
Atmospheric water triggers supramolecular gel formation of novel low molecular weight maslinic and oleanolic triterpenic derivatives†
Karina
Vega-Granados
a,
Gloria Belén
Ramírez-Rodríguez
 b,
Rafael
Contreras-Montoya
a,
Francisco J.
Ramírez
c,
Luis
Palomo
c,
Andrés
Parra
a,
Jose M.
Delgado-López
b,
Modesto T.
Lopez-Lopez
de and
Luis
Álvarez de Cienfuegos
*ae
b,
Rafael
Contreras-Montoya
a,
Francisco J.
Ramírez
c,
Luis
Palomo
c,
Andrés
Parra
a,
Jose M.
Delgado-López
b,
Modesto T.
Lopez-Lopez
de and
Luis
Álvarez de Cienfuegos
*ae
aDpto de Química Orgánica, Facultad de Ciencias, Universidad de Granada (UGR), 18071-Granada, Spain. E-mail: lac@ugr.es
bDpto de Química Inorgánica, Facultad de Ciencias, (UGR), Spain
cDpto de Química Física, Universidad de Málaga, (UMA), 29071-Málaga, Spain
dDpto de Física Aplicada, Facultad de Ciencias, (UGR), Spain
eInstituto de Investigación Biosanitaria ibs.GRANADA, (UGR), Spain
First published on 12th July 2019
Abstract
Pentacyclic triterpenes have a rigid lipophilic backbone decorated with polar functional groups such as hydroxyl, carbonyl and carboxyl, which make them attractive natural amphiphiles. The ability of this type of compound to self-assemble into complex nanostructures has recently gained interest due in part to the abundant existence of renewable feedstocks. Herein, we present two new maslinic and oleanolic triterpene pentacyclic derivatives that have the ability to self-assemble into long fibers giving rise to homogeneous gels. Quite remarkably, these molecules are able to form gels by slowly capturing atmospheric water from their DMSO and DMF solutions. To the best of our knowledge, this mechanism of gel formation has not been previously reported for other LMWGs and highlights the versatility of these compounds to form gels as a response to different external stimuli. The micro- and macroscopic properties of the resulting gels have been studied in detail by TEM, SEM, NMR, VCD, FTIR, XRD and rheology. We have demonstrated that gels prepared by capturing atmospheric water in DMSO are more homogeneous and stiffer than those obtained by direct addition of water to the organic solution.
Introduction
Low molecular weight gelators (LMWGs) are a particular class of compounds that have the ability to efficiently self-assemble into supramolecular structures being able to retain solvent molecules giving rise to gels.1–3 Since the self-assembly nature of these molecules is promoted by non-covalent bonds, mainly van der Waals forces, the resulting gels, in most of the cases, are reversible, being able to respond to external stimuli, such as temperature, pH, sonication, chemical additives, etc.4–6 As such, promising applications have been anticipated for this kind of material as templates for the growth of inorganic particles, biomaterials, catalysts, sensors and optoelectronic devices.7–10To date, it is not possible to anticipate the structural requirements for a given molecule to behave as an LMWG or to predict in which solvent or solvents it will form a gel. Intensive efforts have been focused on discovering new LMWGs and trying to link their chemical composition with their capacity to self-assemble.11,12 Among the types of structures studied, LMWGs based on steroids have been particularly successful.13 Pioneering works of Weiss and co-workers in cholesterol-based organic gelators showed that structures comprising an aromatic moiety (A) connected to a steroidal group (S) via a linker (L) displayed an effective gelation ability.1 Derivatives based on the A(LS)2- or LS2-type structures have also shown an effective gelation ability.14–16 Bile acid derivatives are also another group of steroid derivatives that have been studied in-depth showing the capacity to form organo- and hydrogels.13 In both cases, van der Waals interactions between the rigid polycyclic steroid backbones favour the self-association of the molecules and promote the anisotropic growth in long fibers needed to form gels. In the last decade, efforts have been focused on studying the self-assembly properties of another structural-related family of natural compounds, the triterpenes.17 Triterpenes are vastly present in plants and, similar to steroids, contain a polycyclic rigid skeleton. The interest in these compounds lies in their broad diversity of therapeutic properties, such as anti-tumor, anti-virus, anti-inflammatory and anti-microbial.18–20 Moreover, some of these derivatives can be easily isolated in large quantities from their natural feedstock making them a very attractive alternative, not only in the search for new therapeutics, but also in the field of material science since they can evolve into novel nanomaterials obtained from renewable sources. In this regard, the self-assembly ability of these triterpenes has been reported in isolated natural compounds21–24 and also in derivatives.25–27 They have been able to form chiral helical fibers,28 nanorods27 and vesicles22,24,25 in organic and mixtures of organic and aqueous solvents.17 Most of these compounds are able to form supramolecular aggregates by a heating and cooling cycle or by a change in the proportion of solvent mixtures. The versatility of these supramolecular aggregates has been exploited in the generation of advanced materials,29,30 in catalysis,31 and in drug carriers.32
Considering this, we focused our attention on pentacyclic triterpenic maslinic (MA) and oleanolic (OA) acids, vastly present in the fruits of Olea europaea, and easily isolated and purified from industrial olive-oil waste (Fig. 1).19 These compounds have a carboxylic group that can be conveniently modified to obtain derivatives. As such, we reasoned that the introduction of an aromatic group through this carboxylic group could generate a structure resembling a similar ALS type of gelator. Trying to do this, when we activated the carboxylic group of MA and OA with N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uranium tetrafluoroborate (TBTU) we obtained the corresponding TBTU activated acids (MATBTU compound 1, and OATBTU compound 2) that, unexpectedly, not only formed stable compounds but were also able to form homogeneous gels in DMSO–H2O, DMF–H2O and compound 1 also in toluene (Fig. 1).
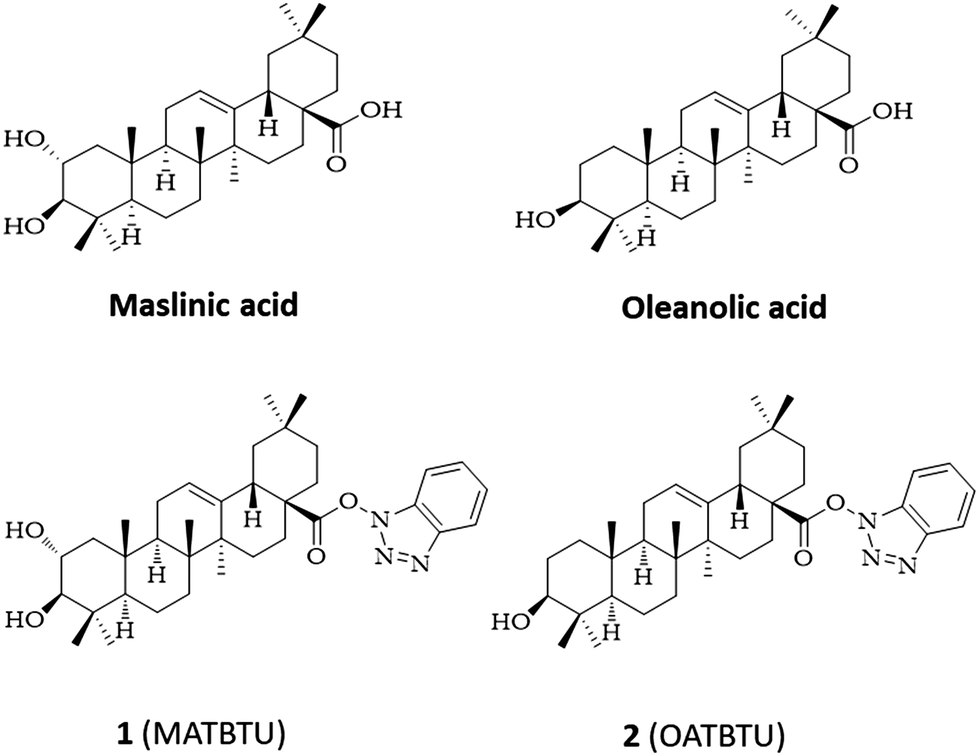 | ||
| Fig. 1 Structures of maslinic and oleanolic acids and their gelator derivatives 1 and 2. | ||
More interestingly, these compounds were able to form homogeneous gels by two different strategies, namely: direct addition of water to their DMSO and DMF solutions or by slowly capturing atmospheric water from their respective solutions. The kinetics of gel formation of both processes, direct addition and slowly capturing water, differ significantly. When water is immediately added to the DMSO solution the gel forms in 5 minutes, whilst gel formation requires much more time when it captures atmospheric water. The gelification time of this second process depends on the gelator concentration. Gels formed in this way are more homogeneous being much stiffer than gels formed by direct addition of water. Additionally, this second process is able to form homogeneous gels with higher gelator concentration giving rise to more rigid self-supported gels. To the best of our knowledge, gel formation by capturing atmospheric water has not been previously described for other LMWGs and highlights the versatility of these compounds to form gels under different types of stimuli. In this case, several requirements have to be achieved by a small compound to be able to gelify. First, the gelator must be able to form a gel at room temperature; second, it must be able to form a gel in the mixture of the resulting combination of solvents, that is, DMSO–H2O or DMF–H2O; third, it must be able to form a gel in the adequate proportion of solvents, that, in this case, is also subjected to the hygroscopic character of the organic solvent. And fourth, to generate homogeneous gels, the kinetics of the self-association of the gelator molecules must be compatible with the rate of atmospheric water dissolution in the organic solvent.
Herein, we have studied in detail the gelification ability of compounds 1 (MATBTU) and 2 (OATBTU) in different solvents as well as the micro- and macroscopic properties of the resulting gels by TEM, SEM, 1H-NMR, FTIR, VCD, XRD, and rheology. Interestingly, compound 1 is able to form gels not only in DMSO–H2O and DMF–H2O but also in neat toluene. In this solvent, compound 1 forms different supramolecular aggregates than those obtained in DMSO–H2O and DMF–H2O. A model of the supramolecular packing of compound 1 in toluene has been proposed using XRD data and theoretical computation.
Experimental
Reagents and materials
Oleanolic (3β-hydroxyolean-12-en-28-oic acid, OA) and maslinic (2α,3β-dihydroxyolean-12-en-28-oic acid, MA) acids were isolated from solid wastes resulting from olive-oil production, which were extracted in a Soxhlet extractor with hexane (Merck, ref. 1.04374) and EtOAc (Fisher Scientific, ref. E/0900/17) successively.19 Both acids were purified from these mixtures by flash chromatography over silica gel 60 (Merck, ref. 1.09385), eluting with CH2Cl2 (Fisher Scientific, ref. D/1852/17), with increasing amounts of acetone (Fisher Scientific, ref. A/0600/17).33 For the control of flash chromatography and reactions, silica gel 60 aluminum sheets (Merk. ref. 1.16835) were used, the compounds were made visible by spraying a mixture of H2SO4 and AcOH, followed by heating at 120 °C, and finally observed with UV light at 254 nm.Synthesis of the gelators
N,N-Diisopropylethylamine (DIEA) 0.3 mmol (Sigma-Aldrich, ≥99%, ref. D125806), and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) 0.66 mmol (Apollo Scientific, ref. PC0921), were added to each solution of triterpene MA and OA, the solution was prepared with 0.44 mmol in THF (20 mL). The reactions were maintained at room temperature for 12 hours, and then extracted several times with water and CH2Cl2. The organic phase was dried with anhydrous Na2SO4 and the solvent was removed under reduced pressure. Finally, each residue was purified by column chromatography using hexane/EtOAc as eluents, obtaining the compounds 1 (92% yield) and 2 (90% yield).34 For compound 11H-NMR: (CDCl3 500 MHz): 8.05 (m, 1H, TBTU group), 7.52 (m, 1H, TBTU group), 7.38 (m, 2H, TBTU group), 5.38 (dd, 1H, J1 = J2 = 3.6 Hz, H-12), 3.69 (ddd, 1H, J1 = 4.4, J2 = 10.0, J3 = 14.4 Hz, H-2), 3.01 (d, 1H, J = 10.0 Hz, H-3), 2.98 (dd, 1H, J1 = 4.0, J2 = 13.5 Hz, H-18), 1.22, 1.05, 1.00, 0.98, 0.97, 0.85, 0.84 (s, 3H each, methyl groups); 13C-NMR (CDCl3, 100 MHz): 16.8 (CH3), 16.9 (CH3), 17.5 (CH3), 18.5 (CH2), 23.3 (CH2), 23.7 (CH), 23.7 (CH3), 25.9 (CH3), 28.2 (CH2), 28.8 (CH3), 30.8 (C), 32.6 (CH2), 32.9 (CH2), 33.1 (CH3), 33.8 (CH2), 38.4 (C), 39.3 (C), 39.7 (C), 41.7 (CH), 42.1 (C), 45.6 (CH2), 46.6 (CH2), 47.7 (CH), 47.8 (C), 55.5 (CH), 69.0 (CH), 84.1 (CH), 108.3 (CH TBTU), 120.7 (CH TBTU), 123.9 (CH), 124.8 (CH TBTU), 128.6 (CH TBTU), 128.9 (C TBTU), 142.3 (C), 143.7 (C TBTU), 173.7 (COOH); ESI-HRMS m/z calcd for C36H52N3O4 [M + 1]+ 590.3958, found 590.3954.For compound 21H-NMR: (CDCl3, 500 MHz): 8.03 (m, 1H, TBTU group), 7.50 (m, 1H, TBTU group), 7.37 (m, 2H, TBTU group), 5.36 (dd, 1H, J1 = J2 = 3.5 Hz, H-12), 3.20 (dd, 1H, J1 = 4.0, J2 = 11.0 Hz, H-3), 2.97 (dd, 1H, J1 = 4.5, J2 = 14.0 Hz, H-18), 1.21, 0.99, 0.98, 0.96, 0.89, 0.84, 0.78 (s, 3H each, methyl groups); 13C-NMR: (CDCl3, 125 MHz): 15.5 (CH3), 15.7 (CH3), 17.4 (CH3), 18.4 (CH2), 23.2 (CH2), 23.6 (CH), 23.6 (CH3), 25.8 (CH3), 27.3 (CH2), 28.2 (CH2), 28.2 (CH3), 30.7 (C), 32.5 (CH2), 32.9 (CH2), 33.0 (CH3), 33.8 (CH2), 37.1 (C), 38.7 (CH2), 38.9 (C), 41.7 (CH), 42.0 (C), 45.6 (CH2), 47.7 (CH), 47.7 (C), 55.3 (CH), 79.0 (CH), 108.3 (CH TBTU), 120.6 (CH TBTU), 124.0 (CH), 124.7 (CH TBTU), 128.6 (CH TBTU), 128.9 (CH TBTU), 142.2 (C), 143.6 (CH TBTU), 173.7 (COOH); ESI-HRMS m/z calcd for C36H52N3O3 [M + 1]+ 574.4009, found 574.4020.
Gelification protocol
To prepare gels in DMSO–H2O or DMF–H2O mixtures, the desired amount of powder of 1 or 2 was weighed in a sample tube and DMSO or DMF was then added to reach the final concentration. The mixture was gently warmed until a clear solution was obtained and it was left to reach room temperature. To prepare gels by capturing water, gelator solution in open vials was left exposed to the lab environment for different times depending on the gel concentration (Table S1, ESI†). For gels prepared by direct addition of water, water was added to the solution and the gelation was completed in 5 minutes in the case of DMSO–H2O and 90 minutes in the case of DMF–H2O.Gels of 1 containing toluene were prepared by adding toluene to reach a gelator concentration of 1.6% (w/v). The mixture was gently warmed to dissolve the gelator and the gels were obtained when the solution reached room temperature, approx. 5 minutes.
Scanning electron microscopy (SEM)
Samples containing DMSO–H2O or DMF–H2O were freeze dried. Samples containing toluene were dried by air-drying exposing a gel portion to the lab environment overnight and by ‘critical point drying’ (CPD). For CPD, a gel portion was sequentially soaked in different mixtures of toluene and ethanol, increasing the ethanol proportion until all the toluene was replaced by ethanol. Then, the sample was placed in the chamber of the Polaron CPD 7501 CPD equipment. The chamber was sealed and cooled and the solvent was removed with supercritical CO2. The sample was covered with carbon with the high vacuum thermal evaporator EMITECH K975X. All samples dried, as described above, were studied by using an FEI Quanta 400 ESEM electron microscope.Transmission electron microscopy (TEM)
TEM images were recorded by using a LIBRA 120 PLUS (Carl Zeiss) electron microscope. For all samples, a little portion of gel was dispersed in water using an ultrasonic bath and a drop of the aqueous suspension was placed on a carbon-coated copper grid (300-mesh). The samples were dried at room temperature overnight.1H-nuclear magnetic resonance (1H-NMR)
NMR gel samples at 1% (w/v) were prepared at room temperature by adding D2O (15%, 0.15 mL) to a solution of 1 or 2 in DMSO-d6 (0.85 mL) contained in a 5 mm NMR tube.NMR spectra corresponding to the relaxation experiments were recorded in a 600 MHz (Agilent VNMRS software VnmrJ-4.2A) instrument at 25 °C (gel state) and at 100 °C (solution state). T2 measurements were performed using the Carr–Purcell–Meiboom–Gill (CPMG2) sequence with dephasing times of 0.4 ms. A minimum of nine points was obtained for each of the relaxation experiments, and a good fit of the data to an exponential decay was obtained in all reported data.
Fourier transform infrared (FTIR) spectroscopy
FTIR spectra were recorded in a Tensor 27 (Bruker) spectrometer equipped with an Attenuated Total Reflection (ATR) accessory (diamond, 1 reflection, 45°). The hydrogels were pressed against the diamond crystal. The spectra are the results of averaging 25 scans collected in the range between 4000 and 500 cm−1 with a resolution of 2 cm−1. A spectrum of air was used as a background.Vibrational circular dichroism (VCD)
VCD spectra were measured by means of a Bruker PMA50 optical bench coupled to the Vertex 70 spectrometer. In the PMA50 instrument, the infrared radiation is focused by a BaF2 lens, passing an optical filter (3800–600 cm−1 range) and a ZnSe photoelastic modulator (PM, 50 kHz frequency). The light beam is finally collected by a D313/QMTC detector with non-dichroic BaF2 windows. The previous calibration of the PM at a fixed wavenumber is required before recording a VCD spectrum. A spectral region of 600 cm−1 centred on each calibration wavenumber is then available. Typically, calibrations at 1300 cm−1 allowed us to obtain VCD signals over the infrared region in which we are interested. Every VCD spectrum was the result of averaging a minimum of 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans (12 h acquisition times) at a spectral resolution better than 4 cm−1.
000 scans (12 h acquisition times) at a spectral resolution better than 4 cm−1.
VCD spectra were recorded from gels formed in DMSO–H2O at a concentration of 3 × 10−3 M. The gels were placed in a variable path liquid cell of 0.015 mm length and KBr windows. Measurements and spectral treatments were carried out using the OPUS 6.5 spectroscopic software.
DFT calculations
The Gaussian’16 suite of programs35 was used to carry out Density Functional Theory (DFT) quantum chemical calculations. The hybrid exchange–correlation functional b3lyp was used in all calculations. It combines the Becke's three parameter (b3) gradient-corrected exchange functional and the Lee–Yang–Parr (LYP) non-local correlations.36–38 Ground state structures were calculated using the cc-pvdz double-zeta Dunning's correlation consistent basis sets,39 which includes polarization functions by definition. The theoretical infrared and VCD spectra were obtained from the DFT intensities in combination with the calculated vibrational wavenumbers uniformly scaled by 0.98. Every band was represented by a Gaussian function of 10 cm−1 half-height width.X-Ray diffraction (XRD)
The X-ray powder diffraction (XRPD) patterns of the samples were collected on a Bruker D8 Discover diffractometer μMR (Bruker, Karlsruhe, Germany) equipped with a DECTRIS PILATUS3 R 100K-A hybrid photon counting (HPC) 2D detector. Measurements were carried out using Cu Kα radiation (λ = 1.5406 Å) generated at 50 kV and 1 mA. Patterns were recorded in the 2θ range from 3 to 45° with a step size (2θ) of 0.02. As prepared gels were deposited on a zero-diffraction silicon plate.Rheological characterization
For the characterization of the dynamic rheological properties of the gels, we used a controlled-stress Haake Mars III rheometer, provided with two different sets of geometries (parallel plates of 35 mm of diameter and coaxial cylinders of 16 mm and 17 mm of internal and external diameter, respectively). We subjected all samples to oscillatory shear strains and measured the resulting oscillatory shear stresses at a temperature of 25 °C. From these measurements, we obtained the viscoelastic moduli of the samples: storage modulus (G′) and loss modulus (G′′). For obtaining a complete characterization of the rheological properties in the regime of oscillatory shear strain, we performed two separate experiments. First, amplitude sweeps, of the fixed frequency of oscillations and stepwise increasing shear strain amplitude, which allowed the determination of the extension of the linear viscoelastic regime (LVR). Then, frequency sweeps of fixed amplitude of shear strain within the LVR and stepwise increasing frequency were performed. In addition, we determined by rheological methods the temperature (Tgel) at which the gel–sol transition of the gels took place. For this aim, we placed the gels in the measuring system of the rheometer at room temperature and increased the temperature 2.5 °C min−1 ± 0.97, up to 85 °C. The samples were subjected to oscillatory shear strains of fixed frequency (1 Hz) and fixed amplitude within the LVR and the temperature-evolution of the viscoelastic moduli of the gels was monitored.For each sample and experimental condition, we conducted at least 3 separate measurements. The results shown in this manuscript represent the average of each set of separate measurements.
Results and discussion
Gelation behaviour of the compounds
The gelation behavior of compounds 1 (MATBTU) and 2 (OATBTU) was tested in 37 solvents at a concentration of 1% (w/v), and the results are listed in Table 1. Gelification tests were performed following different strategies. Firstly, compounds were tried to be dissolved at room temperature through the application of sonication. Secondly, compounds were tried to be dissolved by temperature switch, in which the temperature was raised until complete dissolution of the gelator and, thirdly, compounds were tried to be dissolved by solvent switch, mixing the gelator solution with a solvent in which the gelator is not soluble. Of all the solvents tested, compounds 1 and 2 were able to form homogeneous gels at room temperature by solvent switch in DMSO–H2O and DMF–H2O, and 1 in toluene by a temperature switch.| Solvent | 1 | 2 | Solvent | 1 | 2 |
|---|---|---|---|---|---|
| a Concentration of gelator: 1% w/v; G: gels forming at room temperature; S: solution; PG: partial gel; I: insoluble; TG: turbid gel; P: precipitate. | |||||
| Acetone | PG | S | DMSO (85%) H2O (15%) | G | G |
| Ethyl acetate | S | S | Ethanol | PG | PG |
| Acetonitrile | S | S | Diethyl ether | I | S |
| Acetophenone | S | S | n-Hexane | I | PG |
| Benzene | PG | S | n-Heptane | I | PG |
| n-Butanol | S | S | Isooctane | I | S |
| Butanone | S | S | Isopropanol | PG | S |
| Butyl glycolate | S | S | Methanol | PG | I |
| Chlorobenzene | P | S | Methyl acetate | S | S |
| Chloroform | S | S | 2-Methylfuran | PG | S |
| Cyclohexane | P | PG | 1-Methyl-2-pyrrolidone | S | S |
| Cyclopentane | P | PG | Nitromethane | S | S |
| Cyclopentanone | S | S | Octane | I | PG |
| 1,2-Dichloroethane | S | S | Pentane | I | P |
| Diisopropyl ether | I | S | 1-Propanol | S | S |
| Dichloromethane | S | S | Tetrahydrofuran | S | S |
| DMA | S | S | Tetrahydropyran | S | S |
| Methylformamide | S | S | Toluene | TG | S |
| DMF (75%) H2O (25%) 1. DMF (86%) H2O (14%) 2 | G | G | |||
In DMSO–H2O compounds 1 and 2 were able to form gels in concentrations ranging from 1% to 10% (w/v) giving rise to homogeneous gels of increasing opacity (Fig. 2). At all gelator concentrations the tested amount of absorbed water was always the same. By weighing the vials containing the DMSO gelator solution and the resulting gels we quantified that the amount of absorbed water was 15% (v/v). This amount of water agrees with the water that DMSO solvent is able to absorb at room temperature.40
 | ||
| Fig. 2 Gels of 1 and 2 in DMSO–H2O at increasing concentrations of gelators. From left to right: gels 1 to 3 are at 1% (w/v) (10 mg mL−1), 4 to 6 are at 4% (w/v) (40 mg mL−1) and 7 to 9 are at 8% (w/v) (80 mg mL−1). | ||
Similarly, both compounds were also able to form homogeneous gels in DMF–H2O although the range of concentrations was restrained from 4% to 10% (w/v). At a conc. of 10% (w/v), the gel was opaque and gelator precipitation appeared. In this case, the amount of absorbed water was not the same for both gelators, see Table S2, ESI.† Compound 1 absorbed much more water than compound 2, as indicated in Table 1 and Table S2 (ESI†).
Compound 1 was also able to gelify toluene although, in this case, gelification only occurred at a unique concentration of 1.6% (w/v) giving a whitish opaque gel. It is interesting to note that although these compounds are rather hydrophobic (1clogP = 5.62 and 2clogP = 6.86) they are able to gelify very polar organic solvent mixtures, such as DMSO–H2O and DMF–H2O. Surprisingly, toluene, although classified as a non-polar solvent, has a large permanent quadrupole moment and is able to form not only π–π aromatic interactions but also polar–π interactions with hydrogen bond donors. Although compound 1 differs from compound 2 only in the presence of an additional hydroxyl group (Fig. 1), this small change is sufficient to influence the ability of these compounds to form supramolecular aggregates in a variety of organic solvents. While compound 1 was able to partially gelify highly polar and aromatic solvents, compound 2, on the contrary, was able to partially gelify non-polar solvents such as lineal hydrocarbons (Table 1).
Scanning electron microscopy (SEM) imaging
In order to study the impact of the solvent in the structure of the resulting network, the SEM images of xerogels were taken. Xerogels were prepared by lyophilizing or air-drying gels of 1 and 2 at 1.6% (w/v) in DMSO–H2O (85%/15%) and DMF–H2O (75%/25% for 1 and 86%/14% for 2). Gels were prepared by capturing atmospheric water. Additionally, xerogels from 1 prepared in toluene at the same concentration were obtained by CPD and air-drying methods.The SEM images of xerogels of the two compounds obtained from gels of DMSO–H2O (Fig. 3A and B) and DMF–H2O (Fig. 3C and D) presented a similar morphology. Both samples showed a congested mesh of interwoven helical fibers with diameters ranging, preferentially, from 50 to 100 nm, although fibers with higher diameters were also observed. These results indicated that gelators 1 and 2 gave rise to similar aggregates in these two solvents. In this case, the slightly different gelator structures practically have no influence on the final supramolecular structures formed. This might be explained by the similar character between DMF and DMSO, since both are polar aprotic solvents of similar dielectric constant. The high polarity of these solvents can favour the hydrophobic aggregation of these gelators through the triterpene backbone.
 | ||
| Fig. 3 SEM images of xerogels 1 and 2 in DMSO–H2O (A and B, respectively) and DMF–H2O (C and D, respectively). Xerogel of 1 in toluene after supercritical drying (E) and air-drying (F). | ||
On the other hand, very different aggregates were obtained from the xerogels of 1 in toluene. The SEM images of the CPD xerogel (Fig. 3E) showed well-defined thick rods of larger diameters ranging from 100 to 400 nm. In order to check if the different sample preparation protocol had an influence in the final morphology, samples prepared by air-drying, similarly as prepared by DMF–H2O and DMSO–H2O, were also studied (Fig. 3F). In this case, the morphologies were identical to the sample prepared by CPD being very different to the morphologies obtained in the other solvents. It is not unusual for this type of gelator to present different morphologies in different solvents. Similar results have been obtained with cholesterol and triterpene derivatives14,27 and highlight the dynamic non covalent nature of the self-aggregation process. In this case, the different polarity of toluene from mixtures of DMSO–H2O and DMF–H2O justifies the different morphology of the resulting supramolecular aggregates.
Transmission electron microscopy (TEM) imaging
A similar trend was also observed in the TEM images (Fig. S1, ESI†). In this case, only nanorods were observed for both compounds. The lengths and diameters of these nanorods were significantly affected by the solvent. At such, nanorods of smaller aspect ratio were formed in DMSO–H2O having diameters from 10 to 60 nm (Fig. S1A and B, ESI†). Nanorods formed in DMF–H2O showed larger dimensions with diameters ranging from 20 to 80 nm (Fig. S1C and D, ESI†). Not surprisingly, and resembling the structures observed by SEM, the most different nanorods were observed for 1 in toluene (Fig. S1E, ESI†). In this case, nanorods were the largest aspect ratio, reaching diameters larger than 500 nm. Similar results have been observed in a pyridinium–oleanolate derivative in which the solvophobic effect between the solvents and the gelator is mainly responsible for the aggregation and growth of the gelator molecules.27 In this case, toluene, being a non-polar solvent, promotes a stronger aggregation of 1 giving rise to nanorods of larger aspect ratio.VT-1H-nuclear magnetic resonance (NMR) studies
The temperature-dependent 1H-NMR spectra of 1 and 2 in DMSO-d6 containing 15% of D2O are shown in Fig. 4. At room temperature (25 °C) the gel showed broad peaks of the molecule with unresolved J-couplings. The intensity of the signals suggested that only a fraction of the gelator molecules was observed by NMR since the gel network was NMR silent as a result of the large correlation time of the assemblies. As the temperature increased from 25 °C to 100 °C the spectra became much better resolved, all resonances sharpen and the spectra looked like the monomer suggesting that Tgel was surpassed. | ||
| Fig. 4 Temperature dependence of the 1H-NMR spectra of 1 and 2 in DMSO-d6/D2O. | ||
It can be noticed that there was practically no variation in the chemical shifts of all signals in the gel and sol states, although there was a modification of the tumbling rates (T2 values) upon gel formation (Table 2). This variation in the values of T2 cannot be ascribed to possible changes in the viscosity of the media since it is known that supramolecular gelators do not affect the tumbling rates41 and the viscosity of the solvent.42 This phenomenon has already been observed by other authors in closely related gelator structures such as cholesterol stilbene derivatives43 and suggests that there are mobile gel regions that can be observed by NMR. An alternative explanation of this effect has been proposed by Escuder and Miravet.41 These authors suggest that in these systems there is an exchange of the gelator molecules from the gel to the solution fast in the NMR relaxation times (seconds) and slow on the NMR time scale (milliseconds). In this way, chemical shifts of the molecules would remain unchanged going from sol to gel since only free molecules would be observable while aggregates would not.
| Gelator | Temperature (°C) | Integration peaks (ppm) | T 2 (s) |
|---|---|---|---|
| 1 | 100 (sol) | 7.43–7.38 | 1.0045 |
| 1 | 100 (sol) | 5.34–5.31 | 0.3989 |
| 1 | 25 (gel) | 7.44–7.39 | 0.2376 |
| 1 | 25 (gel) | 5.31–5.23 | 0.1231 |
| 2 | 100 (sol) | 7.43–7.39 | 1.9865 |
| 2 | 100 (sol) | 1.19–1.16 | 0.4276 |
| 2 | 25 (gel) | 7.45–7.40 | 0.6059 |
| 2 | 25 (gel) | 1.24–1.21 | 0.2638 |
Fourier transform infrared (FTIR) spectroscopy
FTIR spectra of gels of 1 in DMSO–H2O at increasing concentration of MATBTU, along with the spectra of pure MATBTU and DMSO, are shown in Fig. 5a. The spectra of the gels are mainly dominated by the features of the organic solvent. The presence of water molecules in the gels is further confirmed by the stretching (3300–3600 cm−1) and bending (ca. 1650 cm−1) modes of water molecules. In addition, the peak at 1041 cm−1, assigned to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of self-associated DMSO molecules,44 is red-shifted up to ca. 1020 cm−1 in the gels (Fig. 5c). DMSO molecules self-associate through dipole–dipole interactions resulting in chain-like and cyclic structures.44 Water induces the formation of hydrogen-bonded DMSO–H2O dimers, which results in a red shift of the SO stretching.44 Indeed, in aqueous DMSO solutions, the frequency of the SO stretching coincides with that of the methyl rocking modes (ca. 1020 cm−1). For this reason, the overall intensity of the region around 1020 cm−1 is increased in the presence of water,44 as we observed in our FTIR spectra (Fig. 5c). In fact, the higher shift was found in the 1% (w/v) gel, which contains the higher water/gelator ratio (Fig. 5a and c). Therefore, the formation of the DMSO–H2O dimers, also occurring in compound 2 in DMSO (Fig. S2, ESI†) could be responsible for triggering the gelification by changing the solvophobic effect between the media and the gelator, not occurring when the gelator is dissolved in pure DMSO. Similar results were also found in mixtures of DMF–H2O (Fig. S3, ESI†).
O stretching mode of self-associated DMSO molecules,44 is red-shifted up to ca. 1020 cm−1 in the gels (Fig. 5c). DMSO molecules self-associate through dipole–dipole interactions resulting in chain-like and cyclic structures.44 Water induces the formation of hydrogen-bonded DMSO–H2O dimers, which results in a red shift of the SO stretching.44 Indeed, in aqueous DMSO solutions, the frequency of the SO stretching coincides with that of the methyl rocking modes (ca. 1020 cm−1). For this reason, the overall intensity of the region around 1020 cm−1 is increased in the presence of water,44 as we observed in our FTIR spectra (Fig. 5c). In fact, the higher shift was found in the 1% (w/v) gel, which contains the higher water/gelator ratio (Fig. 5a and c). Therefore, the formation of the DMSO–H2O dimers, also occurring in compound 2 in DMSO (Fig. S2, ESI†) could be responsible for triggering the gelification by changing the solvophobic effect between the media and the gelator, not occurring when the gelator is dissolved in pure DMSO. Similar results were also found in mixtures of DMF–H2O (Fig. S3, ESI†).
 | ||
| Fig. 5 FTIR spectra of MATBTU (1) hydrogels in DMSO–H2O at different gelator concentrations (a). FTIR spectra of pure MATBTU (green curve) and DMSO (black curve) are also shown. Spectral regions containing the νCO of MATBTU (b) and νSO of DMSO (c). | ||
Vibrational bands assignable to the gelator are visible at higher concentrations of 8 and 15% (w/v) (Fig. 5a). Both 1 and 2 as DMSO–H2O gels showed an intense band at 1808 cm−1, which can be assigned to the stretching vibration of the carbonyl group, ν(CO), according to DFT calculations. In the spectrum of 2 as a pure solid (Fig. S4a, ESI†) this band had a maximum at 1812 cm−1 and exhibited a clear shoulder at lower wavenumber, also visible in the gel. This could suggest the existence of different environments around the carbonyl group.
The water bending modes between 1750 and 1600 cm−1 prevented observing two weak absorptions (which were well predicted by DFT calculations), as the stretching vibrations of CC and benzotriazole moieties (Fig. S4b, ESI†). No differences were predicted for them when comparing 1 and 2. Most of the bands measured between 1500 and 1100 cm−1 were assigned to the bending vibrations of aliphatic C–H bonds, largely methyl and methylene groups. On the basis of DFT calculations, the band at 1382 cm−1 in 1 can also be assigned to a benzotriazole stretching mode. The same band was measured at 1384 cm−1 for gels of 2.
Vibrational circular dichroism (VCD)
The VCD spectra of gels of 1 and 2 in DMSO–H2O are shown in Fig. 6, which also includes the calculated infrared and VCD spectra. VCD spectra of gels made in DMF–H2O and toluene could not be measured due to solvent signal overlapping. We have removed the 1750 and 1600 cm−1 peaks from the spectra as the chiroptical signal from water molecules is not significant. The main outcome was the negative feature observed for the carbonyl stretching mode. In addition, the ν(CO) band of 2 appeared as a doublet, while a single band was observed in the VCD spectrum of 1. This fact is in agreement with the shoulder at ca. 1798 cm−1 measured in the corresponding FTIR band of 2 (Fig. S2, ESI†), which was not observed for 1 (Fig. 5b).
 | ||
| Fig. 6 VCD (above) and infrared (below) spectra of 1 MATBTU (left panel) and 2 OATBTU (right panel) in DMSO–H2O. Red lines: experimental FTIR spectra of gels in DMSO–H2O. Blue lines: calculated b3lyp/cc-pvdz spectra of 1-3H2O and 2-2H2O hydrogen bonding complexes. | ||
The negative VCD feature for the carbonyl stretching mode was correctly predicted by DFT calculations. However, calculations also indicated that the sign of this band was strongly dependent on the dihedral angle between the ester group and the aliphatic polycyclic moiety. Thus, and in order to reach the absolute configuration of these molecules, two different conformations of 1 and 2 were successfully optimized and their VCD spectra were calculated (Fig. S5, ESI†). Although all of the geometrical parameters were allowed to vary independently, only two conformers were successfully stabilized, which differed in the orientation of the ester group with respect to the polycyclic moiety. By looking at their VCD spectra, the only robust difference between the two conformers was the sign of the carbonyl stretching band, which was positive for the A conformers and negative for the B ones.
The optimized parameters were quite similar for 1 and 2. The conformers with negative carbonyl stretching feature, 1A and 2A, were always the most stable. The calculated energy gaps were 2.342 kJ mol−1 for 1 and 2.181 kJ mol−1 for 2. The calculated OCO–C–CH dihedral angles, which define the relative position of the ester group, were −25.2 and −25.7 deg for 1A and 2A, respectively, and 136.3 and 138.0 deg for the less stable conformers, 1B and 2B, respectively. Taking into account the negative sign of the experimental ν(CO) bands, these data indicate that the chiroptical signal comes largely from the A conformer, which can be proposed as the absolute configuration for gels of 1 and 2. In the case of 2, the negative doublet evidences at least two different environments in the gel. Despite the good matching between calculated and experimental FTIR spectra, the VCD spectra of 1 and 2 are barely reproduced in the 1500–1100 cm−1 region. This is largely caused by the high conformational freedom of hydroxyl and methyl groups, which have several bending vibrations in this region, and by the effect of the intermolecular forces into the gel, which cannot be simulated by our gas-phase calculations.
X-ray diffraction (XRD) studies
The X-ray diffraction pattern of MATBTU gel in DMSO–H2O is depicted in Fig. 7. It shows an intense diffuse scattering (dotted lines) from the solvent and non-crystalline material. In addition, some reflections corresponding to d-spacing (d = 2π/Q) of 1.38, 0.97, 0.78, 0.64, 0.61 and 0.50 nm are observed, which verify the lamellar structures of the fibers. This structure is practically maintained after drying the hydrogels, as confirmed by XRD (Fig. S6, ESI†). Moreover, the diffraction patterns of the xerogels of compounds 1 and 2 in DMSO and DMF were very similar (Fig. S6, ESI†). On the other hand, the XRD pattern of the xerogel of 1 in toluene was completely different. It showed better-defined Bragg diffraction peaks indicating that the fibrils contain longer range-order in comparison to the fibrils obtained in DMSO and/or DMF (Table S3, ESI†). We could use DFT calculations to assign reflections to the corresponding intramolecular distances (Table S3 and Fig. S7, ESI†). The preferential dimensions of the optimized compound 1 (MATBTU) are shown in Fig. S7 (ESI†). Values from 0.54 to 0.75 nm correspond to the vertical length of the triterpene skeleton and values from 0.89 to 1.61 nm correspond to the horizontal length of the derivative from different views. The XRD patterns of the compound 1 xerogel from toluene showed similar peaks as DFT calculated corresponding to d-spacings of 2.20, 1.61, 1.48, 1.10, 0.89, 0.75, 0.65 and 0.54 nm. The value measured at 2.20 nm can correspond to the doubling of the molecular length suggesting the formation of a dimer as depicted in Fig. S8 (ESI†), in which the aromatic rings are twisted and interacting by π-stacking. This dimer leaves the triterpene skeletons exposed to the exterior favouring the growth of the structure by intermolecular van der Waals interactions between the triterpene skeletons. | ||
| Fig. 7 XRD pattern of gel 1 in DMSO–H2O. A non-linear fitting has been used to determine the position of the diffraction peaks. The dotted lines represent the diffuse scattering of the DMSO–H2O solvent. | ||
Rheological studies
Amplitude sweeps (see Fig. 8) demonstrated for all samples a typical gel-like behavior, characterized by values of the storage modulus (G′) much larger than the values of the loss modulus (G′′) within the region of low enough shear strain amplitude, for which both G′ and G′′ were approximately independent of the shear strain amplitude. This region of independence of the viscoelastic moduli with the value of the shear strain amplitude is known as a linear viscoelastic region (LVR). At higher values of the shear strain amplitude an initially smooth drop of G′ is observed, accompanied by a smooth increase of G′′, which reflects the onset of the nonlinear viscoelastic regime for which rupture of the gel structure takes place. Then, a maximum in G′′ is obtained, which represents the maximum dissipation of energy associated with the breakage of the gel structure. Results of frequency sweeps also demonstrated a typical behavior of a gel, with G′ almost independent of the frequency and G′′ increasing slightly with this magnitude (Fig. S9, ESI†). | ||
| Fig. 8 Amplitude sweeps: viscoelastic moduli as a function of shear strain amplitude, for oscillatory experiments of constant frequency (1 Hz) and increasing amplitude. (A and B) Gel 1 (MATBTU); (C) gel 1 (MATBTU); (D and E) gel 2 (OATBTU); full symbols represent values of the storage modulus; open symbols values of the loss modulus. Gelator concentration (only gels formed by capturing atmospheric water): ■□: 1%; ◆◇: 1.6%; ●○: 2%; ▲△: 3%; ▼▽: 4%; ★☆: 8%. Gelator concentration (only gels formed by direct addition of water): ▶▷: 1%. | ||
When gels were formed by capturing atmospheric water in DMSO, homogeneous gels containing higher concentrations of the gelators were possible. With respect to gel 1 (MATBTU), measurements performed at increasing gelator concentration (Fig. 8A) showed a monotonic increase of the values of both G′ and G′′, and thus of the robustness of the samples. Even at the lowest gelator concentration of 1% (w/v), the gels were very stiff showing values of G′ higher than 105. On the other hand, when toluene was used as a solvent (Fig. 8C), much lower (about two orders of magnitude) values of both G′ and G′′ were obtained (compare parts A and C of Fig. 8). For gel 2 (OATBTU) a similar trend of concentration-stiffness dependence was also observed. In this case, this gelator was able to form stiffer gels than compound 1, reaching values of G′ close to 107 at 8% (w/v). These high values of G′ are not usual for these type of LMWGs which tend to be 4 orders of magnitude lower.16
On the other hand, when gels were formed by direct addition of water, data demonstrated a weaker nature than for gels formed by capturing atmospheric water, see the comparison in Fig. 8B and E, both gels were measured at 1% (w/v). This result highlights the influence that the kinetics of gel formation has in the final macroscopic properties of the gels.
With respect to experiments aimed at the determination of Tgel, differences were also observed based on the gelification protocol. For gels formed by capturing atmospheric water in DMSO, the typical trend is shown, as an example for gel 2 (OATBTU) at a gelator concentration of 1% (w/v) in Fig. S10, ESI.† As observed, the values of G′ and G′′ did not change appreciably at low temperature (below approx. 35 °C). At this low-temperature range G′ was much higher than G′′, as expected for a gel. On the other hand, at a higher temperature, both G′ and G′′, but especially the former, decreased strongly, until finally, they leveled off at temperatures above approx. 75 °C. At this high-temperature range G′ was not appreciably higher than G′′, which definitely indicates that the samples were not a gel at these temperatures. As the transition temperature from a gel-like sample to a liquid-like sample (Tgel), we took in this work the minimum temperature at which G′ and G′′ attained values only 10% higher than those values corresponding to the average of the high-temperature plateaus. The obtained values for gel 1 (MATBTU) and gel 2 (OATBTU) at 1% (w/v) of gelator concentration were respectively 75.1 ± 2.2 °C and 86 ± 4 °C when they were formed by capturing atmospheric water, and 59.2 ± 2.7 °C and 63.1 ± 0.6 °C when they were formed by direct addition of water. As expected, a lower Tgel was obtained for gels formed by direct addition of water, in connection with their weaker nature, evidenced also as discussed above by smaller values of the viscoelastic moduli.
Conclusions
To sum up, in this work we have studied the capability of two new triterpene derivatives (MATBTU and OATBTU) to form supramolecular gels. These two new compounds have some particularities not previously described for similar structural-related gelators, which make them highly relevant in the field of LMWGs. First of all, they have the ability to form gels by slowly capturing atmospheric water giving rise to homogeneous gels showing extremely high values of G′ up to 107 Pa. As previously commented, it is not trivial for a small compound to gelify upon slow interaction with an external agent since, as it has been described by most LMWGs, gelification implies nucleation and subsequent 1D growth.45 In this case, the slow gelification process subject to capturing atmospheric water suggests that these two gelators must have a great tendency to grow into 1D fibers needed to form the gels. This assumption is confirmed by the long fibers and nanorods observed by SEM and TEM imaging. Quite remarkable is also the ability of these gelators to form homogeneous gels even at higher concentrations up to 10% (w/v), which can also be justified by the great tendency of these molecules to self-assemble into fibers and by the slow gelification process. Nevertheless, NMR studies suggest that part of these gelator molecules can be free in solution exchanging with the gel network at room temperature. Notable also is the combination of solvent mixtures at which these molecules are able to form a gel. As shown by FTIR results, water is able to disrupt DMSO–DMSO interactions giving rise to DMSO–H2O dimers that can be responsible for triggering gel formation. This particular interaction between water and DMSO favors the formation of a more homogeneous environment in which the self-association of the gelator is promoted. Although gelators 1 and 2 only differ in an extra hydroxyl group, results have shown that these two structures interact differently. Primary results can be extracted by the solvents of different nature (Table 1) that can partially gelify. Significantly, only compound 1 is able to gelify toluene and results showed that it probably does so by following a different aggregation pattern, as observed by XRD diffraction and TEM and SEM images. Nevertheless, VCD measurements supported by DFT calculations suggest that both compounds have a preferred and similar conformer in solution.Rheological measurements have revealed that these DMSO-gels, when formed by capturing atmospheric water, are very stiff, even at lower concentrations, having high values of Tgel. When water is directly added to the solution lower values of G′ and G′′ and Tgel are obtained. Weaker gels from 1 are obtained in toluene. The rheological data seem to be in agreement with the conclusions extracted with the other techniques and point to the ability of these two gelators to form very homogeneous gels when capturing atmospheric water.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the Ministry of Economy and Competitiveness of Spain is acknowledged through the following project: FIS2017-85954-R (Agencia Estatal de Investigación, AEI, Spain, co-funded by Fondo Europeo de Desarrollo Regional, ERDF, European Union). K. V. G. thanks CONACYT (Consejo Nacional de Ciencia y Tecnología) GOBIERNO DEL ESTADO DE BAJA CALIFORNIA 2015 for her fellowship. G. B. R. R. thanks the Ministry of Economy and Competitiveness for a ‘‘Juan de la Cierva” contract. R. C. M. thanks the ‘‘Plan Propio de Investigación’’ from the UGR for the “Ayuda Puente” contract. J. M. D. L. thanks the Ministry of Economy and Competitiveness for a ‘‘Ramon y Cajal’’ contract (#RyC-2016-21042). We also thank the ‘‘Unidad de Excelencia Química aplicada a Biomedicina y Medioambiente’’ (UGR) for support and CIC personnel of the University of Granada for technical assistance.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 2002, 97, 3133–3160 CrossRef.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- R. Contreras-Montoya, A. B. Bonhome-Espinosa, A. Orte, D. Miguel, J. M. Delgado-López, J. D. G. Duran, J. M. Cuerva, M. T. Lopez-Lopez and L. Á. De Cienfuegos, Mater. Chem. Front., 2018, 2, 686–699 RSC.
- H. Wang and Y. Cheng, Mater. Chem. Front., 2019, 3, 472–475 RSC.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- B. Escuder, F. Rodríguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044–1054 RSC.
- M. Conejero-Muriel, J. A. Gavira, E. Pineda-Molina, A. Belsom, M. Bradley, M. Moral, J. D. D. G. L. Durán, A. Luque González, J. J. Díaz-Mochón, R. Contreras-Montoya, Á. Martínez-Peragón, J. M. Cuerva and L. Álvarez De Cienfuegos, Chem. Commun., 2015, 51, 3862–3865 RSC.
- R. Contreras-Montoya, G. Escolano, S. Roy, M. T. Lopez-Lopez, J. M. Delgado-López, J. M. Cuerva, J. J. Díaz-Mochón, N. Ashkenasy, J. A. Gavira and L. Álvarez de Cienfuegos, Adv. Funct. Mater., 2019, 29, 1–10 Search PubMed.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- P. G. Argudo, R. Contreras-Montoya, L. Álvarez de Cienfuegos, J. M. Cuerva, M. Cano, D. Alba-Molina, M. T. Martín-Romero, L. Camacho and J. J. Giner-Casares, Soft Matter, 2018, 14, 9343–9350 RSC.
- H. Svobodová, V. Noponen, E. Kolehmainen and E. Sievänen, RSC Adv., 2012, 2, 4985–5007 RSC.
- M. Xue, K. Liu, J. Peng, Q. Zhang and Y. Fang, J. Colloid Interface Sci., 2008, 327, 94–101 CrossRef CAS PubMed.
- J. Peng, K. Liu, X. Liu, H. Xia, J. Liu and Y. Fang, New J. Chem., 2008, 32, 2218–2224 RSC.
- M. Xue, D. Gao, X. Chen, K. Liu and Y. Fang, J. Colloid Interface Sci., 2011, 361, 556–564 CrossRef CAS PubMed.
- B. G. Bag and R. Majumdar, Chem. Rec., 2017, 17, 841–873 CrossRef PubMed.
- E. E. Rufino-Palomares, A. Perez-Jimenez, F. J. Reyes-Zurita, L. G. Salguero, K. Mokhtari, A. Herrera-Merchan, P. P. Medina, J. Peragón and J. A. Lupianez, Curr. Org. Chem., 2015, 19, 919–947 CrossRef CAS.
- M. Medina-O’Donnell, F. Rivas, F. J. Reyes-Zurita, A. Martinez, J. A. Lupiañez and A. Parra, Eur. J. Med. Chem., 2018, 148, 325–336 CrossRef PubMed.
- N. Blanco-Cabra, K. Vega-Granados, L. Moya-Andérico, M. Vukomanović, A. Parra, L. Álvarez de Cienfuegos and E. Torrents, ACS Infect. Dis., 2019 DOI:10.1021/acsinfecdis.9b00125.
- B. G. Bag and S. S. Dash, Nanoscale, 2011, 3, 4564–4566 RSC.
- B. G. Bag and K. Paul, Asian J. Org. Chem., 2012, 1, 150–154 CrossRef CAS.
- B. G. Bag and S. S. Dash, Langmuir, 2015, 31, 13664–13672 CrossRef CAS PubMed.
- B. G. Bag, S. N. Hasan, S. Ghorai and S. K. Panja, ACS Omega, 2019, 4, 7684–7690 CrossRef CAS.
- B. G. Bag, R. Majumdar, S. K. Dinda, P. P. Dey, G. C. Maity, V. A. Mallia and R. G. Weiss, Langmuir, 2013, 29, 1766–1778 CrossRef CAS PubMed.
- B. G. Bag, G. C. Maity and S. K. Dinda, Org. Lett., 2006, 8, 5457–5460 CrossRef CAS PubMed.
- Y. Gao, J. Hao, J. Wu, X. Zhang, J. Hu and Y. Ju, Langmuir, 2016, 32, 1685–1692 CrossRef CAS PubMed.
- Y. Gao, J. Hao, J. Wu, X. Zhang, J. Hu and Y. Ju, Nanoscale, 2015, 7, 13568–13575 RSC.
- J. Lu, J. Hu, Y. Song and Y. Ju, Org. Lett., 2011, 13, 3372–3375 CrossRef CAS PubMed.
- Y. Gao, J. Hao, J. Liu, Y. Liang, F. Du, J. Hu and Y. Ju, Mater. Chem. Front., 2019, 3, 308–313 RSC.
- R. Majumdar, S. Tantayanon and B. Gopal Bag, Chem. – Asian J., 2016, 11, 2406–2414 CrossRef CAS PubMed.
- B. G. Bag, S. Das, S. N. Hasan and A. Chandan Barai, RSC Adv., 2017, 7, 18136–18143 RSC.
- A. Martinez, A. Perojil, F. Rivas, A. Parra, A. Garcia-Granados and A. Fernandez-Vivas, Phytochemistry, 2015, 117, 500–508 CrossRef CAS PubMed.
- M. Medina-O’Donnell, F. Rivas, F. J. Reyes-Zurita, A. Martinez, S. Martin-Fonseca, A. Garcia-Granados, R. M. Ferrer-Martín, J. A. Lupiañez and A. Parra, Eur. J. Med. Chem., 2016, 118, 64–78 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 16, Revis. B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- T. J. Waybright, J. R. Britt and T. G. McCloud, J. Biomol. Screening, 2009, 14, 708–715 CrossRef CAS PubMed.
- B. Escuder, M. LLusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS PubMed.
- D. Miguel, S. P. Morcillo, A. Martín-Lasanta, N. Fuentes, L. Martínez-Fernández, I. Corral, M. J. Ruedas-Rama, D. J. Cárdenas, L. Álvarez De Cienfuegos, A. Orte and J. M. Cuerva, Org. Lett., 2015, 17, 2844–2847 CrossRef CAS PubMed.
- D. C. Duncan and D. G. Whitten, Langmuir, 2000, 16, 6445–6452 CrossRef CAS.
- V. M. Wallace, N. R. Dhumal, F. M. Zehentbauer, H. J. Kim and J. Kiefer, J. Phys. Chem. B, 2015, 119, 14780–14789 CrossRef CAS PubMed.
- X. Huang, P. Terech, S. R. Raghavan and R. G. Weiss, J. Am. Chem. Soc., 2005, 127, 4336–4344 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H-NMR spectra of compounds and supplementary figures. See DOI: 10.1039/c9qm00397e |
| This journal is © the Partner Organisations 2019 |