
Self-healable gradient copolymers†
Jing
Cui
a,
Zhe
Ma
a,
Li
Pan
 *a,
Chun-Hua
An
c,
Jing
Liu
c,
Yu-Feng
Zhou
d and
Yue-Sheng
Li
*ab
*a,
Chun-Hua
An
c,
Jing
Liu
c,
Yu-Feng
Zhou
d and
Yue-Sheng
Li
*ab
aTianjin Key Laboratory of Composite and Functional Materials, School of Materials Science and Engineering, Tianjin University, Tianjin 300350, China. E-mail: lilypan@tju.edu.cn; ysli@tju.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China
cState Key Laboratory of Precise Measurement Technology & Instruments, School of Precise Instruments & Optoelectronics Engineering, Tianjin 300072, China
dSchool of Materials Science & Engineering, Zhengzhou University, Zhengzhou 450002, China
First published on 15th January 2019
Abstract
Self-healing materials typically suffer from poor mechanical performance in terms of practical applications. Herein, a self-healable copolymer with a gradient distribution of hard segments on ionic polymer chains is designed. By the optimization of the monomer sequences and physical cross-linking, the copolymer exhibits excellent mechanical properties with Young's modulus up to 286 MPa and toughness over 33 MJ m−3, which represents a significant improvement compared with traditional self-healing materials. Imidazolium in this copolymer not only generates strong dynamic ionic associations and imparts mending ability, but also provides ionic conductivity for potential device applications. Environment-insensitive self-healing in the presence of moisture, water and artificial sweat is achieved. Strain sensors with rapid response (<114 ms) and high durability (no performance decrease after 7000 cycles of tensile test) are fabricated using the gradient copolymers, opening an avenue for high-performance wearable devices using polymeric materials.
Introduction
Utilizing reversible breakage and recombination of covalent or non-covalent bonds,1–3 one can conveniently impart self-healing properties to polymers. However, there is a trade-off between mechanical strength and self-healing performance. Low mechanical strength is typically obtained in order to maintain good healing ability. This is a significant barrier for self-healing materials to be widely used in practical applications in the future.4 Recently, a strategy for improving mechanical properties employed in self-healing polymers has been reported, via incorporation of noncovalent sacrificial bonds.5–9 Through reversible bond rupture, sacrificial bonds can efficiently dissipate large amounts of energy to toughen the polymeric materials. Various self-healable polymeric networks based on hydrogen bonds,5–7 metal–ligand interactions,8 and ionic bonds9 have been proposed. Bao and co-workers reported a supramolecular polymeric material with high stretchablity (17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000%) and toughness (∼30000 J m−2) that is cross-linked by quadruple hydrogen bonds.5 However, its mechanical stretchablity was decreased to only about 400% when a gold film electrode was deposited. Therefore, integrating diverse merits, such as conductivity, transparency, mild requirement of healing conditions, environmental tolerance and synchronous restoration of mechanical/electrical properties, into a single entity is highly desirable but remains a great challenge.
000%) and toughness (∼30000 J m−2) that is cross-linked by quadruple hydrogen bonds.5 However, its mechanical stretchablity was decreased to only about 400% when a gold film electrode was deposited. Therefore, integrating diverse merits, such as conductivity, transparency, mild requirement of healing conditions, environmental tolerance and synchronous restoration of mechanical/electrical properties, into a single entity is highly desirable but remains a great challenge.
In an attempt to improve the mechanical properties of a self-healable polymer, we have previously tried tuning interaction intensities between the imidazolium and counter ions of a series of imidazolium-based norbornene polymerized ionic liquid (PIL) blends. Homopolymer of PILs with different counter ions (CH3SO3−, CF3SO3−, CF3(CF2)3SO3−, FSI−, and Tf2N−) were first synthesized via ring-opening metathesis polymerization, respectively, and then PIL blends with excellent mechanical performance and self-healing capability could be achieved simultaneously via varying the ratio of PILs with high/low glass transition (Tg) and/or counter ion.10 To construct a promising mechanically robust self-healing material in a simple synthesis pathway, a bottom-up design by controlling the compositions, monomer sequences and connections at the molecular level is of crucial importance.
In nature, self-healable materials with high mechanical strength and toughness play an important role in biological systems. For example, mussel byssus is used for attaching to rocks.11 Superior mechanical performance of mussel byssus is derived from the gradual collagen composition change along the fiber.12,13 This gradient structure results in a continuous alteration of elasticity modulus, minimizing interfacial stresses, and improving energy dissipation, thereby increasing mechanical toughness.12,14 For synthetic polymer materials, a gradient copolymer is a unique class in which chemical composition changes continuously along the copolymer chain.15–17 Therefore, inspired by the gradient structure of mussel byssus, we recently designed and synthesized a series of novel self-healing copolymers consisting of gradient distributed large sterically hindered hard segments and soft units suspended with imidazolium. The hard domains containing a stiff bulky bridged phenyl unit not only ensure the elasticity, but also contribute greatly to the modulus and stiffness of copolymers. Imidazolium cations and bis(trifluoromethylsulfonyl)amide (Tf2N−) anions are selected for dynamic supramolecular interaction to construct a reversible physically cross-linked network which can realize self-healing function. Meanwhile, ion sites only embed in soft segments, which are favorable to dynamic ionic association and endow the copolymer with more flexible interactions and healing ability. This elaborate molecular design provides tunable mechanical performance and environmentally tolerated self-healing properties. Besides, the above gradient copolymer can be directly used for flexible strain sensors, exhibiting fast response, high reproducibility and excellent stability.
Results and discussion
Material synthesis and characterization
Ring-opening metathesis polymerization (ROMP) has been proven to be a powerful and broadly applicable method for synthesizing ionic polymer materials.18,19 In this work, a one-pot ROMP strategy was carried out to prepare gradient copolymers using the Grubbs third-generation catalyst, RuCl2(3-bromopyridine)2(H2IMes)(CHPh) (G3, H2IMes = N,N-dimesityl-4,5-dihydroimidazol-2-ylidene). Two monomers, 3-bicyclo[2.2.1]hept-5-en-2-ylmethyl-1-decyl-3H-imidazolium bis(trifluoromethylsulfonyl)amide (BDI) and exo-1,4,4a,9,9a,10-hexahydro-9,10(1′,2′)-benzeno-l,4-methanoanthracene (HBM), with distinct difference in reactivity are chosen to construct soft and hard sequences, respectively (Fig. 1). Through the classical Fineman–Ross method,20 the reactivity ratios of HBM (rHBM) and BDI (rBDI) were estimated to be 0.11 and 1.50, respectively (Fig. S10, ESI†). As a result, the rBDI × rHBM value is less than 1, indicating that the synthesized copolymers have gradient composition distributions. The gradient copolymers (GCPs) 1–4 with various HBM contents of 4.7, 11.1, 20, and 23 mol% were successfully synthesized. For comparison, four corresponding block copolymers (BCPs) 1–4 were prepared in the way of sequential feeding and one BDI homopolymer (termed as HP) was also synthesized.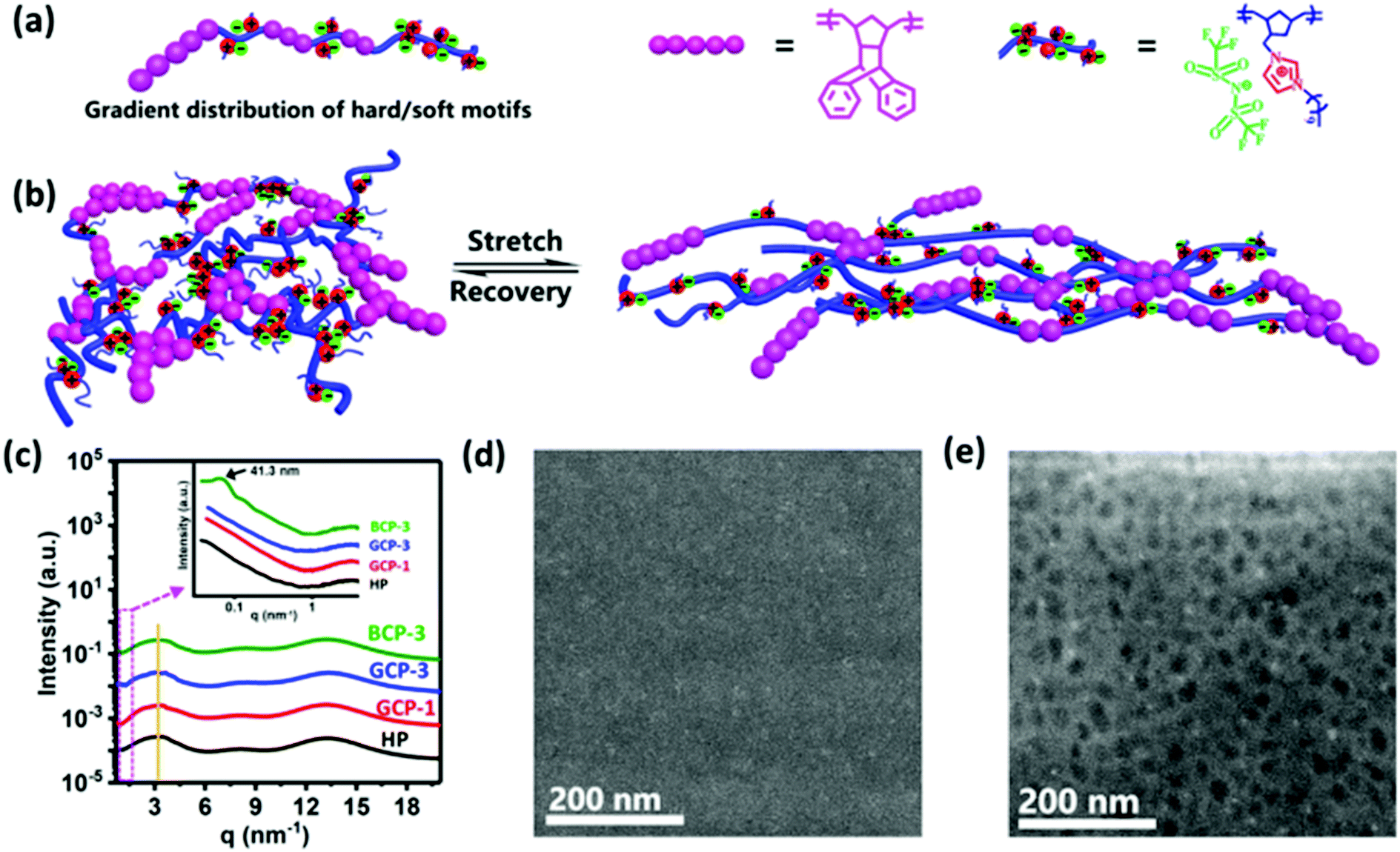 | ||
| Fig. 1 Design and structure analysis of gradient ionic copolymers that harmoniously combined high mechanical strength, toughness and self-healing properties. (a) Chemical structures of ionic copolymers with gradient-distributed hard (HBM)/soft (BDI) motifs. (b) The mechanism of gradient copolymers for elastic response, in which ionic aggregates serve as physical cross-linkings that reversibly break and reform during stretching to dissipate energy, toughening materials. (c) X-ray scattering patterns of HP, gradient and block copolymers at room temperature. Yellow solid line represents the Bragg spacing between ionic aggregates, which was calculated to be ∼2 nm. Inset: Magnified patterns of the corresponding ionic materials. Bragg spacing of ∼41.3 nm caused by hard motifs was observed in BCP-3, while other samples did not exhibit a phase interface. (d) High angle annular dark field-scanning transmission electron microscope (HAADF-STEM) image of GCP-3 without phase separation. (e) HAADF-STEM image of BCP-3 with a HBM motif size of ∼35 nm. Due to the greater incoherent scattering of electrons by heavy nuclei, the higher atomic numbers of fluorine, sulfur, nitrogen and oxygen (9, 16, 7 and 8 respectively) soft motifs compared to carbon and hydrogen (6 and 1 respectively) in the hard domain ensures that the soft motifs lead to brighter regions in a STEM-HAADF image. Conversely, the dark regions represent hard domains. | ||
The gradient copolymers were clearly characterized by 1H NMR where broad resonance signals in the range of 5.0–5.7 ppm are ascribed to olefinic protons in the ring-opened structure (Fig. S1–S8, ESI†). The molar ratios of BDI and HBM incorporated into the polymers agree well with the initial feed ratios by 1H NMR spectra analyses, indicating that the monomers were quantitatively converted into gradient copolymers. Furthermore, the two-dimensional nuclear Overhauser effect spectroscopy (NOESY) spectra of the gradient copolymers revealed spatial proximity (5 Å) among the protons of HBM and BDI units (Fig. S9, ESI†). This is different from that of a block copolymer analogue, which did not exhibit any signal or Overhauser data. According to the modified methodology developed by Matyjaszewski's group,20 we have successfully determined the molecular weights of the gradient copolymers by gel permeation chromatography (GPC) using N,N-dimethylformamide (DMF) containing 50 mM of LiBr as the eluent. High molecular weights (Mn = 89–98 × 103) and narrow molecular weight distributions (Mw/Mn < 1.4) are observed (Table S1, ESI†). As the HBM fraction is varied from 4.7 to 23 mol%, the glass transition temperature (Tg) increases from 27.9 to 63.0 °C according to dynamic mechanical analysis (Fig. S11 and Table S1, ESI†).
Gradient materials with a gradual change of composition do not possess a sharp interface as in block materials,21 as measured by small angle X-ray scattering (SAXS) and high angle annular dark field-scanning transmission electron microscopy (HAADF-STEM). None of the gradient copolymers or the homopolymer (HP) exhibited any scattering peaks by SAXS measurement (Fig. 1c and Fig. S14a, ESI†). For the block copolymers, mcirophase separation was observed (HBM unit >4.7 mol%). When the content of HBM sequence increases from 11.1 through 20 to 23 mol%, the domain spacing (d = 2π/q) increases from 30 through 41.3 to 90.2 nm. These distinct structural differences between gradient and block copolymers were also confirmed by HAADF-STEM. The GCP-3 image presented the shallow contrast of the gradual phase interfaces, indicating the uniform distribution of hard and soft motifs (Fig. 1d). By contrast, distinct phase interfaces with an average spherical size of ∼35 nm were observed for BCP-3 (Fig. 1e). The roughness and morphology of the materials were also tested by atomic force microscopy (AFM). The root-mean-square roughness (RMS) is 3.1 nm for GCP-3 and 1.95 nm for BCP-3 (Fig. S13, ESI†). GCP-3 forms continuous films without phase separation. However, for the BCP-3 film, the HBM hard domain is dispersed in the continuous soft matrix.
In the polymer matrix, imdazolium with Tf2N− counterions aggregates to form multiplets or clusters, which can be reformed in a short relaxation period after crack damage and is responsible for the self-healing process. To identify the presence of such ionic aggregates, X-ray scattering measurement was carried out. All the ionic polymers exhibited three scattering peaks at the same position regardless of the monomer sequence distribution and HBM content (Fig. 1c and Fig. S14b, ESI†): the peaks at 12–15, 6–9 and 2–4 nm−1 were assigned to the amorphous phase, correlation between the anions and Bragg spacing between ionic aggregates of the ionic polymers, respectively.22–24 The relative broad scattering peaks at 2–4 nm−1 implied lower and looser ionic associations,25 which shortens the ionic relaxation time, thereby promoting the self-healing process.
Mechanical performance
The mechanical properties of materials are usually influenced by molecular weight, monomer ratio and sequence distribution. First, the mechanical performance of the gradient copolymers with the same HBM content (20 mol%) but different molecular weight (Mn, ca. 50000, 100000, 200000, 400000 and 500000, respectively) was investigated. As observed, the mechanical strength reached the maximum when the molecular weight of the copolymer exceeded 100000 (Fig. S17, ESI†). Such high molecular weights were speculated to favor chain entanglement and maintain high mechanical strength. For gradient copolymers, the gradual composition transition enhances the miscibility of stiff and soft phases (Fig. 1d). Therefore, gradient materials are not susceptible to radial stress, which is the major advantage compared to block materials. Tensile stress–strain curves in Fig. 2a revealed that the stiffness (Young's modulus) and toughness of the gradient copolymers are superior to those of HP and those of the corresponding block copolymers with the same HBM content. No yielding point was observed in either the homopolymer or block copolymers, whereas the gradient copolymers exhibited a distinct yield phenomenon and their Young's modulus (determined from the strain <5%) rapidly increased from 22 to 286 MPa with increasing HBM content. However, the toughness of the gradient copolymers did not show a monotonous increase with increasing HBM content. The critical toughness reached a maximum (33.2 MJ m−3) at 23 mol% HBM unit. GCP-4 with the highest HBM incorporation presented a Young's modulus as high as 286 MPa while its toughness still remains up to 30 MJ m−3. To the best of our knowledge, such high concurrent values of Young's modulus and toughness have not yet been achieved for self-healing materials based on dynamic interactions (Fig. 2b and Table S4, ESI†).3,6,7,26–29 The significant improvement in mechanical properties mainly stems from the synergetic contribution of gradual distribution of stiff HBM domains and the physical cross-linking via dynamic ionic associations between the polymeric backbones. On the one hand, the gradient change of stiff HBM segments favors the stress transfer, alleviating stress concentrations and dissipating the energy at the interfaces, thereby improving mechanical properties.21,30 On the other hand, ionic groups easily aggregate into large clusters and contribute greatly to intermolecular binding capacity (Fig. 1b). During stretching, ionic groups on the stressed chains are detached from ionic aggregates to dissipate mechanical energy, enhancing the toughness and stretchability of copolymers. Moreover, some detached ionic groups probably reenter other ionic aggregates to further prevent slipping of polymeric networks.31 Therefore, both high strength and toughness of materials can be simultaneously reached, which was a challenge in the previous reports.
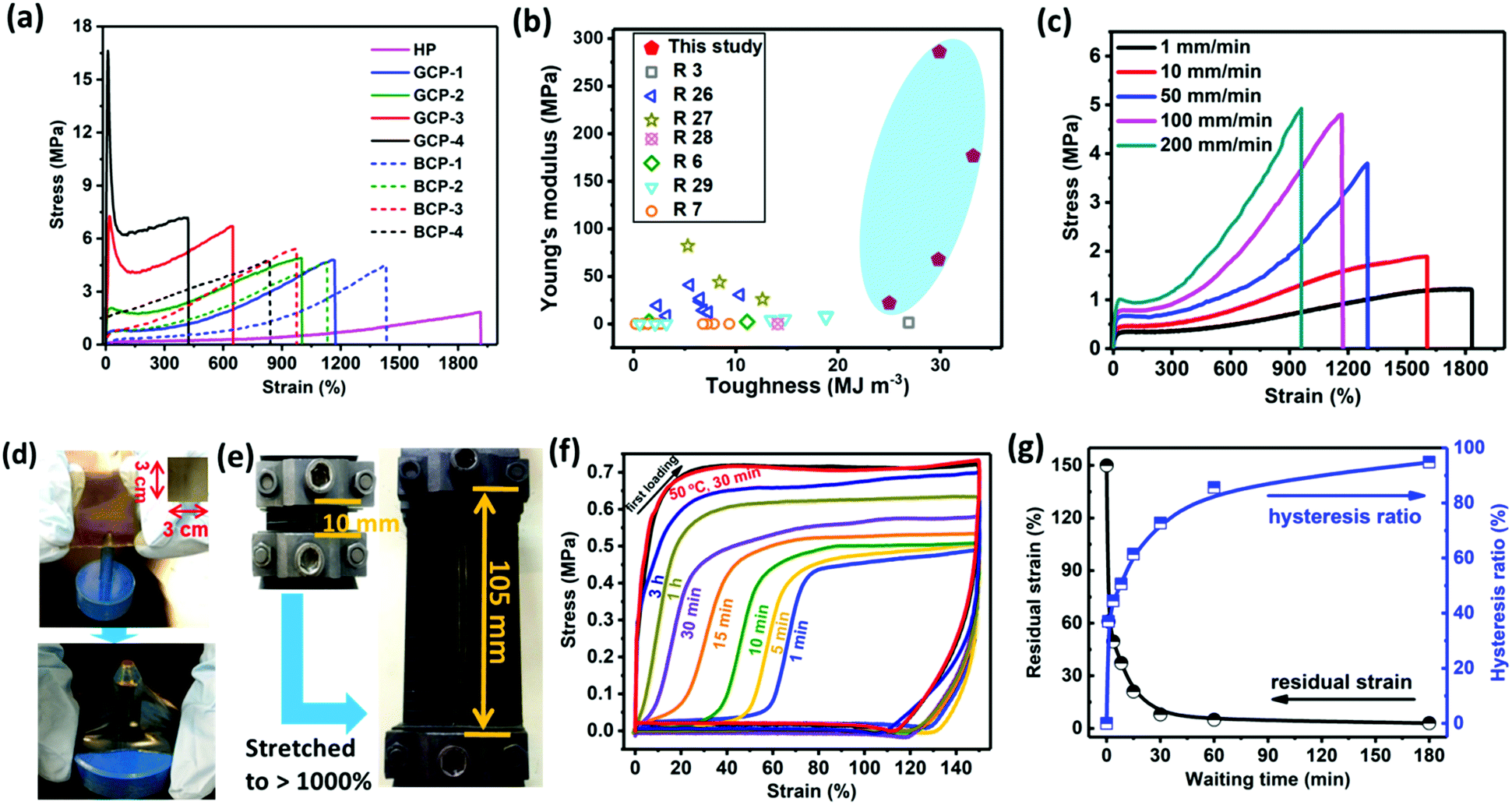 | ||
| Fig. 2 The mechanical performance of the ionic copolymers. (a) Stress–strain curves of ionic homopolymer, and gradient and block copolymers (strain rate = 100 mm mm−1 min−1). (b) A comparison of Young's modulus and toughness for this work with previously developed robust and tough self-healing materials. (c) Stress–strain curves of GCP-1 at different speeds. (d) The square GCP-1 sample (the size was 3 × 3 × 0.02 cm3) withstood a large uneven deformation without fracture after a metal cylindrical cut-off knife (diameter = 0.5 cm) poked the center of the sample film. (e) The GCP-1 containing a notch was stretched to 10.5 times longer than its initial length. (f) Recovery of GCP-1 for different waiting times performed by cyclic tensile tests. (g) Waiting time dependence of the residual strain and hysteresis ratio (area ratio of the second hysteresis loop to the first) for the GCP-1 sample. | ||
The stretching speed-dependent mechanical behavior indicated that both the Young's modulus and fracture stress of GCP-1 increased with increasing stretching rate, while the stretchability decreased (Fig. 2c). At low deformation rates (1 and 10 mm mm−1 min−1), the dynamic dissociation and recombination of ionic groups are capable of transferring stress and dissipating energy, allowing ionic copolymers to have high elongation. This behavior is analogous to that of typical covalent cross-linked elastomers. When the stretching rate exceeds 50 mm mm−1 min−1, there is no sufficient time to reform ionic aggregates after their dissociation within the timescale of the tensile deformation. In this case, ionic aggregates serve as a strong cross-link to enhance fracture stress.31 Therefore, the polymeric network became stiff and a noticeable yield point was also observed.
A great amount of ionic physical cross-linking and the gradual change in composition permit gradient copolymers to dissipate stress from all directions. In Fig. 2d, inhomogeneous deformation of GCP-1 film without any fracture was observed after the sample was poked by a metal cylindrical cut-off knife. Surprisingly, the sample could withstand repeated jabbing (Movie S1, ESI†). Besides, the gradient copolymers were also proved to be notch-insensitive. We cut out a notch in the center of GCP-1 and subsequently stretched the sample, a dramatically blunted notch was presented, and the notched sample could be stretched up to a critical strain of 1050% at a deformation rate of 100 mm mm−1 min−1, demonstrating exceptional toughness of the specimen (Fig. 2e and Movie S2, ESI†).
The gradient copolymers are highly fatigue resistant. After the first loading–unloading cycle, GCP-1 exhibited prominent yielding and hysteresis, indicating a large amount of energy dissipation through ionic disassociations (Fig. 2f). The emergence of a significant residual strain after unloading suggested a plastic deformation of the sample. The residual strain disappeared and the hysteresis ratio almost completely recovered after 3 h of waiting time. Heating at 50 °C greatly promoted self-recovery of the copolymers because of the accelerated dynamic ionic rearrangement and diffusion of polymeric segments. Full mechanical recovery demonstrated that the deformation was caused by disassociation of ionic aggregates and no breaking of main chains occurred. The waiting time-dependent hysteresis ratio, estimated from the hysteresis area change and the residual strain, is shown in Fig. 2g. During unloading, the mechanical recovery involves elastic contraction of the main chain and rearrangement of temporarily reformed ionic aggregates, which are competing with each other. When the deformation exceeds 110%, elastic contraction is dominant leading to a quick mechanical recovery, whereas at a small deformation (<110%), the reformed ionic bonds need time to re-organize, which slows down the recovery of the main chain to its equilibrium state. A complete recovery was observed at a larger loading strain of 1000% (Fig. S15, ESI†), approaching the fracture strain of 1150–1200%. This result suggests that there is no chain sliding until fracture because of strong ionic supramolecular interactions between copolymer chains.
Self-healing ability
The scratch self-healing phenomenon of GCP-1 is detected by optical microscopy at 25 °C (Fig. 3a). A complete self-healing was observed as the scar gradually disappeared with time and became almost invisible after 6 h. Because of dynamic ionic interactions, GCP-1 cut into small species could even be reclaimed by compression-molding at 1 MPa for 1 h at room temperature (Fig. 3b). The reprocessed sample displayed the same mechanical and self-repair properties as the original specimen. More remarkably, the cycle of cutting and reclaiming could be repeated hundreds of times, while the mechanical and self-repair properties remained unchanged. Additionally, the gradient copolymers exhibited high transparency such that a logo was clearly visible through the polymer film (Fig. 3b). Good optical transparency provides these gradient copolymers with diverse promising applications in touch screens, wearable sensors, smart windows, displays, etc., to improve the user experience.32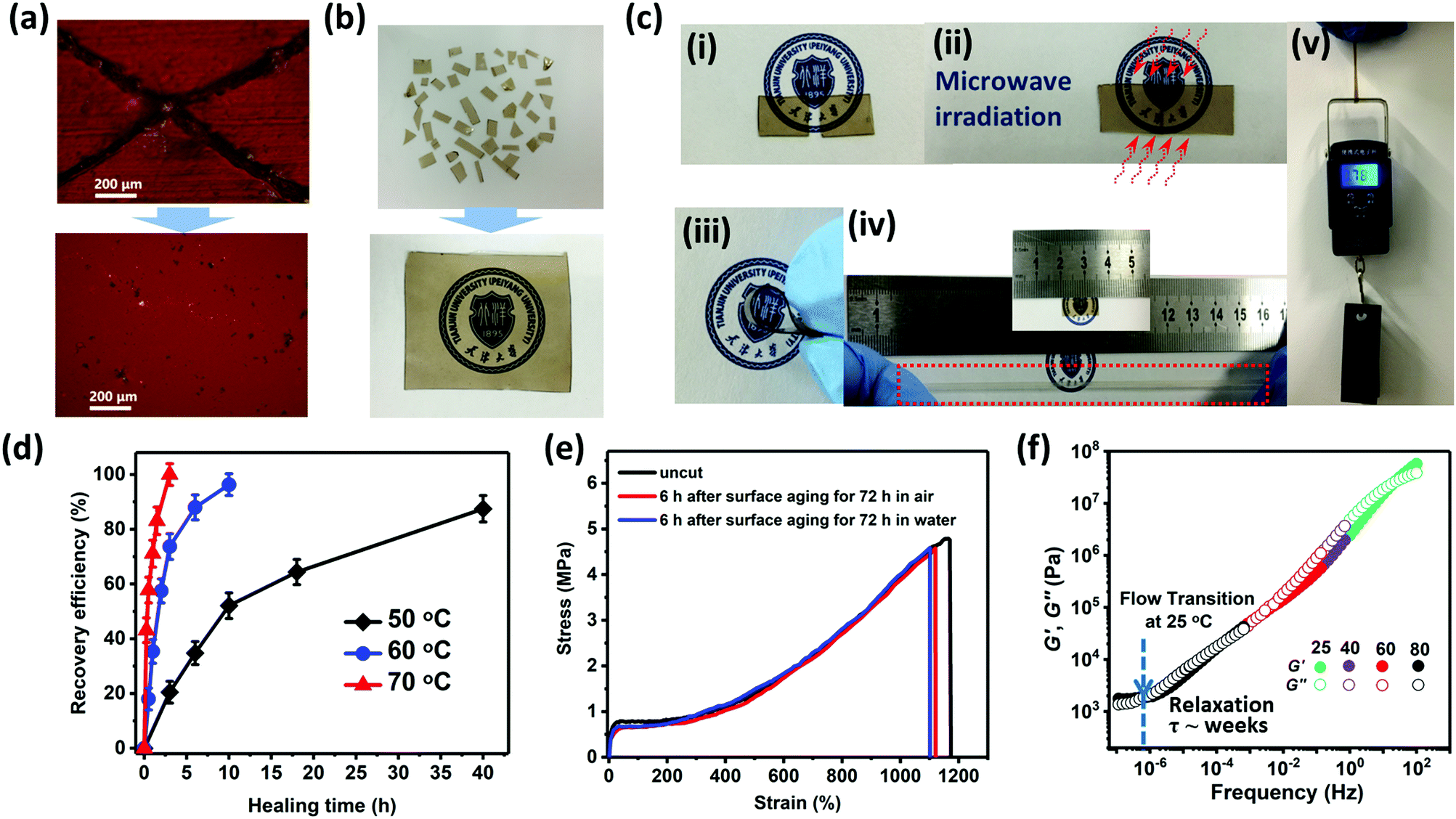 | ||
| Fig. 3 The self-healing capability of ionic copolymers. (a) Optical microscopy images of the X-shaped scratch on the GCP-1 film before and after healing for 6 h at room temperature. (b) Reclaiming the GCP-1 copolymer via compression molding at 1 MPa for 1 h at room temperature. (c) Photographs of the healed GCP-1 copolymer sample with microwave irradiation: (i) cutting; (ii) contacted and irradiated for 50 s; (iii) bending the healed sample; (iv) stretching the healed sample and (v) loaded with 0.785 kg after irradiation. (d) Plots of the recovery efficiency of fracture strain as a function of healing time for GCP-3 at different healing temperatures. (e) Stress–strain curves of the original and healed GCP-1 samples under different conditions. (f) Master curves for GCP-1 at a reference temperature of 25 °C, prepared by the time-temperature superposition treatment of storage modulus (G′) and loss modulus (G′′) values obtained at 25, 40, 60, and 80 °C in frequency-dispersion tests [frequency (ω) = 628 to 6.28 × 10−3 rad s−1] at an applied strain of 0.05 or 0.1%. | ||
To comprehend the detailed healing process, all the cut specimens have been kept in contact for various durations, followed by tensile tests. The stress–strain curves of the repaired samples overlapped with that of the original uncut one (Fig. S18, ESI†), indicating that as the contact time extended, the fracture stress and fracture strain were continuously repaired; however, the Young's modulus and yield stress have already been completely restored after the healed strain exceeded the yield strain. In sharp contrast, most of the previously reported self-healing materials could not reach their original Young's modulus or yield stress unless complete recovery was achieved.33–36 A probable explanation is that ionic aggregates rapidly re-form once the cut samples are contacted, and the interface interactions dominated by ionic associations are strong enough to resist the plastically yielding deformation. Compared to GCP-1 with BCP-1, the self-healing efficiency of the gradient copolymer was superior to that of the block ones (Fig. S18, ESI†). It was speculated that the absence of ionic groups in the self-assembled HBM domain of the block copolymers impedes their self-repair.
The occurrence of healing is explained by the rearrangement of ionic aggregates and the interdiffusion of polymer chains over the surface of the crack. At low HBM content (<5.4%), self-healing of the gradient copolymers was achieved at room temperature in the absence of any external stimuli including plasticizer, solvent or healing agent. As the content of HBM further increased, the rigidity of the polymer chain enhanced but the mobility of the chain decreased. As a result, ion hopping, i.e. ions on the polymer chain hopping from one aggregation to another was restricted, the repair process accordingly slowed down. Higher repair temperature is required to speed up the healing of samples. As observed, GCP-3 took over 40 h to recover its fracture strain to around 90% at 50 °C (Fig. 3d). At 60 °C, a comparable recovery took much less time of about 10 h. Further raising the healing temperature to 70 °C, complete recovery was achieved within only 3 h. The healing process of GCP-3 is remarkably accelerated approximately 13 times just through elevating the temperature by 20 °C. GCP-4 with the highest mechanical strength also possessed excellent self-healing performance. The complete recovery period was shortened from 18 to 4 h through elevating the temperature by 20 °C (from 70 to 90 °C) (Fig. S18, ESI†). The relaxation time (τ) of GCP-1 for the flow transition was evaluated in the range of 107 seconds (∼months), much longer than its complete repair time (∼6 h) at 25 °C (Fig. 3f). This dramatic contrast indicated that high healing efficiency of gradient copolymers is ascribed to the rapid and dynamic ionic associations rather than the flow relaxation of primary chains.
Microwave radiation is also an effective way to enhance recovery efficiency of gradient copolymers. The diffusion of the molecules between two sides of the crack is stimulated by microwaves, which will create connection points that restore the continuity of the material.37 The time for complete healing by microwaves was shortened to 30 s for HP. Even for GCP-4, a complete recovery could be achieved within 150 s (Fig. S17, ESI†). After 50 s of microwave irradiation, the GCP-1 specimen possessed a steady interface capable of sustaining various mechanical forces, including bending, stretching up to a maximum extension and loading up to 0.785 kg, which is more than 4200 times of its own weight (Fig. 3c).
For self-healing materials based on hydrogen bonding interaction, both formation of co-facial interaction partners and combination with water in the environment will destroy the bridges of hydrogen bonding across the fracture interfaces, resulting in the degradation of self-healing capability.34,35 However, the gradient copolymers presented very tenacious mending performance, and their self-healing ability was not significantly affected by moisture or even water (Fig. 3e). Furthermore, because of the hydrophobicity of the counterion (Tf2N−), the repair of the GCP-1 sample in water could be repeated lots of times (Movie S3, ESI†). Besides, the healing process of the separated GCP-1 samples was further tested in artificial sweat. After immersing in artificial sweat for 6 h, the healing efficiency of fracture strain still reached up to 87.5%, very close to the result of healing in air (∼93%).
The applications of gradient copolymers assembled for wearable strain sensors
To obtain self-healable electronic conductors, a common approach is to incorporate conductive fillers into a self-healing network. However, this method always suffers from complex process, sacrificial mechanical performance, and low elastic deformation to curvy or stretchy surfaces.38–40 By contrast, the gradient copolymers consisting of large amounts of ionic groups provide inherent ion conductivity, and can act as intrinsically self-healing wearable sensors without any extra filler incorporation. Ionic conductivity assessed by the impedance for each sample obeys a typical Vogel–Fulcher–Tammann dependence (Fig. S21, ESI†). Here, GCP-1 was selected as a flexible sensor. In addition to the high ionic conductivity (1.88 × 10−4 S m−1 at 30 °C) of GCP-1 and fast self-healing at room temperature, an important reason was that its mechanical toughness is very close to the nature of human skin.41Fig. 4a presents the variation in the current of the sensor before and after healing. After cutting half of the sample, the electrical current dropped by gradient. When the sample was completely cut into two halves, no current flowed through the circuit. The current generated on the fracture surface was connected, but it only partially recovered. The fully healed mechanical performance was simultaneously accompanied by the complete recovery of current at room temperature during 6 h. It is worth noting that the current of the copolymers only displayed tiny variation in the case of long-term voltage application, indicating that the electrical property of the specimen is very stable (Fig. S20, ESI†).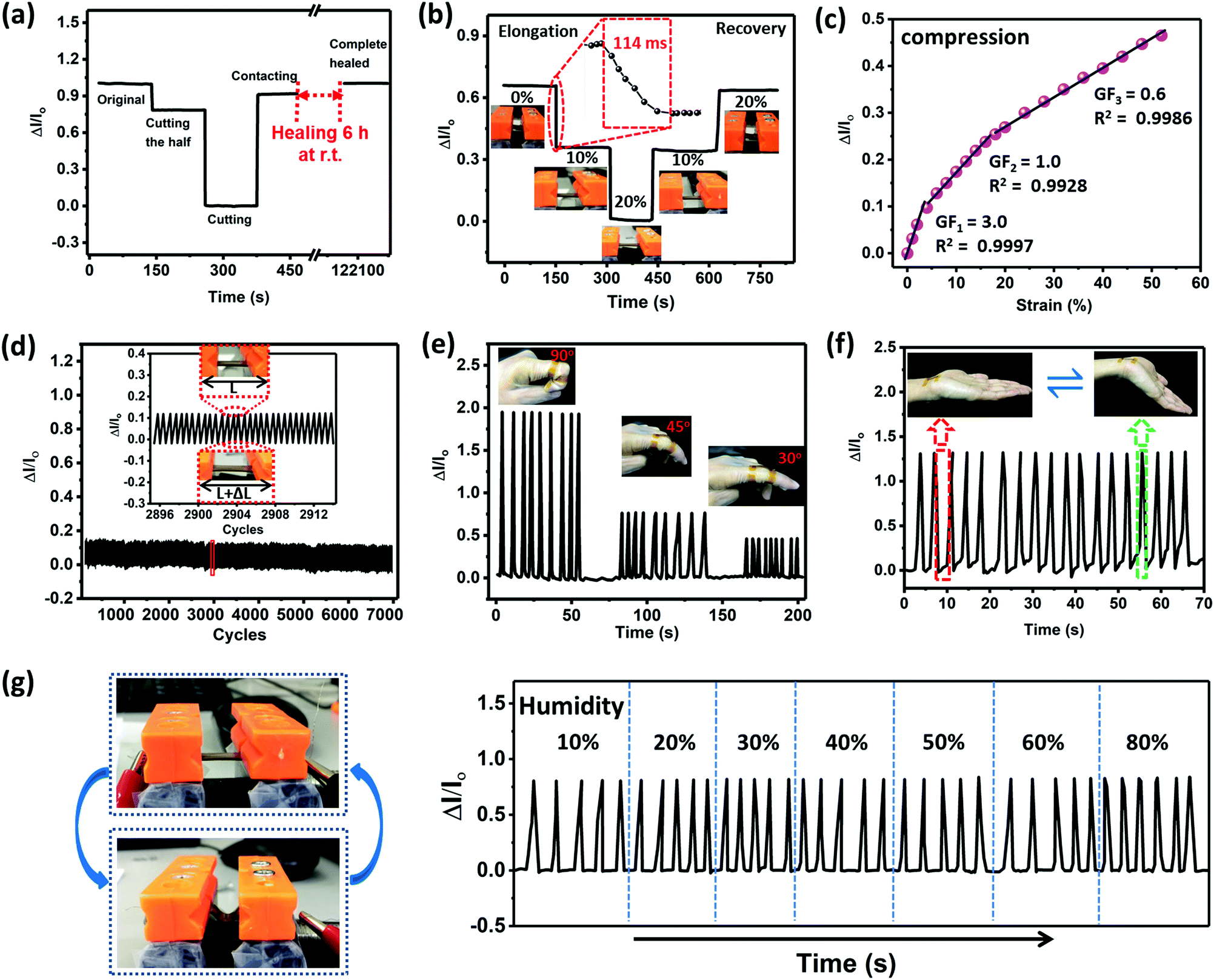 | ||
| Fig. 4 The properties and applications of gradient copolymers assembled for wearable strain sensors. (a) Current changes of the sensor for cutting and healing of GCP-1 (r.t. = room temperature). (b) The relative current as a function of time as a loading–unloading cycle of the GCP-1 at different strains. Inset: Enlarged figure showing the response time of the sensor upon applying a tensile strain from 0 to 10%. (c) The relative current as a function of compressive strain and the linear fittings. (d) The relative current variation under cyclic stretching from 0 to 5% with a frequency of 1.5 Hz over 7000 cycles. (e) Current variation signal measured by using GCP-1 polymer film for finger bending motion with different angles. (f) The relative current change of GCP-1 polymer film for pressing wrist movement. (g) The cyclic bending movement of the sample (strain of 5%) could be monitored under a wide humidity range from 10 to 80%. | ||
The relative current of the sample showed a downward step-like trend during a step-by-step loading–unloading cycle because of the increase in resistance of the stretched conductor (Fig. 4b). The key performance parameters of recently reported strain sensors are compared in Table S5 (ESI†). The response time of the sensor to strain (from 0 to 10%) was only 114 ms, which is comparable to that of human skin (∼100 ms),42 indicating that the sensor can immediately monitor the applied strain without evident hysteresis. In contrast, the current showed an increase with the raise of compressive strain (Fig. 4c). This observation implied the formation of ion transport channels during compression. The gauge factor (GF), a representative parameter for accessing strain sensitivity, was measured to be 3.0 within 5% compressive strain, 1.0 for 5–20%, and 0.6 for compressive strain exceeding 20%, demonstrating the high sensitivity of the soft sensor in a wide sensing range. Fig. 4d presents the variation of current during cyclical stretching from ε = 0% to ε = 5% with a frequency of 1.5 Hz for 7000 cycles. The current changed periodically, implying reproducible and reliable performance of the sensor.
The ionic sensor was fixed on a finger by conductive tapes as a wearable device to detect the joint motion in real-time. When the finger was bent, an increase of current was observed (Fig. 4e). This phenomenon is different from those of the previously reported strain sensors.43–47 The motion of a bending finger involves two opposite movements: the side of the sample attached to the finger is contracted while the other side is stretched. When contraction is dominant, current increases. Furthermore, as the finger bent to different angles from 30 through 45 to 90°, the vibration amplitude of the current was obviously enhanced, exhibiting excellent reliable and highly sensitive sensing response to the bent finger. Similarly, we could monitor the muscle movement of a bent wrist through the changes in current for a long time by attaching the ionic copolymer sensor onto the wrist (Fig. 4f), implying the reproducibility of the strain sensor. Different from the hydrogels, no small molecular component escapes from the matrix of the gradient copolymer during usage, and the strain sensor is able to be used under extra-dry or humid conditions where traditional conductive hydrogel sensors fail to work properly. For example, the sensor responded stably to cyclic mechanical bending motion in a wide humidity range from 10 to 80% (Fig. 4g).
Conclusions
In summary, we have prepared an array of gradient copolymers containing stiff bulky bridged phenyl units as hard motifs and soft imidazolium by a one-pot ROMP. Compared to block copolymers and an ionic homopolymer, the synergistic hard/soft gradient structures and dynamic ionic interactions imparted smooth stress-transfer and recoverable energy dissipation to the copolymer, leading to high mechanical strength, toughness and stretchability. Self-healing performance caused by dynamic dissociation and rearrangement of ionic aggregates can be accelerated by both thermal stimuli and microwave irradiation. Because of the hydrophobic nature of ionic groups, a tenacious reparability has been demonstrated with tolerance to moisture, water and artificial sweat. Furthermore, an intrinsically flexible strain sensor made by the gradient copolymer can accurately monitor the changes of joint motions in either dry or humid environments in real-time. We anticipate that the gradient design of hard and soft segments will allow for a variety of mechanically robust self-healable networks to be investigated using different interactions in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the support of the National Natural Science Foundation of China (No. U1510124 and 21690071). This research was also supported by the Foundation of the State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering (Grant No. 2018-K05). We thank the Research Center of Analysis and Test of Tianjin University of Materials Science and Engineering for help on the material characterization. We also thank Dr Jinyou Lin and Prof. Fenggang Bian from beamline BL16B of Shanghai Synchrotron Radiation Facility (SSRF) for the help with synchrotron X-ray measurements.Notes and references
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334 CrossRef CAS PubMed.
- S.-M. Kim, H. Jeon, S.-H. Shin, S.-A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2017, 30, 1705145 CrossRef PubMed.
- J.-L. Wietor and R. P. Sijbesma, Angew. Chem., Int. Ed., 2008, 47, 8161 CrossRef CAS PubMed.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H.-C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. H. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280 CrossRef CAS PubMed.
- J. A. Neal, D. Mozhdehi and Z. Guan, J. Am. Chem. Soc., 2015, 137, 4846 CrossRef CAS PubMed.
- Q. Chen, L. Zhu, H. Chen, H. Yan, L. Huang, J. Yang and J. Zheng, Adv. Funct. Mater., 2015, 25, 1598 CrossRef CAS.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, Nat. Chem., 2016, 8, 618 CrossRef CAS PubMed.
- I. Jeon, J. Cui, W. R. K. Illeperuma, J. Aizenberg and J. J. Vlassak, Adv. Mater., 2016, 28, 4678 CrossRef CAS PubMed.
- J. Cui, F.-M. Nie, J.-X. Yang, L. Pan, Z. Ma and Y.-S. Li, J. Mater. Chem. A, 2017, 5, 25220 RSC.
- J. H. Waite, Int. J. Adhes. Adhes., 1987, 7, 9 CrossRef CAS.
- J. H. Waite, X.-X. Qin and K. J. Coyne, Matrix Biol., 1998, 17, 93 CrossRef CAS PubMed.
- T. Scheibel, Fibrous Proteins, Landes Bioscience, 2008 Search PubMed.
- J. H. Waite, E. Vaccaro, C. Sun and J. M. Lucas, Philos. Trans. R. Soc., B, 2002, 357, 143 CrossRef CAS PubMed.
- W. Steinhauer, R. Hoogenboom, H. Keul and M. Moeller, Macromolecules, 2013, 46, 1447 CrossRef CAS.
- K. O. Kim and T.-L. Choi, Macromolecules, 2013, 46, 5905 CrossRef CAS.
- Y. Guo, X. Gao and Y. Luo, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 860 CrossRef CAS.
- E. F. Wiesenauer, P. T. Nguyen, B. S. Newell, T. S. Bailey, R. D. Noble and D. L. Gin, Soft Matter, 2013, 9, 7923 RSC.
- T. Suga, M. Sakata, K. Aoki and H. Nishide, ACS Macro Lett., 2014, 3, 703 CrossRef CAS.
- H. He, M. Zhong, B. Adzima, D. Luebke, H. Nulwala and K. Matyjaszewski, J. Am. Chem. Soc., 2013, 135, 4227 CrossRef CAS PubMed.
- K. U. Claussen, T. Scheibel, H.-W. Schmidt and R. Giesa, Macromol. Mater. Eng., 2012, 297, 938 CrossRef CAS.
- H. V. R. Annapureddy, H. K. Kashyap, P. M. De Biase and C. J. Margulis, J. Phys. Chem. B, 2010, 114, 16838 CrossRef CAS PubMed.
- D. S.-D. la Cruz, M. D. Green, Y. Ye, Y. A. Elabd, T. E. Long and K. I. Winey, J. Polym. Sci., Part B: Polym. Phys., 2011, 50, 338 CrossRef.
- W. Wang, W. Liu, G. J. Tudryn, R. H. Colby and K. I. Winey, Macromolecules, 2010, 43, 4223 CrossRef CAS.
- G. J. Tudryn, W. Liu, S.-W. Wang and R. H. Colby, Macromolecules, 2011, 44, 3572 CrossRef CAS.
- D. Mozhdehi, S. Ayala, O. R. Cromwell and Z. Guan, J. Am. Chem. Soc., 2014, 136, 16128 CrossRef CAS PubMed.
- G. A. Williams, R. Ishige, O. R. Cromwell, J. Chung, A. Takahara and Z. Guan, Adv. Mater., 2015, 27, 3934 CrossRef CAS PubMed.
- P. Wang, G. Deng, L. Zhou, Z. Li and Y. Chen, ACS Macro Lett., 2017, 6, 881 CrossRef CAS.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Adv. Mater., 2015, 27, 2722 CrossRef CAS PubMed.
- T. A. Huy, L. H. Hai, R. Adhikari, R. Weidisch, G. H. Michler and K. Knoll, Polymer, 2003, 44, 1237 CrossRef CAS.
- Y. Miwa, J. Kurachi, Y. Kohbara and S. Kutsumizu, Communications Chemistry, 2018, 1, 5 CrossRef.
- C. S. Luo, P. Wan, H. Yang, S. A. A. Shah and X. Chen, Adv. Funct. Mater., 2017, 27, 1606339 CrossRef.
- J.-C. Lai, J.-F. Mei, X.-Y. Jia, C.-H. Li, X.-Z. You and Z. Bao, Adv. Mater., 2016, 28, 8277 CrossRef CAS PubMed.
- J. Hentschel, A. M. Kushner, J. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2012, 51, 10561 CrossRef CAS PubMed.
- C. Bao, Y.-J. Jiang, H. Zhang, X. Lu and J. Sun, Adv. Funct. Mater., 2018, 28, 1800560 CrossRef.
- Y. Chen and Z. Guan, Chem. Commun., 2014, 50, 10868 RSC.
- J. Gallego, M. A. del Val, V. Contreras and A. Páez, Constr. Build. Mater., 2013, 42, 1 CrossRef.
- B. C. K. Tee, C. Wang, R. Allen and Z. Bao, Nat. Nanotechnol., 2012, 7, 825 CrossRef CAS PubMed.
- J. Kang, D. Son, G.-J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. H. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef PubMed.
- C. F. Guo, Q. Liu, G. Wang, Y. Wang, Z. Shi, Z. Suo, C.-W. Chu and Z. Ren, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12332 CrossRef CAS PubMed.
- B. Yu, S.-Y. Kang, A. Akthakul, N. Ramadurai, M. Pilkenton, A. Patel, A. Nashat, D. G. Anderson, F. H. Sakamoto, B. A. Gilchrest, R. R. Anderson and R. Langer, Nat. Mater., 2016, 15, 911 CrossRef CAS PubMed.
- R. S. Johansson and J. R. Flanagan, Nat. Rev. Neurosci., 2009, 10, 345 CrossRef CAS PubMed.
- T. Wang, Y. Zhang, Q. Liu, W. Cheng, X. Wang, L. Pan, B. Xu and H. Xu, Adv. Funct. Mater., 2017, 28, 1705551 CrossRef.
- L. Li, Y. Bai, L. Li, S. Wang and T. Zhang, Adv. Mater., 2017, 29, 1702517 CrossRef PubMed.
- L. Han, K. Liu, M. Wang, K. Wang, L. Fang, H. Chen, J. Zhou and X. Lu, Adv. Funct. Mater., 2017, 28, 1704195 CrossRef.
- S. Gong, W. Schwalb, Y. Wang, Y. Chen, Y. Tang, J. Si, B. Shirinzadeh and W. Cheng, Nat. Commun., 2014, 5, 3132 CrossRef PubMed.
- C. Wang, X. Li, E. Gao, M. Jian, K. Xia, Q. Wang, Z. Xu, T. Ren and Y. Zhang, Adv. Mater., 2016, 28, 6640 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic materials and instruments, synthetic methods, reactivity ratio measurements, NMR, NOESY, DMA, TGA, XRD, mechanical properties, self-healing performance and ion conductivity. See DOI: 10.1039/c8qm00592c |
| This journal is © the Partner Organisations 2019 |