
A carbon quantum dot and rhodamine-based ratiometric fluorescent complex for the recognition of histidine in aqueous systems†
Harupjit
Singh
a,
Jagpreet Singh
Sidhu
b,
Dhiraj Kumar
Mahajan
 c and
Narinder
Singh
*b
c and
Narinder
Singh
*b
aCenter for Biomedical Engineering, Indian Institute of Technology Ropar, Punjab 140001, India
bDepartment of Chemistry, Indian Institute of Technology Ropar, Punjab 140001, India. E-mail: nsingh@iitrpr.ac.in
cDepartment of Mechanical Engineering, Indian Institute of Technology Ropar, Punjab 140001, India
First published on 10th January 2019
Abstract
Histidine is an essential α-amino acid that plays a crucial role in tissue development and helps in the transmission of metallic ions during biological events. However, an abnormal level of histidine in the body is associated with various physiological conditions such as arthritis, liver cirrhosis, kidney diseases, and asthma. Herein, a unique ratiometric fluorescence sensing system has been developed for the recognition of histidine. The sensing system was developed using carbon quantum dots (CQDs) as an energy donor and a rhodamine 6G derivative (HS30) as an energy acceptor unit. Interestingly, upon the addition of Fe(III) into the mixture of CQDs and HS30, the phenomenon of fluorescence resonance energy transfer (FRET) was observed when excited at 350 nm. The emergence of a strong emission peak at 551 nm on the addition of Fe(III) suggested the formation of a ratiometric fluorescent complex “CQDs–Fe–HS30”. The ratiometric behavior of “CQDs–Fe–HS30” was studied by monitoring fluorescence emissions at 425 nm and 551 nm with an excitation wavelength of 350 nm. Furthermore, “CQDs–Fe–HS30” was employed for the recognition of histidine in an aqueous system. Due to the high affinity of histidine to Fe(III), the addition of histidine to an aqueous solution of “CQDs–Fe–HS30” resulted in the displacement of the Fe(III) cation from the complex, and the simultaneous quenching and enhancement of the emission peaks at 551 nm and 425 nm, respectively, was observed. The developed sensing system was successfully employed for a histidine recovery experiment in human urine samples with satisfactory results. Furthermore, the mixture of CQDs and HS30 was successfully utilized to implement an inhibit logic gate with Fe(III) and histidine as inputs and emission at 551 nm as output.
Introduction
Histidine is an indispensable α-amino acid that plays a vital role in the biosynthesis of various proteins.1 The structure of histidine has a highly reactive imidazole functional group that interacts with transition metal ions and controls their transmission in biological bases.2,3 The expression of a low level of histidine is associated with rheumatoid arthritis, liver cirrhosis, and pulmonary dysfunctions, whereas overexpression is linked with cancer, kidney disorders, and Alzheimer's disease.4–9 Therefore, it is critical to develop a simple, highly selective and sensitive method for the assessment of histidine expression for the diagnosis of diseases.Advanced techniques such as gas chromatography and high-performance liquid chromatography are used along with UV-visible or fluorescence spectroscopy for histidine detection.10 However, the typical limitations of these methods are the requirements for sophisticated instrumentation, trained instrument operators, and well-equipped laboratories. Other techniques used for the determination of histidine are electrochemical sensing,11,12 colorimetric sensing,13 and fluorescence spectroscopy.14,15 Amongst all these techniques fluorescent spectroscopy has great advantages concerning its ease of handling, high sensitivity, high throughput, and real-time detection. Therefore, a wide range of single emission point fluorescent probes based on metal complexes, porphyrins, crown ethers, peptides, and polymers has been developed for the selective detection of histidine.11,13–18 However, single emission point fluorescence measurements are severely influenced by external factors such as temperature, the source of excitation, concentration, and the solvent system.19 Consequently, to circumvent these undesirable effects, the focus has shifted to ratiometric sensing. Ratiometric sensing employs the phenomenon of fluorescence resonance energy transfer (FRET) for precise and quantitative analysis under complex conditions. It involves the measurement of fluorescence emission from both interconnected donor and acceptor molecules, followed by a determination of the ratio of the intensities.19–21 Thus, ratiometric measurement overcomes the limitations of a single emission wavelength system and provides higher precision to the analysis.
Recently, CQDs and their composites have been successfully utilized for the sensing of various cations,22–24 anions25–27 and biomolecules.28–31 CQDs possess unique and attractive properties, such as biocompatibility, greater aqueous stability, low toxicity, tuneable emission, and better photostability, which make them ideal candidates for sensing and imaging applications.26,30,32–35 The tuneable emission property of CQDs is beneficial for achieving an effective overlap between the emission spectra of the donor and the absorption spectra of the acceptor to realize efficient FRET. Therefore, for ratiometric sensing of histidine, we preferred CQDs as an energy-donating unit whereas the rhodamine 6G derived “ON–OFF” fluorescent probe HS30 has been utilized as an energy-accepting moiety. HS30 works on the phenomenon of the structural modulation of rhodamine derivatives between the spirolactam form (non-fluorescent) and the open ring form (fluorescent).36 Since Fe(III) shows an affinity to the oxygen atoms of the carboxylic groups present on the surface of CQDs31,37,38 and induces ring opening in rhodamine derivatives,36,39–41 Fe(III) was utilized as a linking unit. The Fe(III) cation not only brought the two fluorophores closer but also changed HS30 from the non-fluorescent form to the fluorescent form and thus resulted in the formation of a FRET system. However, the addition of histidine to an aqueous solution of “CQDs–Fe–HS30” led to the displacement of Fe(III), which resulted in the cancellation of FRET. Therefore, herein, we have developed an effective system for the ratiometric sensing of histidine using a physical mixture of carbon quantum dots (CQDs) and rhodamine 6G derivative (HS30).
Results and discussion
Synthesis of CQDs and HS30
For the synthesis of carbon quantum dots, pyrolysis of citric acid and ethylenediamine was carried out at 180 °C for 30 min as per the previously reported procedure.32 To understand their material properties, CQDs were characterized using powder X-Ray Diffraction (XRD), Transmission Electron Microscopy (TEM) and Dynamic Light Scattering techniques. The uniform dispersion of particles in solution form was observed from the TEM image (Fig. S1, ESI†). The hydrodynamic size was determined using the dynamic light scattering technique, and the average particle diameter was found to be 8 ± 1.2 nm (Fig. S2, ESI†). The XRD pattern exhibited a peak broadening positioned at around 18° (d = 0.47 nm), revealing the amorphous nature of the CQDs (Fig. S3, ESI†). The optical properties of the CQDs were studied using UV-vis absorption spectrophotometry, and the spectrum revealed an absorption band at 350 nm corresponding to different transition states (Fig. 1A).34 A photoluminescence experiment was carried out to confirm the tuneable excitation behavior of the CQDs. Excitation-dependent emission was observed when an aqueous solution of CQDs was excited from 320 nm to 480 nm. As the excitation wavelength was varied, a red shift in emission maxima was observed which is a characteristic feature of the CQDs (Fig. 1B). Thus, CQDs having the characteristic feature of “excitation-dependent emissions” were successfully characterized. The presence of the surface groups was determined using FTIR and the spectrum showed a broad stretching in the range of 2800–3400 cm−1 that signified the presence of –OH and –NH2 groups. Further, an intense FTIR stretch at 1695 cm−1 was due to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of carboxylic acid, and this confirmed that the surface of the CQDs was functionalized with a –COOH functional group (Fig. 1C).
O group of carboxylic acid, and this confirmed that the surface of the CQDs was functionalized with a –COOH functional group (Fig. 1C).
 | ||
| Fig. 1 (A) The UV-vis absorption spectrum of synthesized CQDs shows an absorption band at 350 nm corresponding to different transition states.34 (B) Fluorescence emission spectra of CQDs at different excitation wavelengths varying from 320 to 480 nm. (C) FTIR spectrum of the CQDs. | ||
HS30 was synthesized through the condensation reaction of rhodamine 6G and 1,3-bis(3-aminopropyl)-1,1,3,3-tetramethyldisiloxane in 50 mL of methanol (Scheme 1). The complete synthesis procedure is briefly mentioned in the Experimental section.
 | ||
| Scheme 1 Reaction scheme for the synthesis of compound HS30. | ||
Design of sensing system
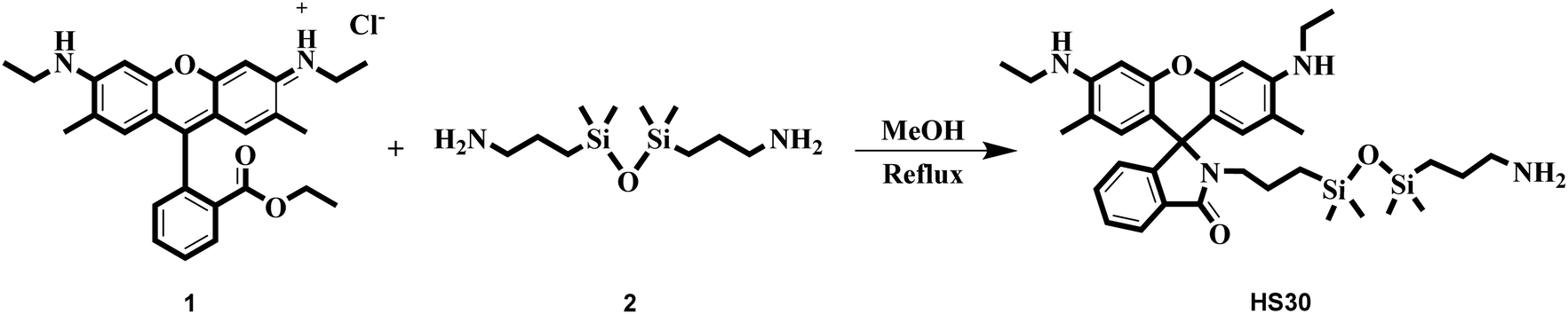
For the design of an effective FRET system, the selection of suitable fluorophores (energy donor and energy acceptor) and the immobilization of these fluorophores at a specific distance are the two key issues.21 Therefore, in this concern, we utilized CQDs as an energy donor and HS30 (a rhodamine 6G derivative) as an energy acceptor molecule. Due to their excitation-dependent emission nature, the emission maxima of CQDs can be easily adjusted to overlap an absorption spectrum of an energy acceptor fluorophore. Such a phenomenon helps in the efficient transfer of energy from the donor to the acceptor unit. Herein, to immobilize these two fluorophores, we used Fe(III) as a linker, which simultaneously interacted with CQDs and HS30 to form a ratiometric chelation complex. Literature reports revealed that carboxylic acid functionalized CQDs have been extensively used for the sensing of metal ions.35,42,43 These CQDs form a chelation complex with a specific metal cation, which results in the fluorescence quenching of CQDs due to the phenomenon of electron transfer. Singhal et al. have studied the interaction between CQDs and the hard acid Eu(III) and observed a decrement in the luminescence intensity on addition of Eu(III) to an aqueous solution of CQDs.34 Similarly, Khan et al. developed a fluorescent probe using carbon dots for the cellular imaging of Fe(III), which is also a hard acid.42 Therefore, in this work, we used this well-established interaction between hard acids and acid-functionalized CQDs for the development of the ratiometric fluorescent system.Rhodamine derivatives have been extensively exploited for the fluorescent sensing of Fe(III) both in an aqueous medium and in cellular imaging.44–47 Therefore, we synthesized a rhodamine derivative HS30 that interacts with the linker cation Fe(III). To understand the interaction between the synthesized ligand HS30 and Fe(III), we performed a fluorescence spectrophotometry experiment. The binding studies were conducted using an aqueous solution of organic nanoparticles (ONPs) of HS30 prepared by the reprecipitation method.47 Various metal nitrate salts (75 μM) were added to a fixed concentration of ONPs (10 μM) and after 15 min, fluorescence emissions were recorded from each sample on excitation at 510 nm. On the addition of Fe(III), strong fluorescence emission enhancement was observed at 551 nm, and no significant changes were observed in the fluorescent emission spectra for all other cations (Fig. 2A and B).
 | ||
| Fig. 2 (A) Changes in emission profile of HS30 (10 μM) in aqueous medium upon the addition of 75 μM of a particular metal nitrate salt. (B) The bar graph shows the selectivity test results, which indicate the selectivity of HS30 to Fe(III). (C) Changes in the fluorescent intensity of HS30 (10 μM) upon successive additions of Fe(III) (0–57 μM). (D) Linear regression graph between the concentration of Fe(III) (0–57 μM) and increase in fluorescence intensity of HS30. | ||
It was observed that the interfering ions did not induce any enhancement, which indicated that HS30 was selective for Fe(III) cations. Additionally, fluorescence titrations were performed by successively adding a small amount of Fe(III) to a fixed concentration of ONPs of HS30 (10 μM) (Fig. 2C). Titration results suggested that HS30 showed a linear response in the 0–57 μM range of Fe(III) with R2 = 0.9859 (Fig. 2D). Therefore, it was confirmed that fluorescent ligand HS30 selectively binds with Fe(III).
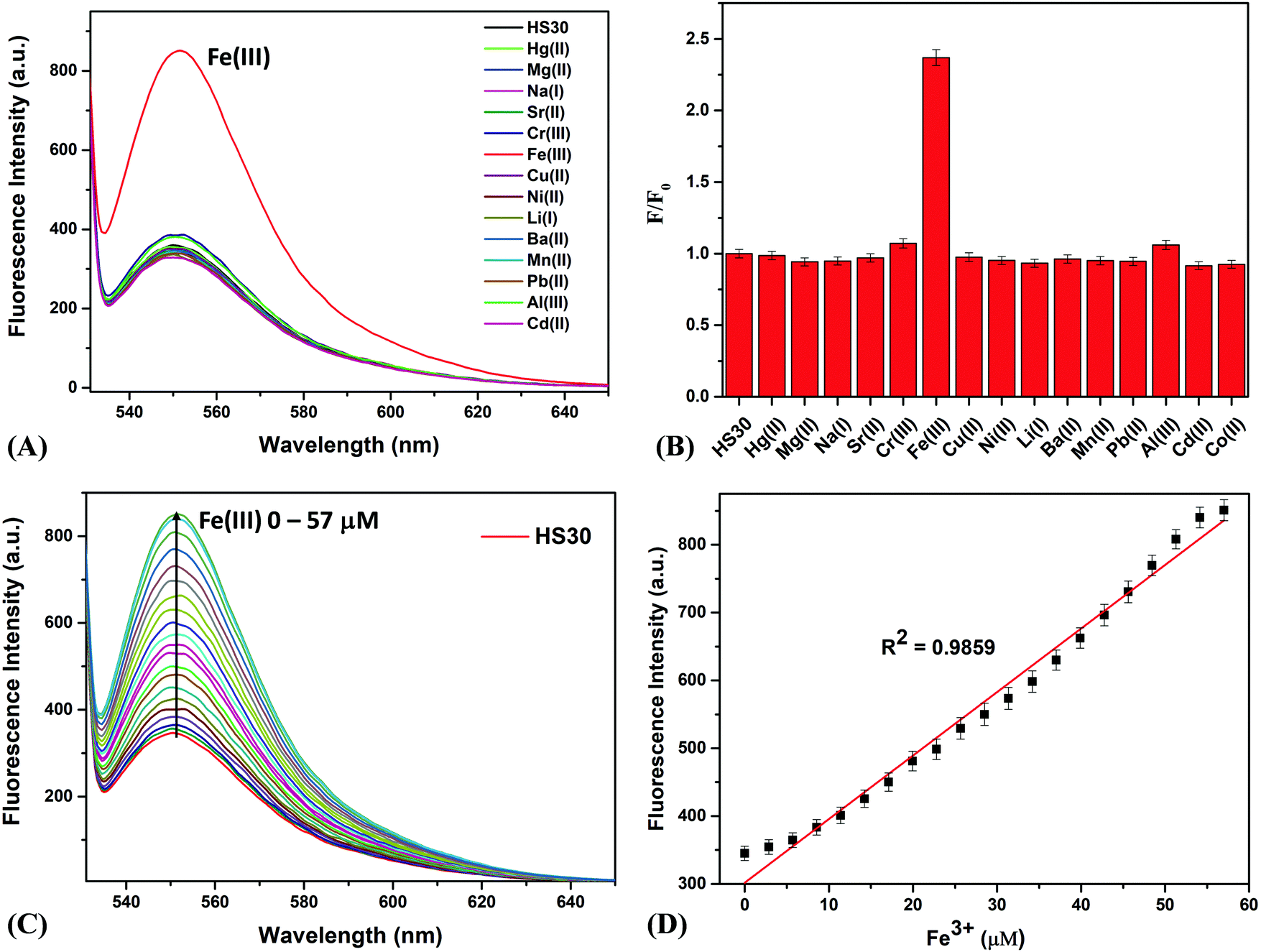
Further, for the development of the composite sensing system “CQDs–Fe–HS30”, CQDs (5 mg) and HS30 (10 mg) with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 were mixed in 1 mL of acetonitrile. Although we also prepared the above mixture with varying mass ratios, the results obtained were not satisfactory. The standard solution was prepared by dissolving 150 μL of the above-described mixture of both fluorophores in 150 mL of ultrapure water. Fluorescence titrations were carried out by adding a small amount of Fe(III) to the standard solution. In the absence of Fe(III), when the standard solution was excited at the absorption wavelength of 350 nm of the CQDs, we observed a fluorescence emission at a single point around 425 nm. Interestingly on continuous addition of Fe(III), a fluorescence emission peak starts to emerge at 551 nm, and simultaneous quenching of the emission peak at 425 nm was observed (Fig. 3A). This observation suggested that the Fe(III) cation linked CQDs and HS30 to form a ratiometric fluorescence complex. A linear change in the fluorescence intensities ratio (I551/I425) was observed with a regression coefficient of 0.9879 (Fig. 3B).
:2 were mixed in 1 mL of acetonitrile. Although we also prepared the above mixture with varying mass ratios, the results obtained were not satisfactory. The standard solution was prepared by dissolving 150 μL of the above-described mixture of both fluorophores in 150 mL of ultrapure water. Fluorescence titrations were carried out by adding a small amount of Fe(III) to the standard solution. In the absence of Fe(III), when the standard solution was excited at the absorption wavelength of 350 nm of the CQDs, we observed a fluorescence emission at a single point around 425 nm. Interestingly on continuous addition of Fe(III), a fluorescence emission peak starts to emerge at 551 nm, and simultaneous quenching of the emission peak at 425 nm was observed (Fig. 3A). This observation suggested that the Fe(III) cation linked CQDs and HS30 to form a ratiometric fluorescence complex. A linear change in the fluorescence intensities ratio (I551/I425) was observed with a regression coefficient of 0.9879 (Fig. 3B).
 | ||
| Fig. 3 (A) Changes in fluorescent intensity of “CQDs–Fe–HS30” (standard solution) upon successive additions of Fe(III) (0–25.7 μM). (B) Linear regression graph between the concentration of Fe(III) (0–25.7 μM) and an increase in the fluorescence intensity ratio (I551/I425). (C) Proposed cartoon sketch of the interaction of HS30 and CQDs with Fe(III). | ||
A plausible mechanism for the interactions of fluorophores and Fe(III) cations is shown in Fig. 3C. Initially, when a standard sample comprising of CQDs and HS30 alone is excited at 350 nm, the emission at 425 nm corresponding to CQDs was observed. However, the addition of Fe(III) resulted in the opening of the spirolactam ring of HS30, forming a chelation complex with the lone pair on the nitrogen atom as well as the negative charge carrying oxygen of HS30.39,41,47 Since Fe(III) has a coordination number of six, it further interacted with the –COOH group present on the surface of the CQDs,37,38 resulting in the formation of a composite system “CQDs–Fe–HS30”. Consequently, the resulting composite system gave a ratiometric emission at 425 nm and 551 nm when excited at 350 nm. Therefore, a FRET-based ratiometric system utilizing the Fe(III) cation linking two fluorophores, i.e., CQDs and HS30, has been successfully developed.
Sensing study of histidine
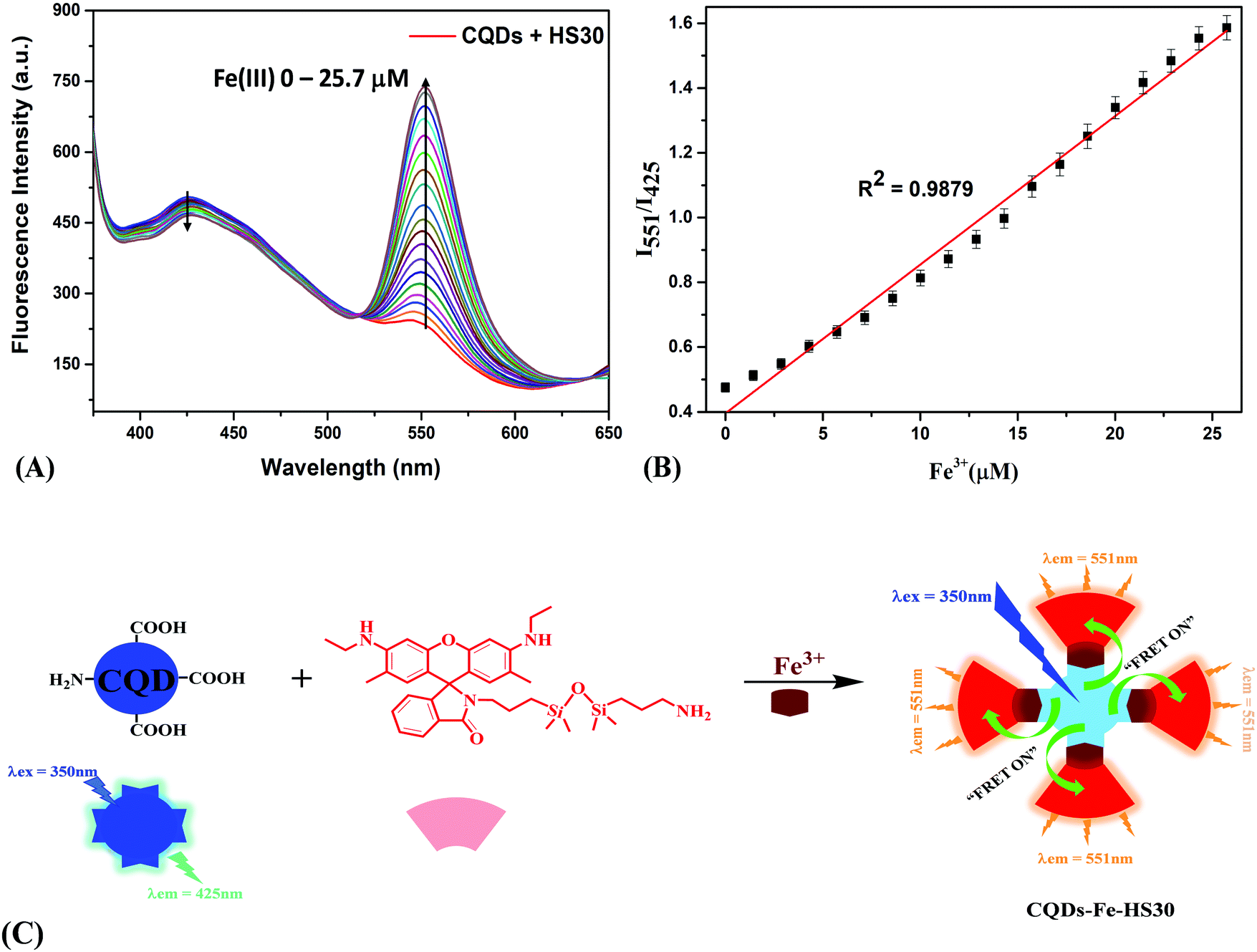
Furthermore, the composite system “CQDs–Fe–HS30” was implemented for the recognition of histidine using the metal displacement approach. Recently a metal displacement assay has emerged as one of the most successful methods for the sensing of various biological analytes.33,48–50 In a cation displacement assay, anions or biomolecules dislocate the cation from the binding cavity, producing a change in the signal intensity. Campen et al. studied the role of L-histidine, L-cysteine and L-lysine in improving iron uptake from ligated rat intestine and the results suggested that all three amino acids form tridentate chelates which enhance iron update.51 Swain et al. studied the role of histidine iron interaction in the enhancement of the bioavailability of nonheme iron and suggested that histidine increases iron bioavailability by forming a stable iron histidine complex.52 Keeping in mind that histidine forms a stable chelation complex with iron and has high affinity to Fe(III) ions,53–55 we utilized this interaction for the displacement of Fe(III) from the sensing complex. Further, the addition of histidine to an aqueous solution of “CQDs–Fe–HS30” resulted in a change in fluorescence emissions at two different wavelengths. Titrations with histidine (0–15.7 μM) were performed, and a quenching of the fluorescence intensity at 551 nm and enhancement of fluorescence intensity at 425 nm were observed (Fig. 4A). The ratio of fluorescence intensity (I551/I425) exhibited a linear relation (R2 = 0.9897) with the increase in histidine concentration from 0 to 15.7 μM (Fig. 4B). Based on the standard deviation (SD) of the response curve and the slope of the calibration curve (S), the limit of detection was calculated (3 × SD/S) to be 1.5 μM. A comparison of the limit of detection of the present work and other reported histidine sensors is shown in Fig. S4 (ESI†). | ||
| Fig. 4 (A) Changes in fluorescent intensity of “CQDs–Fe–HS30” upon successive additions of histidine (0–15.7 μM). (B) Linear regression graph between the concentration of histidine (0–15.7 μM) and a decrease in fluorescence intensity ratio (I551/I425). (C) A plausible mechanism for cation displacement via the histidine molecule resulting in the cancellation of the FRET mechanism. | ||
Thus, the quenching of the fluorescence at 551 nm can be attributed to the cancellation of FRET. The displacement of the linking moiety Fe(III) by histidine from the fluorescent complex moved the two fluorophores apart from each other, which resulted in the elimination of the energy transfer process, as shown in Fig. 4C. Since CQDs and HS30 were no longer held together by Fe(III), we observed an intense peak corresponding to the emission spectra of CQDs at 425 nm whereas a plateau was observed at 551 nm due to the cancellation of FRET. However, when the “CQDs–Fe–HS30” composite system was utilized for the detection of histidine in an aqueous system, in reality, most of the biological samples contained various competing ions and molecules. Therefore, in order to find the selectivity of the sensing system, fluorescence studies were also carried out in the presence of 75 μM of major competing anions (CH3COO−, HSO4−, NO3−, ClO4−, CN−, OH−, F−, Cl− and I−) and biomolecules (glycine, alanine, valine, leucine, isoleucine, tryptophan, arginine, L-glutamine, histamine, spermine, tyramine, cadaverine, citric acid, ascorbic acid and glutamic acid), as shown in Fig. S5 (ESI†). Interestingly, it was observed that the fluorescence intensity ratio (I551/I425) in the case of histidine was greatly quenched, whereas a negligible change was observed in the presence of anions as well as biomolecules. This observation suggested that the composite system is quite selective for the histidine molecule and can be utilized for the detection of histidine in biological samples. This high specificity can be attributed to the greater affinity of the histidine molecules to the Fe(III) ion in comparison to other competing moieties.51,52,54,55 Therefore, the developed composite sensing system can be successfully utilized for the selective sensing of histidine in real samples.
To find applications of the developed system, a histidine recovery experiment was carried out in a human urine sample collected from a healthy volunteer. 500 μL of the urine sample was diluted with 490 μL of ultrapure water, and 10 μL of histidine solution was added to obtain a total volume of 1 mL of spiked urine sample. A total of four samples were prepared and to each sample 10 μL of sensing system (CQDs–Fe–HS30) was introduced. After 30 minutes of incubation, the change in the fluorescence emission was recorded. As shown in Table 1, the quantitative recoveries of spiked histidine in the urine samples lies in the range of 95.2 to 108.5%. Thus, the recovery experiment indicates that the developed sensing system can be utilised for real sample analysis.
| Sample | Spiked amount/μM | Calculated/μM | % Recovery |
|---|---|---|---|
| Sample 1 | 0 | 1.94 ± 0.06 | |
| 3 | 2.85 ± 0.05 | 95.2 | |
| 6 | 6.14 ± 0.09 | 102.3 | |
| 9 | 9.77 ± 0.14 | 108.5 |
Molecular logic gate
Based on the results of the sensing studies performed, it was observed that the mixture of CQDs and HS30 (CQDs + HS30) could be further utilized to implement a molecular logic gate with an inhibit function. An inhibit logic gate (INH) is utilized whenever there is a need to enable or inhibit a logical signal depending upon the status of a particular control input. If we consider Fe(III) and histidine as inputs to CQDs + HS30 and emission at 551 nm as the output, we can implement an inhibit logic operation. Before obtaining an INH, we need to determine the influence of an individual input on the fluorescence emission of the mixture. In the absence of Fe(III), CQDs + HS30 gives no emission at 551 nm and in the presence of Fe(III) strong emission was observed when excited at 350 nm. However, the presence or absence of histidine alone in the CQDs + HS30 mixture did not result in any emission at 551 nm. However, it was observed that in the presence of both histidine and Fe(III), the fluorescence emission of CQDs + HS30 at 551 nm was quenched. Since the presence of histidine in a mixture of Fe(III) and CQDs + HS30 inhibited the output signal and the absence of histidine enabled the output, it apparently acted as a control input. This observation directly leads to truth table characteristics of the INH logic gate (Fig. 5).56,57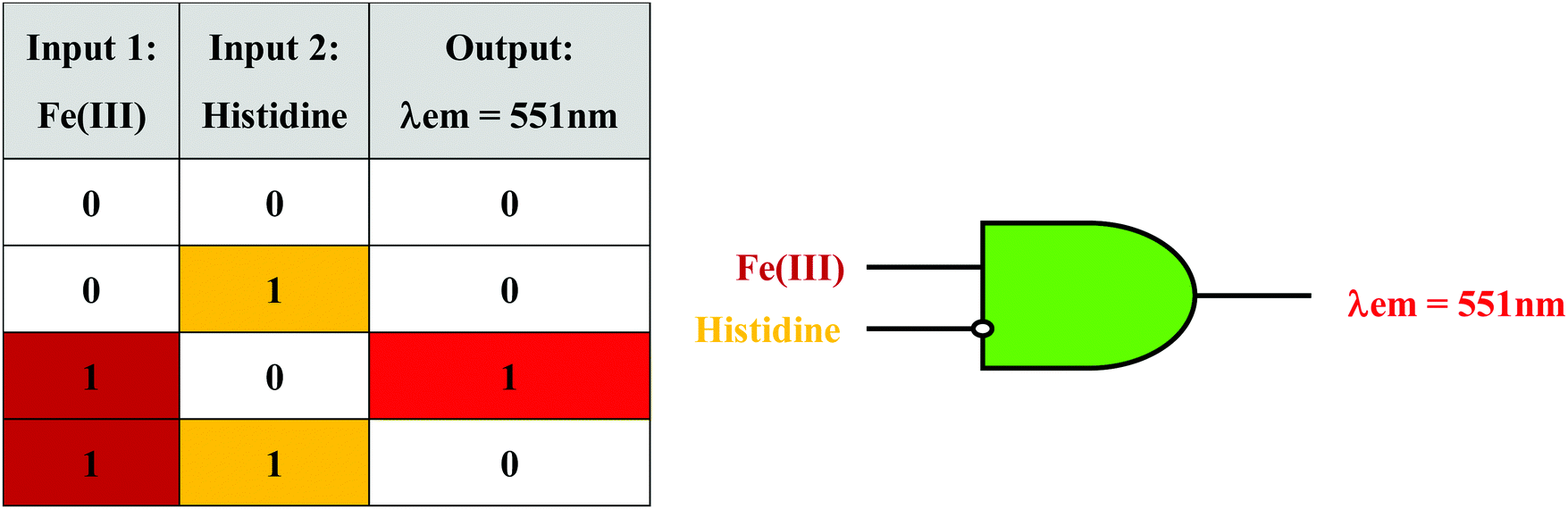 | ||
| Fig. 5 Truth table (left) and combinational logic scheme (right) for an INH logic gate with Fe(III) and histidine as inputs. | ||
Conclusion
In summary, a ratiometric fluorescent sensing system was developed for the sensing of histidine in an aqueous system. In comparison with the tedious and complicated synthesis strategies used for the development of fluorescent probes, we utilized a physical mixture of acid-functionalized carbon quantum dots (CQDs) and a rhodamine derivative (HS30). The addition of Fe(III) to the physical mixture (CQDs + HS30) resulted in a ratiometric response due to the formation of a FRET pair between the CQDs and HS30. Unlike most ratiometric sensors, where two fluorophores are linked by a covalent bond, our system utilized a cation to bring the two fluorophores closer. This ratiometric complex was further employed for the sensing of histidine in an aqueous system. Due to the strong affinity of histidine towards the Fe(III) cation, it displaces the Fe(III) from the ratiometric complex, resulting in the cancellation of FRET. Selectivity studies were carried out using various anions and biomolecules. Furthermore, a histidine recovery experiment in spiked human urine samples produced satisfactory results. Finally, an inhibit logic gate was realized using histidine and Fe(III) as inputs and emission at 551 nm as output. Therefore, this simple and selective sensing system for histidine could be further utilized for sensing histidine-tagged proteins and tracking histidine decarboxylase reactions.Experimental
Materials
All of the chemicals, including citric acid, ethylenediamine, metal nitrate salts, rhodamine 6G, quaternary ammonium salts, biomolecules and 1,3-bis(3-aminopropyl)-1,1,3,3-tetramethyldisiloxane, were purchased from Sigma-Aldrich and used as received.Instruments
1H NMR and 13C NMR measurements were carried on a Jeol JNM-ECS400 spectrophotometer with operation frequencies of 400 and 100 MHz, respectively. The photophysical properties were studied on a Shimadzu UV-2400 spectrophotometer and a PerkinElmer LS 55 fluorescence spectrophotometer. The morphological behaviour of the CQDs was observed using a Hitachi (H-7500) transmission electron microscope. The hydrodynamic diameter of the CQDs was determined with a Metrohm Microtrac Ultra Nanotrac particle size analyzer (dynamic light scattering). FTIR spectra of the CQDs were recorded on a Bruker Tensor 27 spectrophotometer. A PANalytical X’PERT PRO diffractometer was employed to obtain the powder XRD patterns of the CQDs. The mass spectrum was measured on a Xevo G2-XS QTOF mass spectrometer.Synthesis of HS30
In a round bottom flask, 0.5 g of compound 1 (rhodamine 6G) was dissolved in 50 mL of methanol. 1.04 g (4.15 mmol) of compound 2 (1,3-bis(3-aminopropyl)-1,1,3,3-tetramethyldisiloxane) was added into the reaction mixture and stirred at refluxing temperature for 24 hours. The solvent was removed under reduced pressure to obtain an oily residue. The product was extracted with ethyl acetate, and the organic layer was washed with water (30 mL). The organic solvent was evaporated under reduced pressure and the compound was further purified by recrystallization in acetonitrile (yield: 61%).Characterisation of HS30
Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors are thankful to IIT Ropar for providing funds and infrastructure facilities for carrying out the research.References
- X. Zhu, T. Zhao, Z. Nie, Z. Miao, Y. Liu and S. Yao, Nanoscale, 2016, 8, 2205–2211 RSC.
- S. M. Liao, Q. S. Du, J. Z. Meng, Z. W. Pang and R. B. Huang, Chem. Cent. J., 2013, 7, 9–11 CrossRef PubMed.
- R. J. Sundberg, Chem. Rev., 1974, 74, 471–517 CrossRef CAS.
- H. Yoshimatsu, S. Chiba, D. Tajima, Y. Akehi and T. Sakata, Exp. Biol. Med., 2002, 227, 63–68 CrossRef CAS.
- W. W. Hu and Z. Chen, ACS Chem. Neurosci., 2012, 3, 302–311 CrossRef PubMed.
- E. Zampeli and E. Tiligada, Br. J. Pharmacol., 2009, 157, 24–33 CrossRef CAS PubMed.
- D. A. Gerber, J. Clin. Invest., 1975, 55, 1164–1173 CrossRef CAS PubMed.
- M. Watanabe, M. E. Suliman, A. R. Qureshi, E. Garcia-lopez and P. Ba, Am. J. Clin. Nutr., 2008, 87, 1860–1866 CrossRef CAS PubMed.
- S. Seshadri, A. Beiser, J. Selhub, P. F. Jacques, I. H. Rosenberg, R. B. D'agostino, P. W. Wilson and P. A. Wolf, N. Engl. J. Med., 2002, 346, 476–483 CrossRef CAS PubMed.
- J. Meng, W. Zhang, C.-X. Cao, L.-Y. Fan, J. Wu and Q.-L. Wang, Analyst, 2010, 135, 1592–1599 RSC.
- J. Liang, Z. Chen, L. Guo and L. Li, Chem. Commun., 2011, 47, 5476–5478 RSC.
- Z. Zhang, Y. Hu, H. Zhang, L. Luo and S. Yao, Biosens. Bioelectron., 2010, 26, 696–702 CrossRef CAS PubMed.
- C. Wu, D. Fan, C. Zhou, Y. Liu and E. Wang, Anal. Chem., 2016, 88, 2899–2903 CrossRef CAS PubMed.
- D. L. Ma, W. L. Wong, W. H. Chung, F. Y. Chan, P. K. So, T. S. Lai, Z. Y. Zhou, Y. C. Leung and K. Y. Wong, Angew. Chem., Int. Ed., 2008, 47, 3735–3739 CrossRef CAS PubMed.
- Z. Huang, J. Du, J. Zhang, X. Q. Yu and L. Pu, Chem. Commun., 2012, 48, 3412–3414 RSC.
- X. Huang, Y. Lin, J. Chen, Y. Chen, Y. Li and W. Gao, New J. Chem., 2015, 39, 8569–8575 RSC.
- H. Wang, H. Zhang, Y. Chen and Y. Liu, New J. Chem., 2015, 39, 8896–8900 RSC.
- Z. Zhang, L. Wang, G. Li and B. Ye, Analyst, 2017, 142, 1821–1826 RSC.
- T. Kohira, Y. Kurishita, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 13290–13299 CrossRef PubMed.
- J. Du, S. Yu, Z. Huang, L. Chen, Y. Xu, G. Zhang, Q. Chen, X. Yu and L. Pu, RSC Adv., 2016, 6, 25319–25329 RSC.
- E. A. Jares-Erijman1 and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387–1395 CrossRef PubMed.
- Y. Dong, R. Wang, G. Li, C. Chen, Y. Chi and G. Chen, Anal. Chem., 2012, 84, 6220–6224 CrossRef CAS PubMed.
- J. Ju and W. Chen, Biosens. Bioelectron., 2014, 58, 219–225 CrossRef CAS PubMed.
- R. Zhang and W. Chen, Biosens. Bioelectron., 2013, 55, 83–90 CrossRef PubMed.
- Z. Lin, W. Xue, H. Chen and J. M. Lin, Anal. Chem., 2011, 83, 8245–8251 CrossRef CAS PubMed.
- F. Du, F. Zeng, Y. Ming and S. Wu, Microchim. Acta, 2013, 180, 453–460 CrossRef CAS.
- Y. Dong, R. Wang, W. Tian, Y. Chi and G. Chen, RSC Adv., 2014, 4, 3701–3705 CAS.
- D. B. Cordes, S. Gamsey and B. Singaram, Angew. Chem., 2006, 45, 3829–3832 CrossRef CAS PubMed.
- Y. Shi, Y. Pan, H. Zhang, Z. Zhang, M. J. Li, C. Yi and M. Yang, Biosens. Bioelectron., 2014, 56, 39–45 CrossRef CAS PubMed.
- A. H. Loo, Z. Sofer, D. Bousa, P. Ulbrich, A. Bonanni and M. Pumera, ACS Appl. Mater. Interfaces, 2016, 8, 1951–1957 CrossRef CAS PubMed.
- K. Qu, J. Wang, J. Ren and X. Qu, Chem. – Eur. J., 2013, 19, 7243–7249 CrossRef CAS PubMed.
- C. S. Stan, C. Albu, A. Coroaba, M. Popa and D. Sutiman, J. Mater. Chem. C, 2015, 3, 789–795 RSC.
- J. S. Sidhu, A. Singh, N. Garg, N. Kaur and N. Singh, Analyst, 2018, 143, 1853–1861 RSC.
- P. Singhal, B. G. Vats, S. K. Jha and S. Neogy, ACS Appl. Mater. Interfaces, 2017, 9, 20536–20544 CrossRef CAS PubMed.
- W. Lu, X. Qin, S. Liu, G. Chang, Y. Zhang, Y. Luo, A. M. Asiri, A. O. Al-youbi and X. Sun, Anal. Chem., 2012, 84, 5351–5357 CrossRef CAS PubMed.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- P. Song, L. Zhang, H. Long, M. Meng, T. Liu, Y. Yin and R. Xi, RSC Adv., 2017, 7, 28637–28646 RSC.
- N. Dhenadhayalan and K. Lin, Sci. Rep., 2015, 10, 10012 CrossRef PubMed.
- Y. Xiang and A. Tong, Org. Lett., 2006, 8, 1549–1552 CrossRef CAS PubMed.
- S. Ji, X. Meng, W. Ye, Y. Feng, H. Sheng, Y. Cai, J. Liu, X. Zhu and Q. Guo, Dalton Trans., 2014, 43, 1583–1588 RSC.
- S. Bae and J. Tae, Tetrahedron Lett., 2007, 48, 5389–5392 CrossRef CAS.
- W. U. Khan, D. Wang, W. Zhang, Z. Tang, X. Ma and X. Ding, Sci. Rep., 2017, 7, 14866 CrossRef PubMed.
- X.-C. Fu, J.-Z. Jin, J. Wu, J.-C. Jin and C.-G. Xie, Anal. Methods, 2017, 9, 3941–3948 RSC.
- K. Wu, H. Xiao, L. Wang, G. Yin, Y. Quan and R. Wang, RSC Adv., 2014, 4, 39984–39990 RSC.
- W. Chan, RSC Adv., 2016, 6, 74389–74393 RSC.
- A. J. Weerasinghe, C. Schmiesing, S. Varaganti, G. Ramakrishna and E. Sinn, J. Phys. Chem. B, 2010, 114, 9413–9419 CrossRef CAS PubMed.
- A. Singh, S. Sinha, R. Kaur, N. Kaur and N. Singh, Sens. Actuators, B, 2014, 204, 617–621 CrossRef CAS.
- O. Kotova, S. Comby and T. Gunnlaugsson, Chem. Commun., 2011, 47, 6810–6812 RSC.
- F. Gruppi, J. Liang, B. B. Bartelle, M. Royzen, D. H. Turnbull and J. W. Canary, Chem. Commun., 2012, 48, 10778–10780 RSC.
- V. Janowski and K. Severin, Chem. Commun., 2011, 47, 8521–8523 RSC.
- D. Van Campen and E. Gross, J. Nutr., 1969, 99, 68–74 CrossRef CAS PubMed.
- J. H. Swain, L. B. Tabatabai and M. B. Reddy, J. Nutr., 2002, 132, 245–251 CrossRef CAS PubMed.
- B. Khan, S. Ehsan, F. Hassan, J. Kazi and W. A. Shah, Asian J. Chem., 2011, 23, 1969–1972 CAS.
- N. G. Nair, G. Perry, M. A. Smith and V. P. Reddy, J. Alzheimer's Dis., 2010, 20, 57–66 CAS.
- E. Ghiamati and E. Jalaeian, Asian J. Phys. Chem. Sci., 2017, 2, 1–10 Search PubMed.
- M. De Sousa, M. Kluciar, S. Abad, M. A. Miranda, D. Castro and U. Pischel, Photochem. Photobiol. Sci., 2004, 3, 639–642 RSC.
- T. Gunnlaugsson, D. A. MacDonail and D. Parker, Chem. Commun., 2000, 93–94 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00554k |
| This journal is © the Partner Organisations 2019 |