
Hypervalent iodine-based dynamic and self-healing network polymers†
Avichal
Vaish
and
Nicolay V.
Tsarevsky
 *
*
Department of Chemistry, Southern Methodist University, 3215 Daniel Avenue, Dallas, TX 75275, USA. E-mail: nvt@smu.edu
First published on 26th June 2019
Abstract
Linear polymers with multiple carboxylate pendant groups (copolymers of styrene and acrylic acid) participate in ligand-exchange reactions with (diacetoxyiodo)benzene and the reaction yields network polymers with (diacyloxy)iodoarene-type crosslinks. The unreacted carboxylate groups that are present in the polymers can participate in further ligand-exchange reactions with the hypervalent iodine(III) centers at the crosslinks, which makes the materials dynamic and self-healing. The application of strong shear force leads to gel disintegration but when the external force is removed, the gels are regenerated again, as proved by rheology studies. When exposed to UV light or heat (microwave irradiation), the gels are “set” (i.e., become permanent) due to the homolytic cleavage of the weak hypervalent I–O bonds present at the crosslinks, followed by irreversible radical coupling of adjacent polymer-bound C- and/or O-centered radicals.
Introduction
The design and preparation of dynamic polymers formed as a result of reversible covalent interactions is of great interest1–16 since these materials often possess unique properties, which make them attractive for various applications, such as controlled drug delivery systems,17 adaptive surfaces,5 polymeric actuators,18 and self-healing materials.19–28 Self-healing is the process in which a material that had been damaged by mechanical forces can acquire its original properties (and often appearance) without external treatment, such as the application of additional reagents. The process occurs due to the presence of reactive functionalities, which are responsible for the formation of new bonds, typically but not necessarily identical to the ones that had been cleaved during the damage.29–39 If the self-healing is efficient and faster than the propagation of further defects, it improves the reliability of the material, extends its life-time, makes costly repairs unwarranted, and often reduces safety risks by preventing failures caused by the accumulation of cracks or breaks. Electrostatic (ionic) interactions,40,41 hydrogen bonds,42,43 as well as coordination bonds44,45 have all been utilized in the design and preparation of dynamic and self-healing polymers.Many main-group elements form compounds, in which the octet rule is not obeyed, and in which the central polyvalent atom is bonded to some of the “ligands” via 3-center-4-electron, dubbed hypervalent (HV),46 bonds. They are weaker than the “classical” covalent (2-center-2-electron) bonds and are typically rather polar.46–48 As expected based on the number of unpaired electrons in the ground state, iodine is often monovalent in its compounds. However, numerous examples are known, in which the iodine atom is polyvalent, e.g., tri-, penta-, or heptavalent, and participates in the formation of one covalent and several additional HV bonds. HV iodine compounds are primarily used as oxidizing agents49–52 and in other electrophilic and radical reactions, including as initiators for cationic53–55 and/or radical56–59 polymerizations. The HV bonds in the fragment L-I-L in the iodine(III) compound of the type ArIL2 (Ar is aryl, and the ligand L is (pseudo)halide, carboxylate,60 or tetrazolate61) are dynamic in nature62 and the ligands, L can be easily (and typically reversibly) displaced by nucleophiles Nu−. The reaction affords, depending on the amount of added nucleophile and the reaction time, ArI(L)Nu and/or ArINu2 as well as free L−.63 For example, (diacyloxyiodo)arenes ArI(O2CR1)2 (Ar = aryl, R = alkyl or aryl) participate in ligand-exchange reactions with carboxylates R2CO2− or the corresponding acids R2CO2H to yield first the asymmetric compound ArI(O2CR1)(O2CR2) and eventually ArI(O2CR2)2, as well as free R1CO2− or R1CO2H. The thermodynamics and kinetics of this type of reaction involving (diacetoxyiodo)benzene PhI(O2CCH3)2 were studied spectroscopically (with R2CO2H being methacrylic64 or acrylic acid65) and it was shown that the ligand exchange is relatively fast even in dilute solutions. The reaction of PhI(O2CCH3)2 with methacrylic acid was employed to synthesize branched polymers via the generation (in situ) of inimers with methacryloyloxy groups attached to the HV iodine(III) center. More recently, stimuli-responsive and reductively degradable branched polymers were reported containing (diacyloxyiodo)arene groups as structural elements, i.e., at each branching point connecting two linear chains.65 The addition of acetic acid to solutions of these polymers led to fast degradation and it was speculated that the degradation induced by ligand-exchange with free carboxylates could be reversible, i.e., that the materials could be dynamic in nature.
Herein, we report the preparation of dynamic gels by substitution of the acetoxy groups in PhI(O2CCH3)2 by polymer-bound carboxylate groups. The presence of multiple unreacted pendant carboxylate groups, which can participate in ligand-exchange reactions with the HV iodine(III) centers at the crosslinking points, makes the materials both dynamic and self-healing. Further, advantage is taken of the fact that HV iodine(III) compounds ArIL2 decompose homolytically upon irradiation or heating with the formation of the monovalent iodine compound, iodoarene ArI, and the radicals L˙,50,55,58,59,66–68 which can react with a substrate or couple. The coupling reaction between the macromolecular carboxylate radicals and/or the alkyl radicals that are formed by their decarboxylation can reasonably be expected to yield irreversibly the formation of permanently crosslinked polymers. The feasibility of this approach, which converts the dynamic to “set” gels, and which provides flexibility to use the same polymer network for various applications, is demonstrated.
Experimental section
Materials
Styrene (Sty, 99%, Alfa Aesar) and tert-butyl acrylate (tBA, 98%, Aldrich) were purified before the polymerizations by passing through a column filled with basic alumina to remove the inhibitor. The radical initiator, 1,1′-azobis(cyclohexanecarbonitrile) (ACHN, 98%, Aldrich), anisole (99%, Acros), CF3CO2H (99%, Aldrich), PhI(O2CCH3)2 (98%, Acros), glacial CH3CO2H (99.7%, Fisher), NaN3 (99.5%, Aldrich), tri-n-butylphosphine (Bu3P, 95%, Alfa Aesar), indigo carmine (>80%, Fisher), N,N-dimethylacetamide (DMAc, 99%, Alfa Aesar), 1,4-dioxane (99.8%, Alfa Aesar), dichloromethane (99%, Fisher), methanol (MeOH, 99%, Fisher), and tetrahydrofuran (THF, 99.5%, EMD Millipore) were used as received. The chain transfer agent, (2-(((butylthio)carbonothioyl)thio)-2-methylpropanoic acid (BMP), was synthesized by following a reported method.69 Cobalt Blue was prepared by first drying a mixture of cobalt(II) and aluminum hydroxides (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) at 120 °C for 40 h and then calcining the obtained material at 1000 °C for 6 h.70 The deuterated solvents, acetone-d6 (99.9% D, Cambridge Isotope Laboratories) and DMSO-d6 (99.9% D, Cambridge Isotope Laboratories), contained a small amount of tetramethylsilane (TMS) as the chemical shift reference.
:2 molar ratio) at 120 °C for 40 h and then calcining the obtained material at 1000 °C for 6 h.70 The deuterated solvents, acetone-d6 (99.9% D, Cambridge Isotope Laboratories) and DMSO-d6 (99.9% D, Cambridge Isotope Laboratories), contained a small amount of tetramethylsilane (TMS) as the chemical shift reference.
Analyses and characterization methods
Molecular weights and molecular weight distribution (MWD) dispersities (Đ = Mw/Mn) were determined by size exclusion chromatography (SEC) on a Tosoh EcoSEC system equipped with a series of 4 columns (TSK gel guard Super HZ-L, Super HZM-M, Super HZM-N, and Super HZ2000) and using refractive index (RI) and UV detectors. THF was used as the eluent at a flow rate of 0.35 mL min−1 (40 °C). The SEC calibration was based on linear polystyrene standards. The total monomer conversion was determined by 1H NMR spectroscopy, using a Bruker Avance DRX (400 MHz) or a JEOL ECCA (500 MHz) spectrometer, by monitoring the decrease of the sums of intensities of the vinyl peaks (from Sty and tBA) relative to the intensity of the methoxy protons of anisole (internal standard present in the reaction mixtures). Rheological data was obtained by using a Discovery Hybrid HR-3 rheometer (TA instruments) with cone plate geometry (diameter of 40 mm, cone angle 2° 00′ 32′′, truncation gap 53 μm). All rheological measurements were carried out at 25 °C, which was controlled with a Peltier thermo-module unit. The entire plate was covered with sample (2 mL) for consistent results and the measurements were performed 30 min after mixing of the components of the gel. Photochemical curing was conducted by either UVP CL-1000L UV crosslinker (365 nm) or Ushio G20T10 low pressure mercury arc UV lamps (254 nm, 21 W). Thermal curing was carried out in a CEM Discover SP microwave reactor (300 W). The time-lapse video of gel healing was taken by using AmScope optical microscope and MD600E camera.Synthetic procedures
:3 and 3:1, keeping the same total monomer to chain transfer agent (BMP) ratio. Polymerization kinetics as well as the evolution of molecular weights and MWD dispersities with conversion, and individual SEC traces are shown in Fig. S1 in the ESI.†
:1 (v/v)). The reprecipitation procedure was repeated once again. The polymers were then dried under vacuum for a day.
:2 (v/v)) could be examined visually (Fig. 1). The gels derived from Sty1AA0.7 (intermediate fraction of carboxylate groups) were the most homogeneous. Gels that could be studied both visually (Fig. 2) and by rheology (Fig. 3) were prepared by mixing that copolymer solution (0.35 M of carboxylate groups, prepared after dilution of the stock solution with DMAc) with PhI(O2CCH3)2 stock solution (4:1 (v/v)).
 | ||
| Fig. 1 Dynamic gel 15 min after mixing copolymer Sty1AA0.9 (0.73 M with respect to carboxylate groups, 300 μL) and PhI(O2CCH3)2 (0.15 M, 200 μL) solutions (a); gel immediately after addition of DMAc and CH3CO2H (5:1 (v/v), 600 μL) (b); and degraded gel 12 h after the addition of CH3CO2H (c). The precipitate on the bottom of the tube consists of Co Blue. | ||
 | ||
| Fig. 2 Gels prepared by mixing solutions of Sty1AA0.7 (0.35 M with respect to carboxylate groups, 400 μL) and PhI(O2CCH3)2 (0.15 M, 100 μL) in DMAc with trace amount of indigo carmine present in one of the gels (a); the gels cut into two pieces (b); reattached semicircular segments taken from the two pieces of gels after 30 min (c); and effect of stretching on the self-healed gel (d). | ||
 | ||
| Fig. 3 G′ and G′′ values of a gel prepared by using Sty1AA0.7 and PhI(O2CCH3)2 in DMAc (a) on strain amplitude sweep; (b) on angular frequency sweep; (c) in time-dependent strain sweep measurement, in which, strain was gradually increased from 0.1% to 1300% and then back to 1% strain for 300 s; (d) in continuous step strain measurement, gel was subjected to 1300% strain for 30 s, then go back to 1% strain in the linear regime for 120 s, and 3 cycles were carried out. | ||
The stability of some of the prepared gels with respect to monocarboxylic acids (CH3CO2H), azides, and phosphines was examined by adding various amounts of the mentioned reagents to the swollen gels, as described in the Results and Discussion section.
In some cases, to achieve better visibility of the gels, a small amount of indigo carmine was added, but, due to the tendency of this dye to change color and fade over time in the presence of the components of the gels, most of the colored gels were prepared using Co Blue (selected because of its stability in the presence of many reagents, including oxidants70).
Larger amounts of gels were prepared by mixing 400 μL of PhI(O2CCH3)2 (0.15 M) solution with 1600 μL of Sty1AA0.7 (0.35 M) solution in DMAc in a 10 mL glass beaker. After 10 min of mixing, the gel was cured by exposing it to two low pressure mercury arc UV lamps (254 nm, 21 W) for 90 min. The light source was kept ca. 5 inches above the surface of the gel. After curing, the gel was washed twice with 1 mL of DMAc to remove the byproducts (e.g., iodobenzene and the products of its photochemical decomposition) and the gel was subjected to rheological examination.
Results and discussion
The reversible addition–fragmentation chain transfer (RAFT) polymerization71–74 of Sty and tBA (Scheme 1) was performed to prepare three different random copolymers – Sty1tBA0.9, Sty1tBA0.7, and Sty1tBA0.5, where the numbers indicate the molar ratios of the monomer units in the final isolated copolymers (determined by 1H NMR spectroscopy). The polymerizations were carried out in bulk at 85 °C for 40–50 h, depending on the monomer feed ratio. ACHN was chosen as initiator because of its relatively long half-life time at the conditions of the experiment (the reported value is 16 h at 85 °C in toluene75). BMP was used as the chain transfer agent, which controls well the polymerization of both acrylates and styrenes. As the polymerizations proceeded, the number-average molecular weights (Mn,app) increased and the MWDs narrowed (Fig. S1†). After purification of the random copolymers, the tert-butyl ester groups were removed using CF3CO2H. The apparent molecular weights of all three copolymers after the deprotection were similar (83700–88000 g mol−1).
 | ||
| Scheme 1 Synthesis of carboxylate group-containing copolymers by copolymerization of Sty and tBA under RAFT polymerization conditions, followed by deprotection of the tert-butyl ester groups in the presence of CF3CO2H. | ||
Preparation of dynamic gels
(Diacyloxyiodo)arenes can participate in ligand-exchange reactions with various nucleophiles, including carboxylates. This reaction was taken advantage of and dynamic networks were prepared by displacing the acetoxy ligands in PhI(O2CCH3)2 by the pendant carboxylate groups in poly(Sty-co-AA), as shown in Scheme 2. Three different poly(Sty-co-AA) copolymers (designated Sty1AA0.9, Sty1AA0.7, and Sty1AA0.5, as explained in Scheme 1) were prepared by removal of the tert-butyl ester groups in the poly(Sty-co-tBA) precursors by treatment with CF3CO2H. A dynamic gel was then prepared by mixing 200 μL of PhI(O2CCH3)2 (0.15 M) and 300 μL of Sty1AA0.9 (0.73 M with respect to carboxylate groups) solutions in DMAc, with or without addition of trace amounts of indigo carmine or Co Blue (used for better visualization). Once the gel was formed, the HV iodine center could exchange ligands with unreacted carboxylate acid groups, or external nucleophiles. The formation of network polymers and their transformations in the presence of monocarboxylic acids, or upon irradiation or heating are presented in Scheme 2. The Sty1AA0.5 copolymer did not form a gel with PhI(O2CCH3)2, most likely due to insufficient amount of accessible carboxylate pendant groups in the copolymer.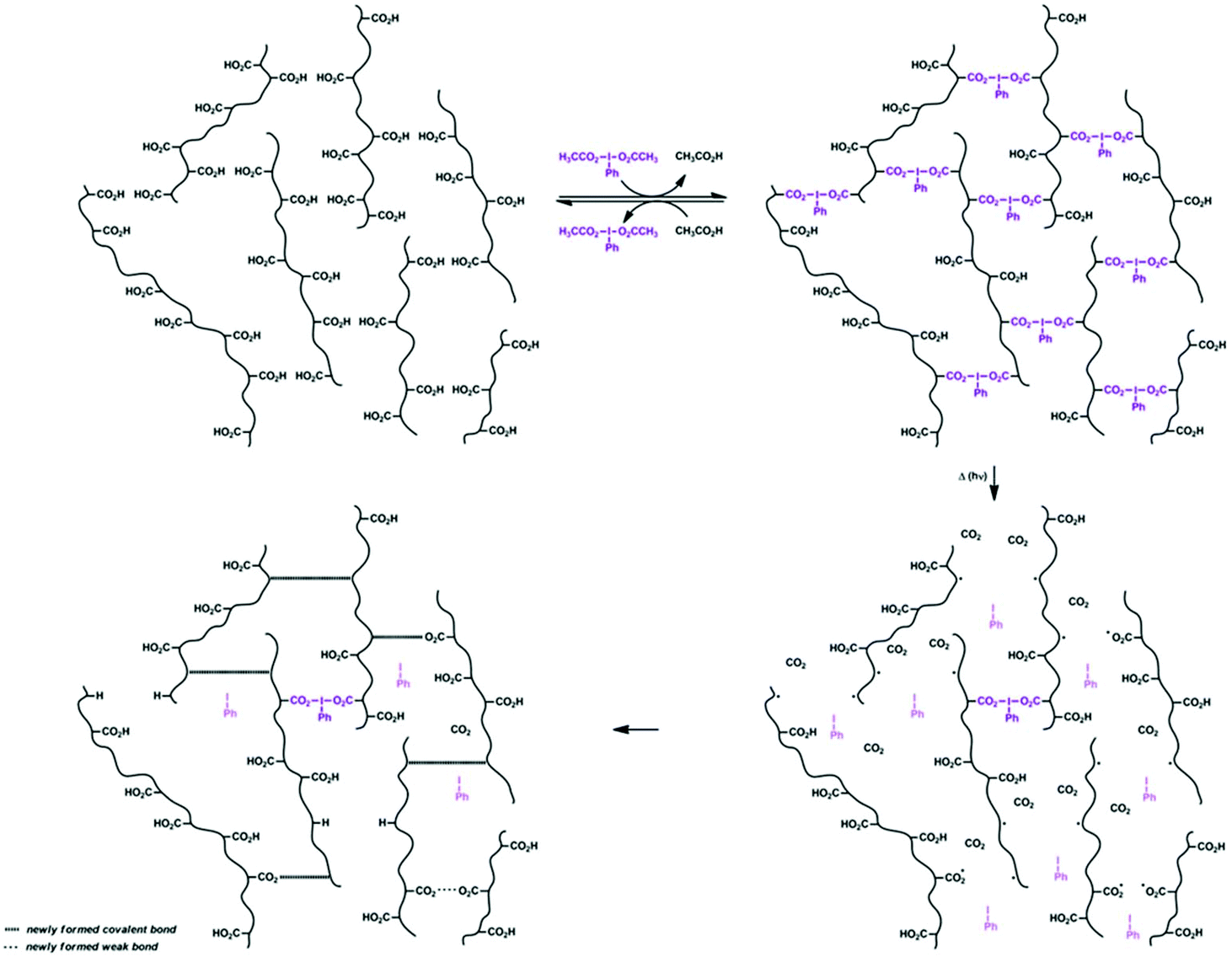 | ||
| Scheme 2 Preparation of dynamic gels via carboxylate ligand-exchange reactions with PhI(O2CCH3)2, curing of the gels using light or heat, and conversion of the dynamic to permanent network following the coupling of some of the radicals formed during the curing step. | ||
To confirm the dynamic nature of the network polymers, CH3CO2H (100 μL, 1.75 mmol, 58 eq. vs. the originally employed PhI(O2CCH3)2) was added after washing the gel twice with DMAc and, as expected, the gel completely disintegrated in 12 h to yield the initial copolymer and PhI(O2CCH3)2, as shown in Fig. 1 and clarified in Scheme 2 (top part).
The same experiment was repeated with azide as a nucleophile and the gel degraded, as shown in Fig. S3a and b (ESI).† This reaction yields the very unstable PhI(N3)2,76 which decomposes readily with the formation of iodobenzene PhI and the highly reactive azide radicals.77 The dynamic gel also disintegrated after addition of reducing agents, such as Bu3P, which reduces iodine(III) to iodine(I),78i.e., PhI (Fig. S3c and d†). This reductive degradation in the presence of Bu3P was recently demonstrated to occur in closely-related branched polymers with (diacyloxyiodo)benzene groups at the branching points.65
Self-healing and rheological properties of dynamic gels
After establishing the dynamic nature of the gels, their ability to self-heal was examined. In a preliminary experiment, two disk-shaped gels were prepared by mixing 400 μL of Sty1AA0.7 (0.35 M) solution and 100 μL of PhI(O2CCH3)2 (0.15 M) solution in DMAc, one of which contained a minuscule amount of indigo carmine dye to provide color difference, in a cylindrical container. Gelation occurred within ca. 10 min and the gels were transparent in appearance (Fig. 2). Then, the disk-shaped gels were cut in halves and the pieces were swapped and kept in contact for 30 min without any external pressure. The free carboxylate groups and HV crosslinks present on the surface of one piece of cut gel could participate in ligand-exchange with HV iodine centers on the other piece, which led to re-formation of the network. The cut between the two original semi-cylindrical pieces became invisible after 30 min and the healed gel did not break at the conjoining line even upon stretching. The self-healing was also monitored under an optical microscope and a time-lapse video showing healing of the cut was recorded and can be found in the ESI.†The self-healing was then quantified by rheological measurements (Fig. 3). The elastic response of the crosslinked network was analyzed first by strain amplitude sweep to determine the linear viscoelastic regime of the gel. In this experiment (Fig. 3a), the storage and loss moduli (G′ and G′′, respectively) were recorded as a function of the strain (γ) at a constant angular frequency (ω = 10 rad s−1). It was noticed that, in the viscoelastic region, i.e. 0.1% to ca. 500% strain, G′ was constant and greater than G′′ but, above a critical strain (γc ≈ 500%), it rapidly began to decline, which suggested the collapse of the crosslinked network. When the strain was increased to over 1200%, the values of G′ became lower than G′′, which suggested that virtually no crosslinking between the polymer chains had been retained. At this point, the gel behaved like a viscoelastic liquid rather than a solid. In another experiment, the rheological behavior of the gel was assessed by subjecting it to increasing angular frequency (ω = 0.1–100 rad s−1) at a constant strain (1%), which was in the viscoelastic regime. The G′ values (ca. 230 Pa) were independent of ω and larger than the G′′ values over the entire range of angular frequencies (Fig. 3b), which affirmed the re-formation of gel.79,80
After the preliminary rheological experiments, self-healing was confirmed by time-dependent strain sweep and continuous step-strain measurements. In the time-dependent strain sweep experiment (Fig. 3c), at first the strain was increased gradually, from 0.1 to 1300%, to break the network and then the strain was reduced to 1% for 300 s at a constant angular frequency (ω = 10 rad s−1). Interestingly, the gel exhibited a swift recovery of the mechanical strength after large amplitude oscillatory breakdown. The gel recovered ca. 85% of its original G′ values in less than 20 s, which demonstrated efficient and rapid self-healing. Furthermore, continuous step-strain measurements were conducted to quantify the reproducibility of the self-healing. In this experiment, the gel was subjected to 1300% strain for 30 s and then the strain was decreased to 1% for 120 s. This cycle was repeated three times. When nonlinear, large amplitude oscillations (γ = 1300% at ω = 10 rad s−1) were applied to the gel, the values of G′ decreased to ca. 10 Pa, resulting in viscoelastic fluid-like behavior (tanδ (G′′/G′) ≈ 1.5). However, when the strain was decreased to 1%, the values of G′ recovered promptly (within 20 s) to ca. 100 Pa and G′ values became greater than G′′ values, showing quasi-solid-like behavior (Fig. 3d). Although the G′ values were not completely restored after each cycle, the recovery of the gel was reproducible for at least three cycles. This was the conclusive evidence of self-healing nature of the gel.
Conversion of dynamic to permanent gels
The HV I–O bonds in ArI(O2CR)2 can dissociate homolytically upon irradiation or heating to yield acyloxy radicals (RCO2˙) or the products of their decarboxylation (R˙), along with iodoarene ArI. This property of the HV bonds enabled the conversion of the dynamic to permanently crosslinked gel by exposing the former to UV light or heat. During this treatment, macromolecular acyloxy and eventually alkyl radicals are generated (bottom of Scheme 2). Several types of bonds can be formed by combination of the radicals: weak O–O bonds (coupling of acyloxy radicals), which can be cleaved homolytically again to acyloxy radicals, or the stable C–C or C–O bonds, which are actually responsible for the formation of permanent networks. The efficiencies of all these combination reactions depend on the flexibility of the polymer chains in the medium and the ease of diffusion of the macromolecular radicals. If these radicals cannot diffuse fast enough to couple, transfer reactions with the solvent are likely to occur, which would minimize and might even completely prevent the formation of permanent crosslinks.To show the conversion of dynamic to set, non-dynamic, network, an experiment was carried out by exposing the dynamic gel, prepared by mixing 200 μL of PhI(O2CCH3)2 (0.15 M) solution and 300 μL of Sty1AA0.9 (0.73 M with respect to carboxylate groups) solution in DMAc along with trace amount of Co Blue, to UV light (365 nm) for 30 min in a glass tube (Fig. 4). After curing, 500 μL of DMAc was added to the tube along with 100 μL of glacial CH3CO2H. It was noticed that although some fraction of the gel degraded, a significant portion remained intact even after 24 h. This suggested that the dynamic gel was at least partially converted to permanently crosslinked network. However, due to the moderate efficiency of radical coupling, and inconsistent curing throughout the gel, not all the dynamic linkages were converted to permanent bonds. In addition, transfer reactions to the solvent could have occurred. The gel was then washed again with DMAc until all the side products and extra Co Blue were removed. Then, 1 mL of DMAc was added, followed by 100 μL of glacial CH3CO2H and the gel was left for 1 week. Even after that period, the gel did not degrade but swelled to a larger degree than the original dynamic precursor (Fig. 4d), owing to low crosslinking density after the curing. The main text of the article should appear here with headings as appropriate.
 | ||
| Fig. 4 (a) Gel 15 min after mixing of Sty1AA0.9 (0.73 M with respect to carboxylate groups, 300 μL) and PhI(O2CCH3)2 (0.15 M, 200 μL) solutions in DMAc. (b) Gel after 30 min of UV curing and addition of CH3CO2H in DMAc (1:5 (v/v), 600 μL). (c) Washed gel 24 h after the addition of CH3CO2H and DMAc, and (d) the same swollen gel after 1 week. | ||
To confirm the formation of permanently crosslinked network, continuous step-strain measurements were carried out on the UV-light-cured gel. In this experiment, a dynamic gel was formed by mixing 400 μL of PhI(O2CCH3)2 (0.15 M) solution with 1600 μL of Sty1AA0.7 (0.35 M) solution in DMAc. Subsequently, the gel was cured by using two UV lamps (254 nm, 21 W) for 90 min, followed by continuous step-strain measurement. The gel was subjected to high-amplitude oscillations (γ = 1300%, ω = 10 rad s−1) for 30 s, resulting in an increase in the tanδ value, which showed quasi-liquid-like behavior (Fig. 5). Moreover, G′ did not recover to even close to its original value of 230 Pa. The average G′ value measured under low amplitude oscillations (γ = 1%, ω = 10 rad s−1) was only 33 Pa, which clearly suggested the scarcity of dynamic bonds, as expected. The data indicated that the major fraction of dynamic HV bonds were converted to permanent crosslinks (since the gel did not degrade in the presence of CH3CO2H). The self-healing efficiency was significantly decreased after the second and third cycle of high-amplitude oscillations due to unrecoverable damage to the dynamic bonds and crosslinked network during the first damage.
 | ||
| Fig. 5 Continuous step-strain measurement of the UV cured gel, in which, gel was subjected to 1300% strain for 30 s, then go back to 1% strain in the linear regime for 120 s, and 3 cycles were done. | ||
Similar conversion to permanent gel was achieved by microwave heating, but the gel was either completely degraded or not properly cured even after trying several different conditions. This is due to the high dielectric constant of DMAc,81 because of which, this medium attains high temperature even at low power of the microwave irradiation and in a short time. The steep increase in temperature did not provide sufficient time for the gel to set. Therefore, a lower dielectric constant solvent, 1,4-dioxane,81 was chosen, which proved to be optimal also because it dissolved well both PhI(O2CCH3)2 and the Sty-AA copolymers, and swelled the final network polymer. The thermal curing of these gels was markedly improved compared to similar systems using DMAc as the medium as shown in Fig. S4 (ESI).†
Although the conversion of dynamic into permanent networks has been reported previously,14 the approach presented here has the advantage of using readily accessible monomers and crosslinkers. In principle, any polymer containing carboxylate groups can be employed to prepare dynamic networks, which can be readily converted into permanent ones by simple irradiation or thermal treatment.
Conclusions
Dynamic gels were prepared using ligand exchange reaction between PhI(O2CCH3)2 and the free pendant carboxylate groups in copolymers of Sty and AA. The gels exhibited self-healing, were confirmed by rheological measurements. The dynamic gel prepared by mixing 4:1 volume ratio Sty1AA0.7 (0.35 M) and PhI(O2CCH3)2 (0.15 M) solutions in DMAc, respectively, showed 85% recovery even after 1300% strain in less than 20 s over several cycles. Furthermore, the conversion of dynamic to permanent gels following exposure to UV light or microwave heating was demonstrated. The conversion of dynamic gel to permanent crosslink network was visually confirmed by adding CH3CO2H to the gel, which did not degrade the network even after a week. The formation of permanent gel was confirmed by continuous step-strain measurements, during which the gel showed poor recovery due to conversion of majority of dynamic bonds to permanent bonds by irreversible radical coupling reaction between the macromolecular carboxylate radicals and/or the alkyl radicals, which are product of their decarboxylation, produced by the photochemical or thermal cleavage of the (diacyloxyiodo)benzene bridging groups.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support provided by National Science Foundation (NSF Award Career 1455200). The authors also thank to Prof. Danieli Rodrigues and Mr Danyal Siddiqui from University of Texas at Dallas for providing help with the rheological measurements.References
- J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- J. R. Stuart, Angew. Chem., Int. Ed., 2002, 41, 899–952 Search PubMed.
- J.-M. Lehn, Aust. J. Chem., 2010, 63, 611–623 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS.
- M. C. Stuparu, A. Khan and C. J. Hawker, Polym. Chem., 2012, 3, 3033–3044 RSC.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- J.-M. Lehn, in Hierarchical macromolecular structures: 60 years after the Staudinger Nobel Prize I, ed. V. Percec, Springer, Cham, 2013, vol. 261, pp. 155–172 Search PubMed.
- N. Roy, B. Bruchmann and J.-M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 8350–8369 CrossRef CAS PubMed.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817 CrossRef CAS PubMed.
- H. Frisch, D. E. Marschner, A. S. Goldmann and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2018, 57, 2036–2045 CrossRef CAS PubMed.
- F. García and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551–3577 CrossRef.
- S. Ulrich, Acc. Chem. Res., 2019, 52, 510–519 CrossRef CAS.
- Q. Zhang, C. Huang, F. Xia and J. Su, in Electroactive Polymer (EAP) Actuators as Artificial Muscles: Reality, Potential, and Challenges, ed. Y. Bar-Cohen, The Society of Photo-Optical Instrumentation Engineers, 2004, ch. 4, pp. 95–150 Search PubMed.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- S. Billiet, X. K. D. Hillewaere, R. F. A. Teixeira and F. E. Du Prez, Macromol. Rapid Commun., 2012, 34, 290–309 CrossRef PubMed.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383 CrossRef CAS PubMed.
- M. W. Urban, Nat. Chem., 2012, 4, 80–82 CrossRef CAS PubMed.
- G. L. Fiore, S. J. Rowan and C. Weder, Chem. Soc. Rev., 2013, 42, 7278–7288 RSC.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrínyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- C. Barner-Kowollik and F. Georg Schmidt, Macromol. Chem. Phys., 2012, 213, 129–130 CrossRef CAS.
- M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 1–2 CrossRef CAS.
- Y. Yang and M. W. Urban, Adv. Mater. Interfaces, 2018, 5, 1800384–1800380 CrossRef.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- M. D. Hager, P. Greil, C. Leyens, S. van der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430 CrossRef CAS.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- M. W. Urban, Angew. Chem., Int. Ed., 2014, 53, 3775–3775 CrossRef CAS.
- Y. Yang, Polym. Chem., 2013, 5, 126–148 CAS.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- E. B. Stukalin, L.-H. Cai, N. A. Kumar, L. Leibler and M. Rubinstein, Macromolecules, 2013, 46, 7525–7541 CrossRef CAS.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- M. W. Urban, D. Davydovich, Y. Yang, T. Demir, Y. Zhang and L. Casabianca, Science, 2018, 362, 220–225 CrossRef CAS PubMed.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- H. Jiang, L. Duan, X. Ren and G. Gao, Eur. Polym. J., 2019, 112, 660–669 CrossRef CAS.
- S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed.
- M. Enke, D. Döhler, S. Bode, W. H. Binder, M. D. Hager, U. S. Schubert and S. van der Zwaag, Adv. Polym. Sci., 2016, 273, 467–112 Search PubMed.
- M. Häring and D. D. Díaz, Chem. Commun., 2016, 52, 13068–13081 RSC.
- L. Shi, P. Ding, Y. Wang, Y. Zhang, D. Ossipov and J. Hilborn, Macromol. Rapid Commun., 2019, 40, 1800837 CrossRef PubMed.
- J. I. Musher, Angew. Chem., Int. Ed. Engl., 1969, 8, 54–68 CrossRef CAS.
- R. J. Hach and R. E. Rundle, J. Am. Chem. Soc., 1951, 73, 4321–4324 CrossRef CAS.
- G. C. Pimentel, J. Chem. Phys., 1951, 19, 446–448 CrossRef CAS.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- G. Georgiev, E. Kamenska, L. Christov, I. Sideridou-Karayannidou, G. Karayannidis and A. Varvoglis, Eur. Polym. J., 1992, 28, 207–211 CrossRef CAS.
- A. Yoshimura, M. S. Yusubov and V. V. Zhdankin, Org. Biomol. Chem., 2016, 14, 4771–4781 RSC.
- A. Maity and D. C. Powers, Synlett, 2019, 30, 257–262 CrossRef CAS.
- J. V. Crivello, Adv. Polym. Sci., 1984, 62, 1–48 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., 1999, 37, 4241–4254 CrossRef CAS.
- A. Vaish and N. V. Tsarevsky, in Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jaekle, Wiley, 2018, pp. 483–514 Search PubMed.
- G. Georgiev, S. Spyroudis and A. Varvoglis, Polym. Bull., 1985, 14, 523–526 CrossRef CAS.
- G. S. Georgiev, Polym. Bull., 1999, 43, 223–230 CrossRef CAS.
- G. S. Georgiev, E. B. Kamenska, N. V. Tsarevsky and L. K. Christov, Polym. Int., 2001, 50, 313–318 CrossRef CAS.
- R. Kumar, Y. Cao and N. V. Tsarevsky, J. Org. Chem., 2017, 82, 11806–11815 CrossRef CAS PubMed.
- A. Varvoglis, Chem. Soc. Rev., 1981, 10, 377–407 RSC.
- R. Kumar, A. Vaish, T. Runčevski and N. V. Tsarevsky, J. Org. Chem., 2018, 83, 12496–12506 CrossRef CAS PubMed.
- K.-Y. Akiba and Y. Yamamoto, Heteroat. Chem., 2007, 18, 161–175 CrossRef CAS.
- A. R. Fox and K. H. Pausacker, J. Chem. Soc., 1957, 295–301, 10.1039/jr9570000295.
- H. Han and N. V. Tsarevsky, Polym. Chem., 2012, 3, 1910–1917 RSC.
- H. Han, R. Kumar and N. V. Tsarevsky, Macromol. Rapid Commun., 2019, 40, 1900073 CrossRef.
- T. Muraki, H. Togo and M. Yokoyama, Rev. Heteroat. Chem., 1997, 17, 213–243 CAS.
- H. Togo and M. Katohgi, Synlett, 2001, 565–581 CrossRef CAS.
- A. D. Asandei, O. I. Adebolu, C. P. Simpson and J.-S. Kim, Angew. Chem., Int. Ed., 2013, 52, 10027–10030 CrossRef CAS PubMed.
- G. Gody, D. A. Roberts, T. Maschmeyer and S. Perrier, J. Am. Chem. Soc., 2016, 138, 4061–4068 CrossRef CAS.
- A. Roy, in Artists’ Pigments: A Handbook of Their History and Characteristics, ed. B. H. Berrie, National Gallery of Art, Washington, 2007, vol. 4, pp. 151–177 Search PubMed.
- G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. A. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, in Fundamentals of Controlled/Living Radical Polymerization, ed. N. V. Tsarevsky and B. S. Sumerlin, RSC, Cambridge, 2013, ch. 6, pp. 205–249 Search PubMed.
- E. T. Denisov, T. G. Denisova and T. S. Pokidova, Handbook of Free Radical Initiators, Wiley, Hoboken, 2003 Search PubMed.
- F. Cech and E. Zbiral, Tetrahedron, 1975, 31, 605–612 CrossRef CAS.
- N. V. Tsarevsky, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 966–974 CrossRef CAS.
- S. Makowiec and J. Rachon, Heteroat. Chem., 2003, 14, 352–359 CrossRef CAS.
- A. Vaish, S. G. Roy and P. De, Polymer, 2015, 58, 1–8 CrossRef CAS.
- M. T. Shaw, Introduction to Polymer Rheology, John Wiley & Sons, Incorporated, Hoboken, United States, 2012 Search PubMed.
- C. Reichardt and T. Welton, in Solvents and Solvent Effects in Organic Chemistry, ed. C. Reichardt and T. Welton, Wiley, 2010, pp. 549–586 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Polymerization kinetics, characterization data, and video of self-healing. See DOI: 10.1039/c9py00664h |
| This journal is © The Royal Society of Chemistry 2019 |