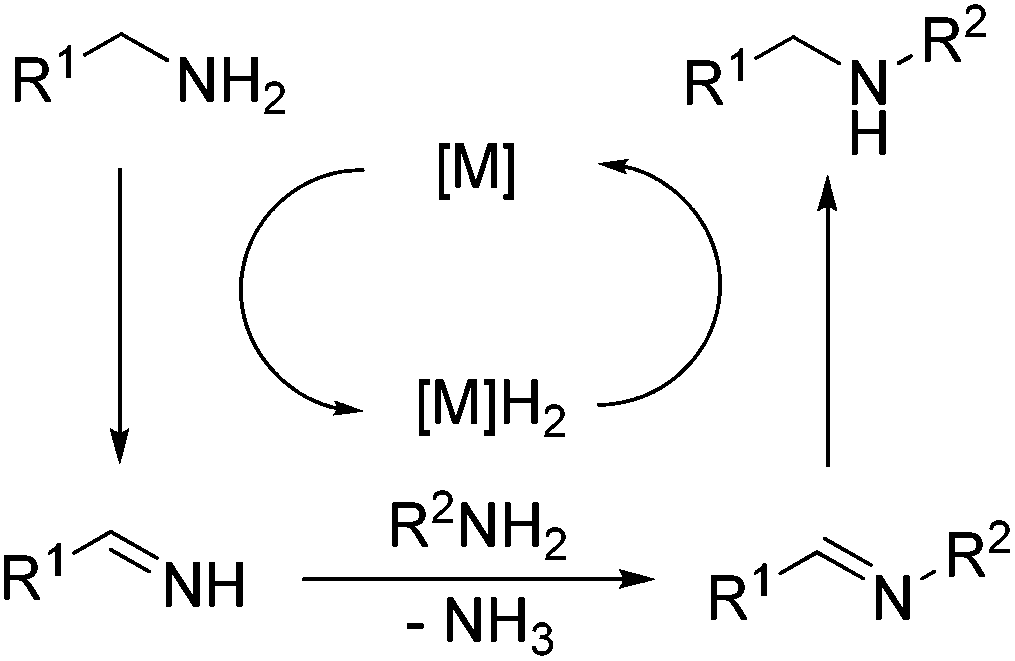
However, the selectivity of this coupling reaction of amines with amines is a challenging issue and highly difficult to realize. With this highly active TTA-Au-NG catalyst in hand, we next explored the borrowing hydrogen reaction of benzylamine and aniline. Experiments showed that the reaction could occur, and the desired product was separated. As shown in Table 5, a wide range of amine substrates and functional groups were tolerated, including chlorine, bromine, tert-butyl, methyl and methoxy substituents at different amine positions with moderate to good yields. Notably, 4-(tert-butyl)aniline was also a suitable substrate for this transformation, with good yields.
Mechanism exploration
Catalyst investigation
The highly active TTA-Au-NG catalyst system being used in the borrowing hydrogen reactions of amines with alcohols, ketones with alcohols, alcohols with alcohols, and amines with amines, in water, motivated us to study the possible mechanism to better explain the catalyst. It was noted that a control experiment revealed that no borrowing hydrogen product was obtained in water when the Au-NG catalyst was used in this reaction (Au-NG: HAuCl4 directly supported on NG; thienylbenzotriazole wasn't employed). Meanwhile, the simple gold salt has no catalytic activity for this reaction in water (Scheme 4). As expected, enhanced catalytic activity was achieved when thienylbenzotriazole was used in the gold catalyst. This supported catalyst was synthesized without additional surfactants or reductants. In addition, another advantage was that thienylbenzotriazole was an effective ligand to synthesis the gold complex, which was the most important factor for the high activity of the TTA-Au-NG catalyst. The possible reason might be the introduction of a sulphur-containing ligand, which may serve as a reductant in situ or form a sulphur-doped nanocomposite.19 Meanwhile, it should be noted that this reaction was greatly promoted by AgNTf2, which probably leads to the question: could silver catalyze or promote this transformation or not? Therefore, several experiments were carried out and the results showed that silver couldn't promote this reaction (Table 1, entries 18 and 19), and high catalytic activity was achieved with the help of non-coordinating anions. It should be noted that a blank experiment revealed that the reaction couldn't happen in the absence of AgNTf2 (Table 1, entry 20). This was in line with previous results20 and is a good example of the application of non-coordinating anions (NCAs).21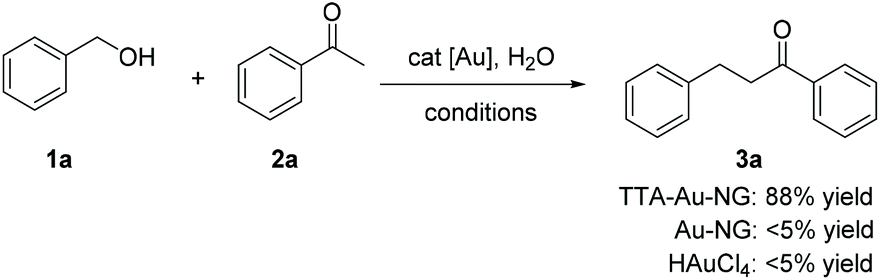 | ||
| Scheme 4 Catalyst exploration experiments. | ||
The capture of intermediates
The trapping of key intermediates is an interesting and effective means to better understand a possible reaction mechanism. Therefore, we set up borrowing hydrogen reactions involving benzyl alcohol and 1-phenylethan-1-ol, respectively, and attempted to isolate the reaction intermediates, respectively. To our delight, it was found that intermediate 8a was obtained in 16% yield, while up to 87% yield of the product (2a) was achieved under the best conditions. This is consistent with our previous research results (Scheme 5).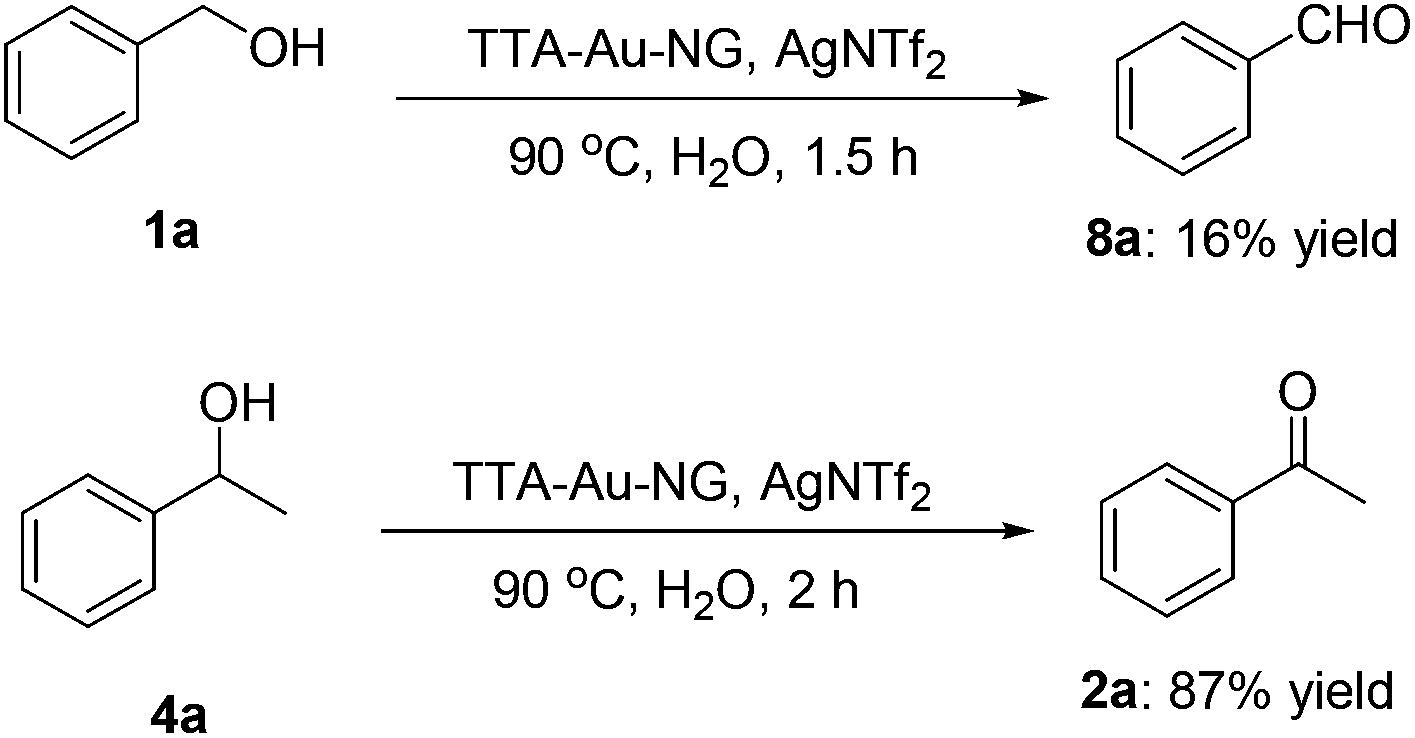 | ||
| Scheme 5 The capture of intermediates. | ||
It is accepted that 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) can be used as a radical scavenger to test reaction intermediates and possible mechanisms. To check the involvement of radicals, a control reaction for a possible radical mechanism was conducted with the addition of one equivalent of TEMPO (Scheme 6). The results showed that this did not prevent the formation of the ketone product 3a, which might rule out the involvement of free-radical intermediates during this borrowing hydrogen process.
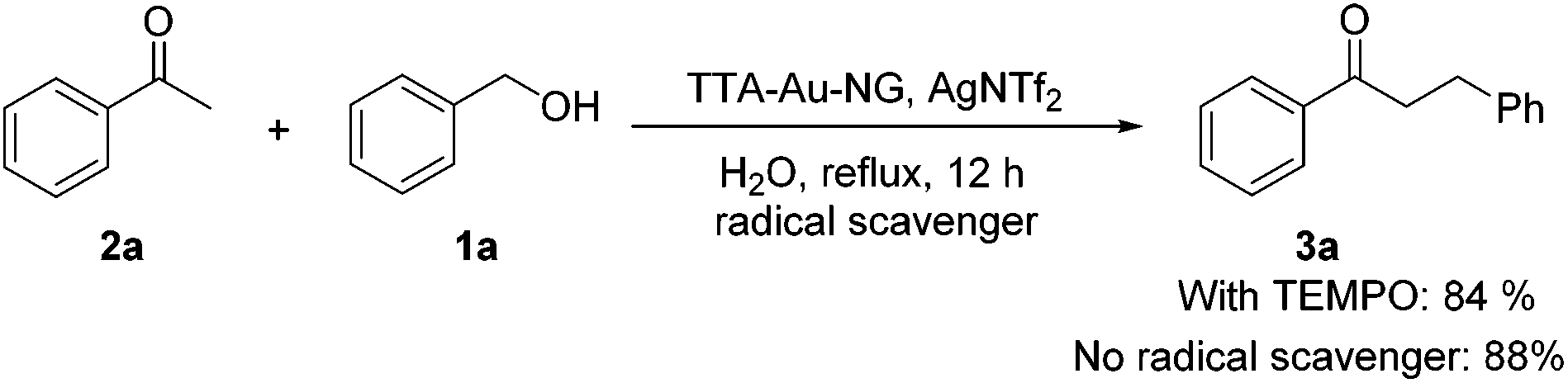 | ||
| Scheme 6 Mechanism exploration experiment. | ||
In addition, the Hammett plot equation was obtained to better understand this reaction process and the electronic effects. Selected substrates were used to build the equation, and the Hammett plot is shown in Fig. 5. The experiments disclosed that the electronic effects of benzyl alcohol had a great impact on this transformation, and a negative charge at the reaction center might be favorable during the transition state.
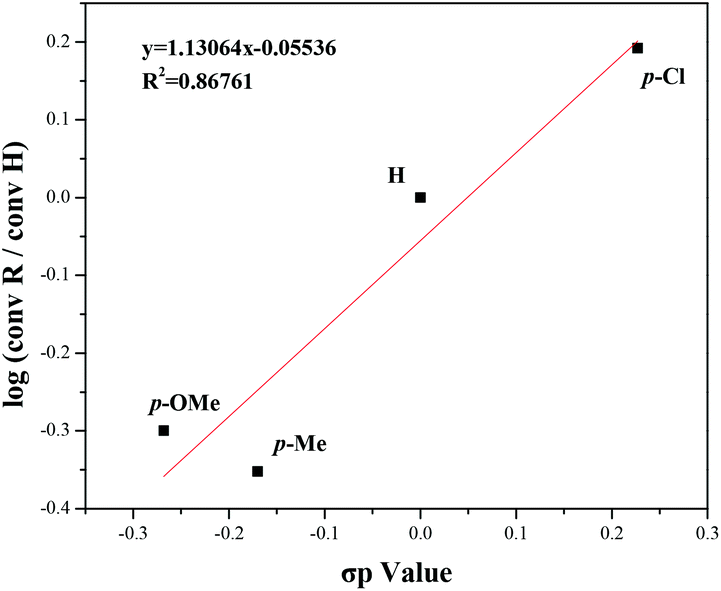 | ||
| Fig. 5 A Hammett plot of para-substituted alcohols. | ||

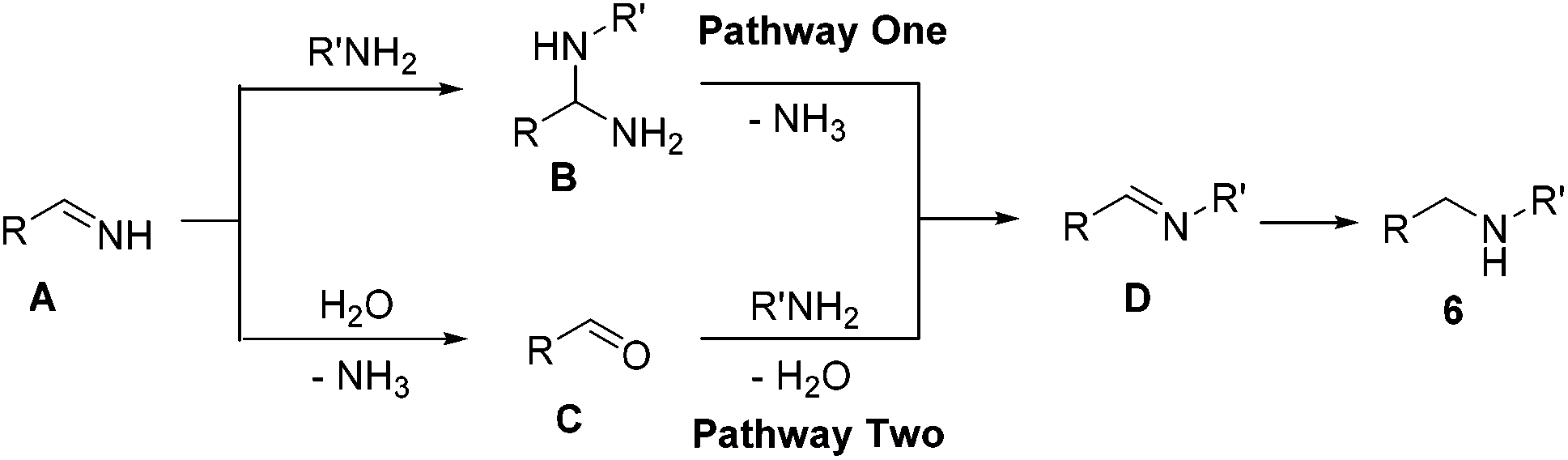 | ||
| Scheme 7 The possible mechanism for the TTA-Au-NG catalyzed reaction of amines with amines. | ||
To better understand this reaction mechanism, several investigations were carried out. The trapping of intermediates is a useful method to study possible pathways. It was pleasing to find that intermediate C was detected during the course of this reaction, which was also examined via mass spectroscopy. However, the intermediate N,1-diphenylmethanediamine (B) couldn't be detected. Therefore, it could basically be decided that the second pathway was the possible mechanism. This was in line with previous results8b and supported by the literature.22
Reusability of the catalyst
Finally, the catalyst TTA-Au-NG was washed and centrifuged several times with water, and dried and recycled for reuse in the borrowing hydrogen reaction. It was found that as the number of cycles increased, the reaction yields gradually decreased (Scheme 8). Despite this, if the recovered TTA-Au-NG catalyst was reused five times, reasonably good yields were achieved.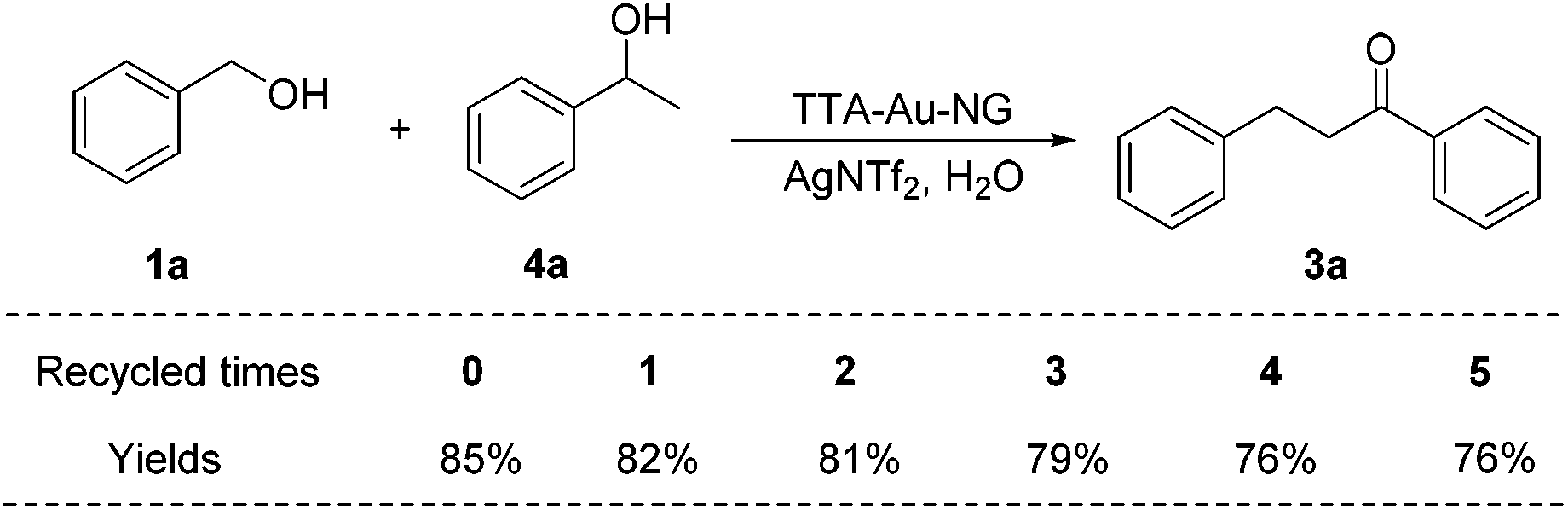 | ||
| Scheme 8 Recycling experiments. | ||
At the same time, we performed XRD testing of the recycled TTA-Au-NG catalyst after five cycles, and the results showed that TTA-Au-NG had excellent stability; the diffraction peak of gold was only slightly reduced, which resulted from the loss of gold nanoparticles (Fig. 6). Meanwhile, the extent of TTA-Au-NG leaching after the borrowing hydrogen reaction of alcohols (1a) and (2a) in water was conducted after reflux through ICP analysis. The experiments indicated that the amount of gold leaching after the reaction, tested via ICP assessment, was about 0.98% (%, w/w). Therefore, the ICP experiments disclosed that the leaching of TTA-Au-NG was almost negligible, which revealed that TTA-Au-NG had fine stability.
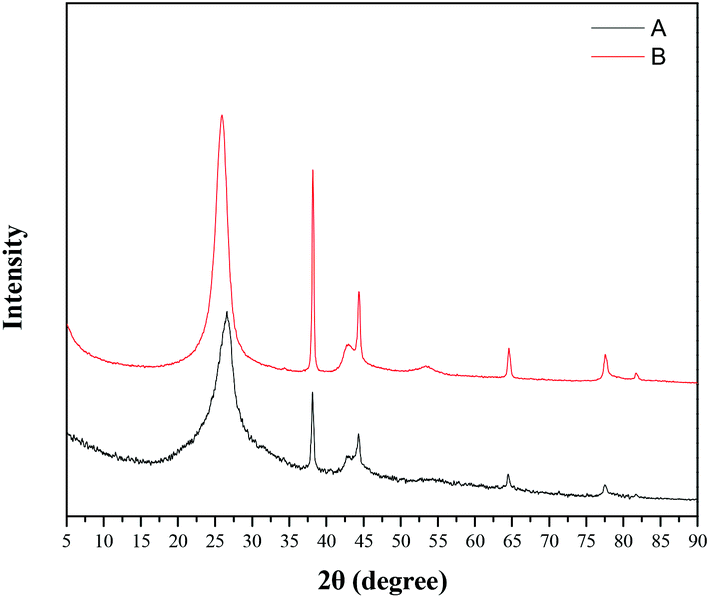 | ||
| Fig. 6 XRD pattern of (B) the fresh TTA-Au-NG nanocomposite; and (A) the TTA-Au-NG nanocomposite after five runs. | ||
Conclusions
In conclusion, we developed a new type of gold nanocomposite, TTA-Au-NG, which was synthesized with thienylbenzotriazole gold as the precursor and supported on nitrogen-doped graphene, with the use of no additional surfactants or reductants. This gold nanocomposite was characterized via X-ray powder diffraction, X-ray photoelectron spectroscopy, energy dispersive X-ray spectroscopy and transmission electron microscopy studies. This kind of gold nanocomposite was proven to be effective for the chemoselective alkylation of ketones with alcohols, alcohols with alcohols, amines with alcohols, and particularly for amines with amines, under water-based and base-free conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support for this work from the National Natural Science Foundation of China (21776111, 21861039), the Fundamental Research Funds for the Central Universities (JUSRP 51627B) and MOE & SAFEA for the 111 Project (B13025).Notes and references
- Recent reviews: (a) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410 CrossRef CAS; (b) G. Chelucci, Coord. Chem. Rev., 2017, 331, 1 CrossRef CAS; (c) F. Huang, Z. Liu and Z. Yu, Angew. Chem., Int. Ed., 2016, 55, 862 CrossRef CAS; (d) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305 RSC; (e) A. Nandakumar, S. P. Midya, V. G. Landge and E. Balaraman, Angew. Chem., Int. Ed., 2015, 54, 11022 CrossRef CAS; (f) B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851 CrossRef CAS; (g) K.-I. Shimizu, Catal. Sci. Technol., 2015, 5, 1412 RSC; (h) Y. Obora, ACS Catal., 2014, 4, 3972 CrossRef CAS; (i) D. Hollmann, ChemSusChem, 2014, 7, 2411 CrossRef CAS.
- (a) A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS; (b) C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS; (c) P. Hu, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 1061 CrossRef CAS; (d) P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 14373 CrossRef CAS; (e) S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5139 CrossRef CAS; (f) L. Neubert, D. Michalik, S. Bähn, S. Imm, H. Neumann, J. Atzrodt, V. Derdau, W. Holla and M. Beller, J. Am. Chem. Soc., 2012, 134, 12239 CrossRef CAS; (g) M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384 CrossRef CAS; (h) J. Schranck, A. Tlili and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 7642 CrossRef CAS PubMed; (i) M. Zhang, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 597 CrossRef CAS; (j) C. S. Lim, T. T. Quach and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 7176 CrossRef CAS.
- (a) F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525 CrossRef CAS; (b) J. R. Frost, C. B. Cheong, W. M. Akhtar, D. F. J. Caputo, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2015, 137, 15664 CrossRef CAS; (c) N. Deibl and R. Kempe, J. Am. Chem. Soc., 2016, 138, 10786 CrossRef CAS PubMed; (d) T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2016, 6, 381 CrossRef CAS; (e) C. Schlepphorst, B. Maji and F. Glorius, ACS Catal., 2016, 6, 4184 CrossRef CAS; (f) B. Emayavaramban, M. Roy and B. Sundararaju, Chem. – Eur. J., 2016, 22, 3952 CrossRef CAS; (g) D. Shen, D. L. Poole, C. C. Shotton, A. F. Kornahrens, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2015, 54, 1642 CrossRef CAS; (h) F. Jiang, M. Achard and C. Bruneau, Chem. – Eur. J., 2015, 21, 14319 CrossRef CAS; (i) M. V. Jimenez, J. Fernandez-Tornos, F. J. Modrego, J. J. Perez-Torrente and L. A. Oro, Chem. – Eur. J., 2015, 21, 17877 CrossRef CAS; (j) A. J. Rawlings, L. J. Diorazio and M. Wills, Org. Lett., 2015, 17, 1086 CrossRef CAS; (k) T. T. Dang, B. Ramalingam and A. M. Seayad, ACS Catal., 2015, 5, 4082 CrossRef CAS; (l) X. Xie and H. V. Huynh, ACS Catal., 2015, 5, 4143 CrossRef CAS; (m) H. Hikawa, T. Koike, K. Izumi, S. Kikkawa and I. Azumaya, Adv. Synth. Catal., 2016, 358, 784 CrossRef CAS; (n) H. Hikawa, R. Ichinose, S. Kikkawa and I. Azumaya, Green Chem., 2018, 20, 1297 RSC.
- (a) B. Xiong, S. Zhang, H. Jiang and M. Zhang, Org. Lett., 2016, 18, 724 CrossRef CAS; (b) Z. Tan, H. Jiang and M. Zhang, Org. Lett., 2016, 18, 3174 CrossRef CAS; (c) B. Xiong, S.-D. Zhang, L. Chen, B. Li, H.-F. Jiang and M. Zhang, Chem. Commun., 2016, 52, 10636 RSC; (d) Z. Tan, H. Jiang and M. Zhang, Chem. Commun., 2016, 52, 9359 RSC; (e) F. Xie, R. Xie, J.-X. Zhang, H.-F. Jiang, L. Du and M. Zhang, ACS Catal., 2017, 7, 4780 CrossRef CAS; (f) H.-J. Pan, T. W. Ng and Y. Zhao, Chem. Commun., 2015, 51, 11907 RSC; (g) Z.-Q. Rong, Y. Zhang, R. H. B. Chua, H.-J. Pan and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 4944 CrossRef CAS; (h) Y. Zhang, C.-S. Lim, D. S. B. Sim, H.-J. Pan and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 1399 CrossRef CAS PubMed; (i) C. S. Lim, T. T. Quach and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 7176 CrossRef CAS PubMed; (j) X. Chen, H. Zhao, C. Chen, H. Jiang and M. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14232 CrossRef CAS.
- (a) F. Li, L. Lu and P. Liu, Org. Lett., 2016, 18, 2580 CrossRef CAS; (b) R. Wang, H. Fan, W. Zhao and F. Li, Org. Lett., 2016, 18, 3558 CrossRef CAS; (c) S. Li, X. Li, Q. Li, Q. Yuan, X. Shi and Q. Xu, Green Chem., 2015, 17, 3260 RSC; (d) Q. Xu, J. Chen, H. Tian, X. Yuan, S. Li, C. Zhou and J. Liu, Angew. Chem., Int. Ed., 2014, 53, 225 CrossRef CAS; (e) X. Cui, X. Dai, Y. Deng and F. Shi, Chem. – Eur. J., 2013, 19, 3665 CrossRef CAS; (f) Q. Wang, K. Wu and Z. Yu, Organometallics, 2016, 35, 1251 CrossRef CAS; (g) T. Liu, L. Wang, K. Wu and Z. Yu, ACS Catal., 2018, 8, 7201 CrossRef CAS.
- (a) C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661 CrossRef CAS; (b) F. Shi, M. K. Tse, X. Cui, D. Gördes, D. Michalik, K. Thurow, Y. Deng and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 5912 CrossRef CAS; (c) B. Gnanaprakasam, J. Zhang and D. Milstein, Angew. Chem., Int. Ed., 2010, 49, 1468 CrossRef CAS; (d) Y. Zhao, S. W. Foo and S. Saito, Angew. Chem., Int. Ed., 2011, 50, 3006 CrossRef CAS; (e) S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5139 CrossRef CAS; (f) L. Neubert, D. Michalik, S. Bähn, S. Imm, H. Neumann, J. Atzrodt, V. Derdau, W. Holla and M. Beller, J. Am. Chem. Soc., 2012, 134, 12239 CrossRef CAS; (g) T. Yan, B. L. T. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed; (h) T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2016, 6, 381 CrossRef CAS; (i) P. Hu, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 1061 CrossRef CAS; (j) N. Deibl and R. Kempe, J. Am. Chem. Soc., 2016, 138, 10786 CrossRef CAS.
- (a) D. Wang, K. Zhao, C. Xu, H. Miao and Y. Ding, ACS Catal., 2014, 4, 3910 CrossRef CAS; (b) Y. Yang, A. Qin, K. Zhao, D. Wang and X. Shi, Adv. Synth. Catal., 2016, 358, 1433 CrossRef CAS; (c) Z. Xu, D.-S. Wang, X. Yu, Y. Yang and D. Wang, Adv. Synth. Catal., 2017, 359, 3332 CrossRef CAS; (d) Q. Wu, L. Pan, G. Du, C. Zhang and D. Wang, Org. Chem. Front., 2018, 5, 2668 RSC.
- (a) R. Huang, Y. Yang, D.-S. Wang, L. Zhang and D. Wang, Org. Chem. Front., 2018, 5, 203 RSC; (b) C. Ge, X. Sang, W. Yao, L. Zhang and D. Wang, Green Chem., 2018, 20, 1805 RSC; (c) Z. Xu, X. Yu, X. Sang and D. Wang, Green Chem., 2018, 20, 2571 RSC.
- (a) A. Leyva-Perez, J. Oliver-Meseguer, J. R. Cabrero-Antonino, P. Rubio-Marques, P. Serna, S. I. Al-Resayes and A. Corma, ACS Catal., 2013, 3, 1865 CrossRef CAS; (b) F. Schroder, M. Ojeda, N. Erdmann, J. Jacobs, R. Luque, T. Noel, L. Van Meervelt, J. Van der Eyckend and E. V. Van der Eycken, Green Chem., 2015, 17, 3314 RSC; (c) J. Mao, J. Zhou, Z. Xia, Z. Wang, Z. Xu, W. Xu, P. Yan, K. Liu, X. Guo and Z. C. Zhang, ACS Catal., 2017, 7, 695 CrossRef CAS; (d) X.-B. Zhao, W. Ha, K. Jiang, J. Chen, J.-L. Yang and Y.-P. Shi, Green Chem., 2017, 19, 1399 RSC.
- (a) W. Lv, Y. Ju, Y. Chen and X. Chen, Int. J. Hydrogen Energy, 2018, 43, 10334 CrossRef CAS; (b) L. Shao, X. Huang, D. Teschner and W. Zhang, ACS Catal., 2014, 4, 2369 CrossRef CAS.
- (a) L. He, X.-B. Lou, J. Ni, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Chem. – Eur. J., 2010, 16, 13965 CrossRef CAS; (b) X. Liu, R.-S. Ding, L. He, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, ChemSusChem, 2013, 6, 604 CrossRef CAS.
- A. Corma, J. Navas and M. J. Sabater, Chem. – Eur. J., 2012, 18, 14150 CrossRef CAS.
- X. Cui, C. Zhang, F. Shi and Y. Deng, Chem. Commun., 2012, 48, 9391 RSC.
- M. J. Climent, A. Corma, J. C. Hernandez, A. B. Hungria, S. Iborra and S. Martinez-Silvestre, J. Catal., 2012, 292, 118 CrossRef CAS.
- J.-F. Soule, H. Miyamura and S. Kobayashi, Chem. Commun., 2013, 49, 355 RSC.
- C.-H. Tang, L. He, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Chem. – Eur. J., 2011, 17, 7172 CrossRef CAS PubMed.
- (a) F. C.-M. Leung and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2018, 10, 15582 CrossRef; (b) H. Yan, C. Tian, L. Wang, A. Wu, M. Meng, L. Zhao and H. Fu, Angew. Chem., Int. Ed., 2015, 54, 6325 CrossRef CAS; (c) H. Yang, H. Li, J. Zhai, L. Sun and H. Yu, Ind. Eng. Chem. Res., 2014, 53, 17878 CrossRef CAS.
- (a) C. Rizescu, I. Podolean, J. Albero, V. I. Parvulescu, S. M. Coman, C. Bucur, M. Puche and H. Garcia, Green Chem., 2017, 19, 1999 RSC; (b) H. Fu, Y. Chen, Z. Ren, Y. Xiao, Y. Liu, X. Zhang and G. Tian, Electrochim. Acta, 2018, 262, 107 CrossRef CAS; (c) C. Zhang, J. Sha, H. Fei, M. Liu, S. Yazdi, J. Zhang, Q. Zhong, X. Zou, N. Zhao, H. Yu, Z. Jiang, E. Ringe, B. I. Yakobson, J. Dong, D. Chen and J. M. Tour, ACS Nano, 2017, 11, 6930 CrossRef CAS.
- (a) J. R. Reimers, Y. Wang, B. O. Cankurtaran and M. J. Ford, J. Am. Chem. Soc., 2010, 132, 8378 CrossRef CAS; (b) G. Zotti, B. Vercelli and A. Berlin, Chem. Mater., 2008, 20, 397 CrossRef CAS; (c) M. K. Corbierre and R. B. Lennox, Chem. Mater., 2005, 17, 5691 CrossRef CAS.
- (a) D. Wang, X. Yu, X. Xu, B. Ge, X. Wang and Y. Zhang, Chem. – Eur. J., 2016, 22, 8663 CrossRef CAS; (b) X. Yu, D.-S. Wang, Z. Xu, B. Yang and D. Wang, Org. Chem. Front., 2017, 4, 1011 RSC.
- (a) C. Reed, Acc. Chem. Res., 1998, 31, 133 CrossRef CAS; (b) E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391 CrossRef CAS; (c) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS.
- H. Huang, J. Huang, Y.-M. Liu, H.-Y. He, Y. Cao and K.-N. Fan, Green Chem., 2012, 14, 930 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qo00941d |
| This journal is © the Partner Organisations 2019 |