
All-small-molecule dynamic covalent hydrogels with multistimuli responsiveness†
Hui
Wang
a and
Yiyun
Cheng
 *ab
*ab
aSouth China Advanced Institute for Soft Matter Science and Technology, South China University of Technology, Guangzhou, 510640, P. R. China
bShanghai Key Laboratory of Regulatory Biology, School of Life Sciences, East China Normal University, Shanghai, 200241, P. R. China. E-mail: yycheng@mail.ustc.edu.cn
First published on 9th January 2019
Abstract
Low molecular weight hydrogels have shown great promise in cell culture, tissue engineering, and controlled drug release. They are generally constructed from small molecules that form fibrous networks in aqueous solutions via supramolecular assembly. Here, we report a class of all-small-molecule dynamic covalent hydrogels via formaldehyde condensation with aminoglycosides, a family of naturally occurring antibiotics consisting of oligosaccharides and multiple amine groups. The abundant hydroxyl groups on aminoglycosides facilitate the formation of a secondary network via intermolecular hydrogen bonding at room temperature. The aminoglycoside hydrogels show excellent pH- and temperature-responsiveness and display controllable stiffness in the presence of different alkali cations and halogen anions. This study provides a facile strategy to construct low molecular weight hydrogels with multistimuli-responsiveness, and modulable and recyclable properties.
Hydrogels are emerging materials in controlled drug release, flexible electronics, diagnostics, cell culture and tissue regeneration.1 They are three-dimensional networks consisting of hydrophilic gelators crosslinked with each other via reversible or irreversible covalent bonds, or non-covalent approaches.2 Hydro-gels can be classified into polymeric hydrogels and low molecular weight hydrogels according to the composition of gelators. Polymeric hydrogels are the most investigated gels and the polymer scaffolds could be synthetic hydrophilic polymers or natural biomacromolecules.3 These hydrogels possess high mechanical properties, controllable microstructures and pores, and thus have shown great promise in various biomedical applications.4
Another class of hydrogels are low molecular weight hydrogels constructed from small molecule gelators such as peptides, peptoids, nucleotides, and other amphiphilic chemicals.5 These gels are usually prepared by heating a suspension of small molecule gelators, followed by cooling down to room temperature. During this process, the gelators self-assemble into fibrous nanostructures via hydrophobic, hydrogen bonding and sometimes π–π interactions, which is followed by nucleation of fiber growth and gelation.6 These hydrogels showed several advantages such as facile construction, availability for bioresponsiveness, predictable degradation, and reduced immunogenicity compared to polymeric hydrogels.7 On the other hand, the noncovalent forces maintaining the gel structure are relatively weak, and this may lead to low stiffness and instability of the supramolecular gels.8 For example, the fibrous nanostructures in low molecular weight gels might be destroyed during injection using a syringe or when encapsulated with hydrophobic drugs.9 The disturbed gels usually need a long period to recover the gel state. This situation is expected to be improved by introducing reversible covalent connectivity into the gel networks, however, the hydrogel constructed from low molecular weight gelators via dynamic covalent bonds is yet to be investigated.
It is reported that paraformaldehyde or formaldehyde react with amines at a temperature above 50 °C and form hemiaminal dynamic covalent networks.10 This chemistry was successfully used to construct organogels between amine-terminated poly(ethyl glycol) and paraformaldehyde.11 To the best of our knowledge, the hemiaminal chemistry has never been used to construct hydrogels, especially all-small-molecule hydrogels. Here, we reported such an all-small-molecule dynamic covalent hydrogel by formaldehyde condensation of aminoglycosides. Aminoglycosides are a class of naturally occurring antibiotics. The saccharide units in aminoglycosides render the molecules with excellent aqueous solubility and secondary crosslinking via hydrogen bonding interactions, while the primary amines provide a perfect reactive site to construct reversible covalent networks.12 Both the interactions contribute to the gel formation.
Aminoglycosides such as tobramycin and neomycin were reacted with formaldehyde to prepare the proposed all-small molecule hydrogels (Fig. 1A). The reaction of tobramycin with formaldehyde at room temperature generates white precipitates due to the formation of methylenimine on aminoglycosides. When heating the suspension to 85 °C for 4 h, the precipitates gradually disappeared. A light yellow and transparent hydrogel is formed by slowly cooling the hot solution to room temperature (Fig. 1B). As shown in Fig. 1C, the storage modulus (G′) of the formed hydrogel is higher than the loss modulus (G′′), which is considered as a sign of gelation.10 The aminoglycoside hydrogel shows an excellent shear-thinning property (Fig. 1D). As a result, the hydrogel can be ejected through a syringe, and recovered immediately after withdrawing the shear force (Fig. 1E). The hydrogel can be used as a transparent coating on a substrate (Fig. 1F), and fabricated into different shapes (Fig. 1G).
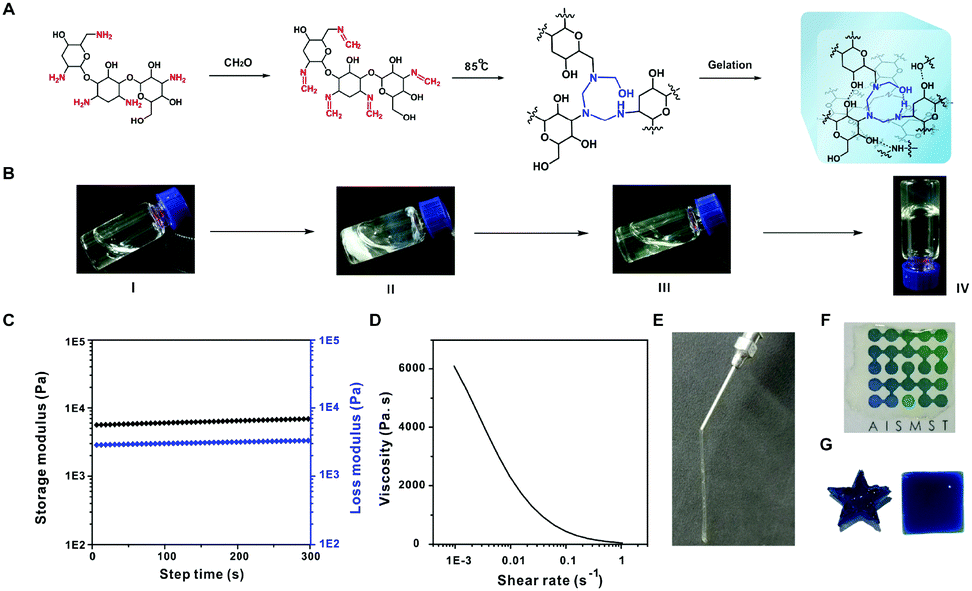 | ||
| Fig. 1 (A) Synthesis of all-small-molecule dynamic covalent hydrogels. (B) Preparation of hydrogels by the proposed reactions. (C) Time-dependent rheology of the hydrogel. (D) Effect of shear rate on gel viscosity. (E) Photograph of the hydrogel ejected from a syringe. (F) Coating of the hydrogel on paper printed with the sign of Advanced Institute for Soft Matter Science and Technology (AISMST). (G) Hydrogels loaded with methylene blue were fabricated into different shapes. | ||
We further investigate the gelation mechanism. As shown in Fig. S1 (ESI†), the band at ∼1598 cm−1 assigned to the N–H stretching vibration disappears when formaldehyde was added, while a new band at 1665 cm−1 for methylenimine is observed. In the representative hydrogel, the molar feeding ratio of formaldehyde to primary amine is 1.87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and thus most of the amines are transformed to methylenimine, which results in increased hydrophobicity and the generation of precipitates. When the suspension is heated to 85 °C, the hemiaminal dynamic covalent networks form between the amine and formaldehyde as previously reported.12 The appearance of the band at ∼1598 cm−1 confirmed the formation of hemiaminal networks (Fig. S1A, ESI†). This process is also confirmed by NMR. The peak around 7.5 ppm corresponds to the characteristic peak for methylenimine (Fig. S2A, ESI†).11b The peaks at 4.2–4.6 ppm were assigned to the methylene protons in the hemiaminal dynamic covalent network (Fig. S2B, ESI†).12 The NMR spectrum of the tobramycin/formaldehyde mixture is not shown due to the insolubility of the yielded products at room temperature. Finally, the formation of hydrogel is observed when the mixture was heated to 85 °C and cooled to room temperature (Fig. 1B). The gel formation is attributed to both the hemiaminal dynamic covalent network, and the intermolecular hydrogen bond interactions.
:1, and thus most of the amines are transformed to methylenimine, which results in increased hydrophobicity and the generation of precipitates. When the suspension is heated to 85 °C, the hemiaminal dynamic covalent networks form between the amine and formaldehyde as previously reported.12 The appearance of the band at ∼1598 cm−1 confirmed the formation of hemiaminal networks (Fig. S1A, ESI†). This process is also confirmed by NMR. The peak around 7.5 ppm corresponds to the characteristic peak for methylenimine (Fig. S2A, ESI†).11b The peaks at 4.2–4.6 ppm were assigned to the methylene protons in the hemiaminal dynamic covalent network (Fig. S2B, ESI†).12 The NMR spectrum of the tobramycin/formaldehyde mixture is not shown due to the insolubility of the yielded products at room temperature. Finally, the formation of hydrogel is observed when the mixture was heated to 85 °C and cooled to room temperature (Fig. 1B). The gel formation is attributed to both the hemiaminal dynamic covalent network, and the intermolecular hydrogen bond interactions.
Since the hydrogen bonding interactions play a critical role in gel formation, the yielded hydrogel should be temperature-responsive due to the destruction of hydrogen bonding at a relatively high temperature (Fig. 2A). As shown in Fig. 2B, both G′ and G′′ of the hydrogel are gradually decreased when the temperature is increased from 25 °C to 65 °C, and the G′ value is lower than G′′ above 42 °C, indicating a phase transition from gel to liquid (Tg). When the heated solution was cooled to room temperature, the G′ was again larger than G′′, suggesting the recovery of the gel state. The temperature-responsive property of the gel was further investigated by a continuous step temperature measurement. The hydrogel can almost recover to its original G′ and G′′ when the temperature is decreased to 25 °C, and this behavior can be repeated for at least five cycles, indicating excellent temperature reversible properties (Fig. 2C). The gel weight is scarcely decreased when it is incubated at 5 °C or 25 °C for 2 h, but is sharply decreased with increasing incubation temperature (Fig. 2D).
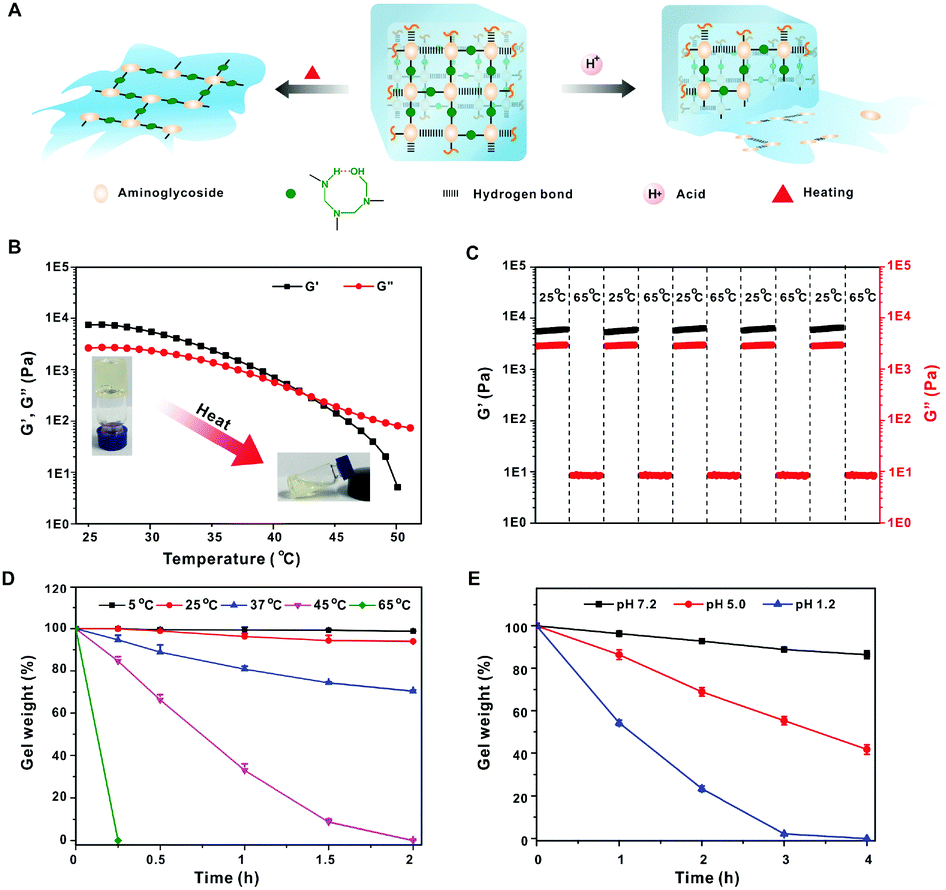 | ||
| Fig. 2 (A) Mechanism of temperature- and pH-responsiveness. (B) Temperature-dependent gel rheology. (C) G′ and G′′ measured at a continuous step temperature between 25 °C and 65 °C. (D) Gel degradation at various temperatures. (E) Gel degradation at various pHs. | ||
It is reported that the hemiaminal dynamic covalent network between amine and formaldehyde is acid-labile. The network can be completely digested to recover the amino monomers in acidic environments.11a As shown in Fig. 2E, the hydrogel is slightly degraded at pH 7.2 for 4 h, but the erosion is significantly accelerated under low pH. Nearly 70% of the tobramycin was released when immersed in pH 5.0 solution for 24 h (Fig. S3, ESI†), while only 15% aminoglycosides were released at pH 7.2. These results suggest that acid can cleave the hemiaminal dynamic covalent network and lead to complete gel erosion.
We also investigated the effect of alkali cations and halogen anions on the gel rheology. The introduction of alkali metal ions before gel formation results in hydrogels with much higher G′ (Fig. 3A). This is due to the formation of metal-hydroxyl coordination that improves the gel stiffness.13 The effect of the alkali ions on the hydrogel stiffness is concentration-dependent, and higher modulus could be achieved with higher ion concentrations (Fig. S4, ESI†). Among the alkali ions, Li+ containing hydrogel shows the highest stiffness, with a G′ value more than 20-fold higher than that of intact hydrogels without metal ions. Surprisingly, the Na+ displays the lowest ability to increase the gel stiffness, which is weaker than that of K+ and Rb+ containing hydrogels. Previous studies have shown that the interaction between alkali and poly(ethyl oxide) decreases with increasing metal ion size in the order Li+ > Na+ > K+ > Rb+ in solid states.14 However, the trend is completely inverse in water due to the hydration effect. The small-sized Li+ is easily hydrated and bound with water molecules, which efficiently shields the electric field of the cation and decreases its binding affinity with oxygen atoms.13a,15 Here, the mixture of tobramycin and formaldehyde was heated to 85 °C for 4 h to prepare the hydrogel. In this case, the solvation shell around the Li+ is destroyed at such a temperature, which facilitates the binding of Li+ with hydroxyl during gel formation. However, the alkali ions might be hydrated when the hot solutions were cooled to room temperature. As a result, the alkali-hydroxyl coordination and hydration effect co-exist in the finally yielded hydrogel. The dehydration effect is especially predominant for Li+, which shows the strongest binding affinity with hydroxyl groups. Therefore, the trend Li+ > Rb+ > K+ > Na+ is observed. Besides the alkali ions, the halogen anions also significantly affect the gel stiffness. The G′ was slightly increased in the presence of 20 mM NaF, while the value was enhanced by more than an order of magnitude for NaI (Fig. 3B). The gel stiffness is in the order of NaF < NaCl < NaBr < NaI. This can be explained by the electronegativity of the halogen anions. F− has the strongest electronegativity among the halogen anions. It may form strong hydrogen bonding interactions with the hydroxyls and destroy the intermolecular hydrogen bonding interactions between hydroxyl groups. Alternatively, the anions with weaker Lewis basicity would result in a stronger coordination interaction of alkali/oxygen.16 Therefore, the I− with the lowest electronegativity and Lewis basicity generates the strongest hydrogel among the halogen anions. Similarly, Tg of the gels increases in the same order with G′, and all the Tg values are higher than those of intact hydrogels without any salts (Fig. 3C). This is due to the predominant effect of the alkali-hydroxyl coordination. Since the Li+ and I− ions display the stiffest hydrogels among the alkali ions and halogen anions, respectively, we could expect that the addition of LiI will generate a hydrogel with the highest G′. As shown in Fig. 3D, a 65.5-fold increase in G′ was achieved when 20 mM LiI was added. These results suggest that the hydrogels possess controllable stiffness in the presence of different cations and anions.
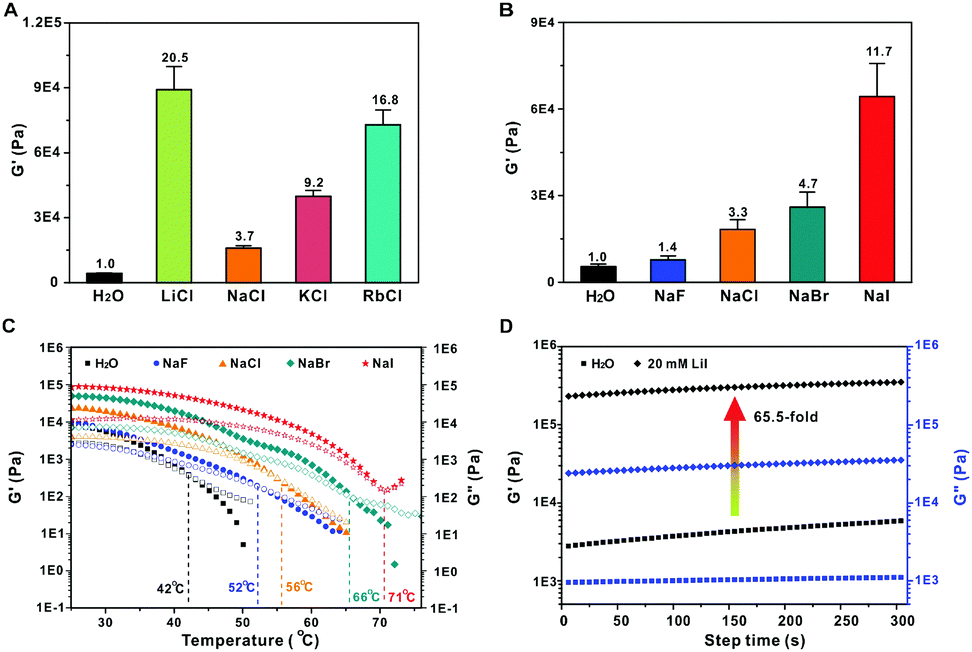 | ||
| Fig. 3 (A) G′ of hydrogels in the presence of 20 mM LiCl, NaCl, KCl and RbCl, respectively. (B) G′ of gels in the presence of 20 mM NaF, NaCl, NaBr and NaI, respectively. (C) Temperature-dependent gel rheology in the presence of different salts. (D) Time-dependent G′ and G′′ in the presence of 20 mM LiI. | ||
Neomycin is another aminoglycoside with a high density of both primary amine and hydroxyl groups. As expected, neomycin could also react with formaldehyde to form a similar hydrogel (Fig. 4A). The hydrogel also exhibits excellent temperature- and acid-responsive behaviours (Fig. 4B and C). Moreover, the gel stiffness can be significantly improved by the addition of salts. The G′ of the neomycin/formaldehyde gel was increased by 12-fold in the presence of 20 mM LiI compared to that without any salt (Fig. 4D).
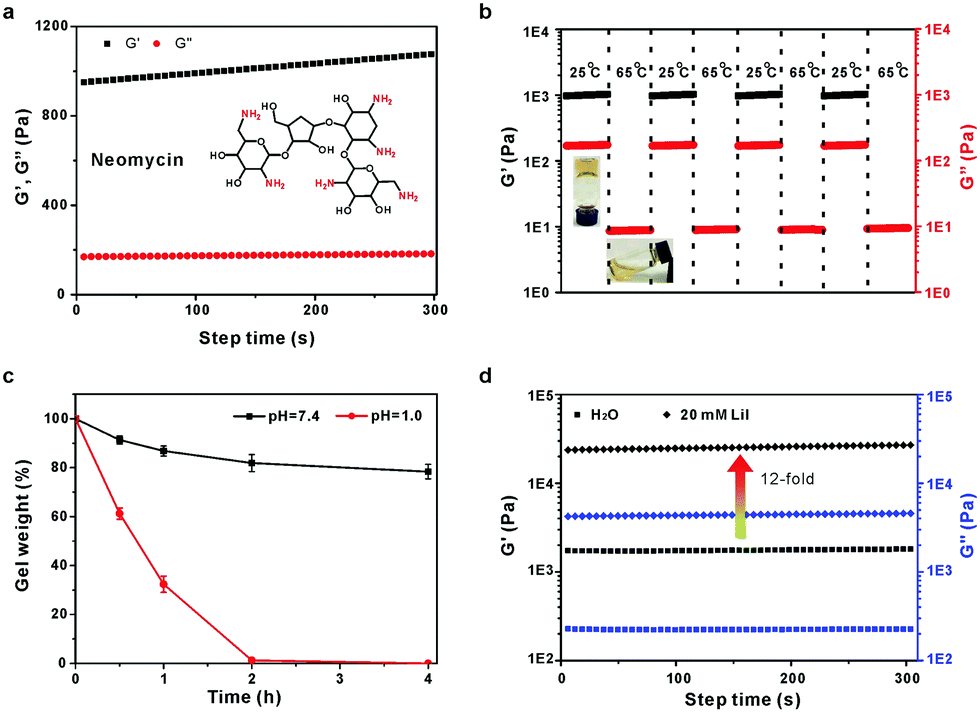 | ||
| Fig. 4 (A) Time-dependent rheology of the neomycin/formaldehyde hydrogel. (B) G′ and G′′ of the hydrogel measured at a continuous step temperature between 25 °C and 65 °C. (C) Degradation of the hydrogel at pH 7.4 and 1.0, respectively. (D) Time-dependent gel rheology in the presence of 20 mM LiI. | ||
Conclusions
In summary, we reported a class of all-small-molecule dynamic covalent hydrogels via formaldehyde condensation of aminoglycosides. The reaction of formaldehyde with aminoglycosides generates hemiaminal dynamic covalent networks which connect aminoglycosides. In addition, the abundant hydroxyl groups on aminoglycosides form hydrogen bonding interactions when the hot solution was cooled to room temperature. The combination of hemiaminal dynamic covalent networks and hydrogen bonding interactions contributes to gel formation. The yielded hydrogels possess many unique properties: (1) high cargo loading: the content of aminoglycosides in the hydrogel is as high as 23% (w/w), (2) totally degradable: the hydrogels could be completely degraded into the small molecules upon multistimuli-responsiveness, such as temperature and acid, and (3) the gel stiffness could be easily modulated by the introduction of alkali cations and halogen anions during gel formation. This study expands the family of low molecular weight hydrogels from supramolecular hydrogels to dynamic covalent hydrogels, and provides a facile strategy to prepare dynamic covalent hydrogels with multistimuli responsiveness. And in the future, we will focus on the all-small-molecule dynamic covalent hydrogels based on biocompatible molecules for use in biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (Grant No. 21725402) is greatly appreciated.Notes and references
- N. Oliva, J. Conde, K. Wang and N. Artzi, Acc. Chem. Res., 2017, 50, 669–679 CrossRef CAS PubMed.
- D. Caccavo, S. Cascone, G. Lamberti and A. A. Barba, Chem. Soc. Rev., 2018, 47, 2357–2373 RSC.
- (a) X. Zhang, Y. Li, Y. E. Chen, J. Chen and P. X. Ma, Nat. Commun., 2016, 7, 10376 CrossRef CAS; (b) J. D. Weaver, D. M. Headen, J. Aquart, C. T. Johnson, L. D. Shea, H. Shirwan and A. J. Garcia, Sci. Adv., 2017, 3, e1700184 CrossRef PubMed; (c) B. Hu, Y. Shen, J. Adamcik, P. Fischer, M. Schneider, M. J. Loessner and R. Mezzenga, ACS Nano, 2018, 12, 3385–3396 CrossRef CAS PubMed.
- (a) N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 26, 85–123 CrossRef CAS PubMed; (b) G. Zabow, S. J. Dodd and A. P. Koretsky, Nature, 2015, 520, 73–77 CrossRef CAS PubMed; (c) Z. Liu, Q. Lin, Y. Sun, T. Liu, C. Bao, F. Li and L. Zhu, Adv. Mater., 2014, 26, 3912–3917 CrossRef CAS PubMed; (d) D. Shi, X. Xu, Y. Ye, K. Song, Y. Cheng, J. Di, Q. Hu, J. Li, H. Ju, Q. Jiang and Z. Gu, ACS Nano, 2016, 10, 1292–1299 CrossRef CAS PubMed; (e) Z. Liu, T. Liu, Q. Lin, C. Bao and L. Zhu, Angew. Chem., Int. Ed., 2015, 54, 174–178 CrossRef CAS PubMed; (f) M. Liu, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nature, 2015, 517, 68–72 CrossRef CAS; (g) K. Iwaso, Y. Takashima and A. Harada, Nat. Chem., 2016, 8, 625–632 CrossRef CAS PubMed.
- (a) A. N. Moore and J. D. Hartgerink, Acc. Chem. Res., 2017, 50, 714–722 CrossRef CAS; (b) M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS PubMed; (c) P. W. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed; (d) E. R. Draper, E. G. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848–852 CrossRef CAS PubMed; (e) M. A. Rahim, M. Bjornmalm, T. Suma, M. Faria, Y. Ju, K. Kempe, M. Mullner, H. Ejima, A. D. Stickland and F. Caruso, Angew. Chem., Int. Ed., 2016, 55, 13803–13807 CrossRef CAS PubMed; (f) G. M. Peters, L. P. Skala, T. N. Plank, H. Oh, G. N. Reddy, A. Marsh, S. P. Brown, S. R. Raghavan and J. T. Davis, J. Am. Chem. Soc., 2015, 137, 5819–5827 CrossRef CAS PubMed; (g) Z. Luo, Q. Wu, C. Yang, H. Wang, T. He, Y. Wang, Z. Wang, H. Chen, X. Li, C. Gong and Z. Yang, Adv. Mater., 2017, 29, 1601776 CrossRef PubMed.
- (a) J. M. Poolman, J. Boekhoven, A. Besselink, A. G. Olive, J. H. van Esch and R. Eelkema, Nat. Protoc., 2014, 9, 977–988 CrossRef CAS PubMed; (b) C. Maity, W. E. Hendriksen, J. H. van Esch and R. Eelkema, Angew. Chem., Int. Ed., 2015, 54, 998–1001 CrossRef CAS PubMed.
- (a) Y. Cai, H. Shen, J. Zhan, M. Lin, L. Dai, C. Ren, Y. Shi, J. Liu, J. Gao and Z. Yang, J. Am. Chem. Soc., 2017, 139, 2876–2879 CrossRef CAS PubMed; (b) H. Wang, Z. Feng, Y. Wang, R. Zhou, Z. Yang and B. Xu, J. Am. Chem. Soc., 2016, 138, 16046–16055 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- M. Singh, S. Kundu, M. A. Reddy, V. Sreekanth, R. K. Motiani, S. Sengupta, A. Srivastava and A. Bajaj, Nanoscale, 2014, 6, 12849–12855 RSC.
- G. O. Jones, J. M. Garcia, H. W. Horn and J. L. Hedrick, Org. Lett., 2014, 16, 5502–5505 CrossRef CAS PubMed.
- (a) J. M. Garcia, G. O. Jones, K. Virwani, B. D. McCloskey, D. J. Boday, G. M. ter Huurne, H. W. Horn, D. J. Coady, A. M. Bintaleb, A. M. Alabdulrahman, F. Alsewailem, H. A. Almegren and J. L. Hedrick, Science, 2014, 344, 732–735 CrossRef CAS PubMed; (b) C. H. Fox, G. M. ter Hurrne, R. J. Wojtecki, G. O. Jones, H. W. Horn, E. W. Meijer, C. W. Frank, J. L. Hedrick and J. M. Garcia, Nat. Commun., 2015, 6, 7417 CrossRef PubMed.
- J. Hu, Y. Quan, Y. Lai, Z. Zheng, Z. Hu, X. Wang, T. Dai, Q. Zhang and Y. Cheng, J. Controlled Release, 2017, 247, 145–152 CrossRef CAS PubMed.
- (a) M. T. Rodgers and P. B. Armentrout, Chem. Rev., 2016, 116, 5642–5687 CrossRef CAS PubMed; (b) Z. Liu, A. Samanta, J. Lei, J. Sun, Y. Wang and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 11643–11653 CrossRef CAS PubMed.
- (a) F. Sagane, T. Abe, Y. Iriyama and Z. Ogumi, J. Power Sources, 2005, 146, 749–752 CrossRef CAS; (b) N. R. Dhumal and S. P. Gejji, Chem. Phys., 2006, 323, 595–605 CrossRef CAS; (c) R. Nadimicherla, R. Kalla, R. Muchakayala and X. Guo, Solid State Ionics, 2015, 278, 260–267 CrossRef CAS.
- (a) L. Chai, R. Goldberg, N. Kampf and J. Klein, Langmuir, 2008, 24, 1570–1576 CrossRef CAS PubMed; (b) C. Ren, W. Tian, I. Szleifer and Y. Ma, Macromolecules, 2016, 44, 1719–1727 CrossRef.
- J. Huang, R. Y. Wang, Z. Z. Tong, J. T. Xu and Z. Q. Fan, Macromolecules, 2014, 47, 8359–8367 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: 10.1039/c8qm00612a |
| This journal is © the Partner Organisations 2019 |