
Synthesis of chain end acyl-functionalized polymers by living anionic polymerization: versatile precursors for H-shaped polymers†
Kazuki
Takahata
 ,
Satoshi
Uchida
,
Raita
Goseki
and
Takashi
Ishizone
*
,
Satoshi
Uchida
,
Raita
Goseki
and
Takashi
Ishizone
*
Department of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1-S1-13 Ohokayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: tishizon@polymer.titech.ac.jp
First published on 20th June 2019
Abstract
A series of α,ω-chain end acyl-functionalized polystyrenes with well-defined structures was synthesized by the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 addition reaction between 1,1-bis(4-(1-adamantanecarbonyl)phenyl)ethylene (1) and difunctional living anionic polystyrenes prepared with lithium naphthalenide or potassium naphthalenide in THF at −78 °C after capping with 1,1-diphenylethylene (DPE). A tailored ω-functionalized polystyrene was similarly obtained by the reaction of 1 and DPE-capped living polystyrene initiated with sec-BuLi. The DPE-capped polyisoprenyl anion and poly(tert-butyl methacrylate) anion were also effective at preparing tailored end-functionalized polymers by the termination method using 1. The electrophilicity of the C
:1 addition reaction between 1,1-bis(4-(1-adamantanecarbonyl)phenyl)ethylene (1) and difunctional living anionic polystyrenes prepared with lithium naphthalenide or potassium naphthalenide in THF at −78 °C after capping with 1,1-diphenylethylene (DPE). A tailored ω-functionalized polystyrene was similarly obtained by the reaction of 1 and DPE-capped living polystyrene initiated with sec-BuLi. The DPE-capped polyisoprenyl anion and poly(tert-butyl methacrylate) anion were also effective at preparing tailored end-functionalized polymers by the termination method using 1. The electrophilicity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in 1 was significantly enhanced by the electron-withdrawing effect of the 1-adamantylcarbonyl groups on the phenyl rings. The reaction of the living anionic polystyrene or poly(2-vinylpyridine) and the four CO terminal groups in the resulting end-functionalized polystyrene afforded H-shaped (co)polymers possessing well-defined structures. The introduced acyl terminal groups showed high reactivity toward nucleophiles, even in the presence of bulky and stiff adamantyl neighboring groups.
C bond in 1 was significantly enhanced by the electron-withdrawing effect of the 1-adamantylcarbonyl groups on the phenyl rings. The reaction of the living anionic polystyrene or poly(2-vinylpyridine) and the four CO terminal groups in the resulting end-functionalized polystyrene afforded H-shaped (co)polymers possessing well-defined structures. The introduced acyl terminal groups showed high reactivity toward nucleophiles, even in the presence of bulky and stiff adamantyl neighboring groups.
Introduction
End-functionalized polymers including telechelic and semitelechelic polymers are industrially important prepolymers for cross-linking reactions, chain extensions, and preparation of block and graft copolymers, and various methods have been developed to prepare these structurally special polymers.1–8 Among the synthetic methods for the end-functionalized polymers, living anionic polymerization is significantly efficient at achieving precise controls on molecular weight and molecular weight distribution of the polymers in addition to quantitative functionalization. In fact, a variety of well-defined ω-functionalized polymers have been synthesized by the termination of living anionic polymers using various electrophilic reagents such as carbon dioxide, epoxides, sultones, silyl chlorides, alkyl halides, and 1,1-diphenylethylenes (DPE).3–5,7–27For the above synthetic method, the chemistry of DPE derivatives is very useful and unique, since it allows a 1:1 addition reaction with strong nucleophiles, such as sec-BuLi and the living anionic polystyrene (PSt), to form the bulky π-conjugated “living DPE anions” due to negligible homopolymerizability.28,29 In the former case, the resulting low molecular weight DPE anions can initiate the polymerization of various monomers to achieve the α-functionalization of the polymer. On the other hand, the latter DPE-capped polymer anion quantitatively gave the ω-functionalized polymer if the reaction was terminated with water or methanol (MeOH). Interestingly, one can also perform the in-chain functionalization of the polymer via the sequential (co)polymerization of suitable monomers with the same DPE-capped polymer anion. In fact, various DPE derivatives having OH, NH2, NMe2, COOH, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, and SiMe2H groups are developed to synthesize well-defined end-functionalized polymers, while the protection of the functional groups is necessary in most cases.3–5,7,8,19–27
CH, and SiMe2H groups are developed to synthesize well-defined end-functionalized polymers, while the protection of the functional groups is necessary in most cases.3–5,7,8,19–27
Acyl groups, such as acetyl groups, are attractive functional groups and allow a variety of organic reactions due to their high reactivity and are also well known as electron-withdrawing groups with a positive Hammett substituent parameter (σp = 0.47).30 Although the CO moiety shows intriguing characteristics, it is very difficult to synthesize polymers possessing ketone moieties by anionic polymerization because of their inherent high reactivity toward nucleophiles and/or bases. Nucleophilic anionic initiators and propagating carbanions readily react with the highly electrophilic carbonyl moiety and/or immediately abstract the highly acidic α-hydrogens (pKa = 25). Therefore, protection of the acyl groups is usually necessary to attain the synthesis of polymers possessing reactive CO groups. For example, a well-defined poly(4-acetylstyrene) was obtained by the living anionic polymerization of the protected 4-acetylstyrene, the corresponding tert-butyldimethylsilyl enol ether, and the subsequent deprotection of the resulting polymer.31,32
In contrast, we have recently succeeded in the direct living anionic polymerization of a styrene (St) derivative carrying the acyl group, 1-adamantyl-4-vinylphenyl ketone (AdK), to form a polymer possessing well-defined chain structures.33 During the polymerization, the CO groups in AdK and poly(AdK) coexist with the propagating poly(AdK) anion stabilized by the electron-withdrawing acyl group (Chart 1). A bulky adamantyl substituent without acidic α-hydrogens might play an important role in realizing the living anionic polymerization of the acylstyrene derivative without protection. Nevertheless, the CO groups in the resulting poly(AdK) still maintain the electrophilicity toward the nucleophilic attack of Grignard reagents such as methylmagnesium iodide. These stimulate us to explore the possibility of the direct anionic end-functionalization using a novel DPE derivative carrying two acyl groups, 1,1-bis(4-(1-adamantanecarbonyl)phenyl)ethylene (1), without the protection procedure. In the case of 1, the π-conjugation system is notably expanded and the electron density of the CC bond is significantly reduced by two electron-withdrawing acyl groups, inducing the high reactivity toward the nucleophilic addition reaction.
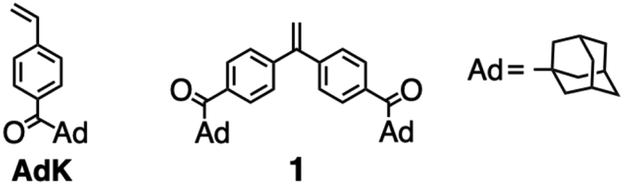 | ||
| Chart 1 Structures of AdK and 1. | ||
In this study, we attempted the direct end-functionalization of PSt, polyisoprene (PIsp), and poly(tert-butyl methacrylate) (PtBMA) under anionic conditions to utilize the high electrophilicity of 1. Tailored semitelechelic and telechelic polymers possessing reactive CO groups were anionically synthesized by the termination method using 1 as a versatile electrophile. Furthermore, the synthesis of H-shaped polymers was examined by the reaction of the resulting telechelic type polymer carrying four acyl end functionalities and the living anionic polymers in order to demonstrate the reactivity of the introduced CO terminal moieties.
Results and discussion
A novel DPE derivative carrying two acyl groups, 1, was synthesized by the four step reactions shown in Scheme 1. A monofunctional Grignard reagent of p-dibromobenzene was reacted with 1-adamantanecarbonyl chloride in the presence of CuCl to give 1-adamantyl-4-bromophenyl ketone. The resulting ketone was then converted into a cyclic acetal by treating with ethylene glycol. The obtained acetal was reacted with magnesium to give the corresponding Grignard reagent. Then, ethyl acetate was reacted with two equivalents of the Grignard reagent of the acetal to afford a tertiary alcohol. The simultaneous acetal deprotection and dehydration of the resulting alcohol under acidic conditions gave a diacyl DPE derivative, 1. After recrystallization from a mixed solvent of CH2Cl2 and hexane, 1 was used for the end-functionalization in the anionic polymerization.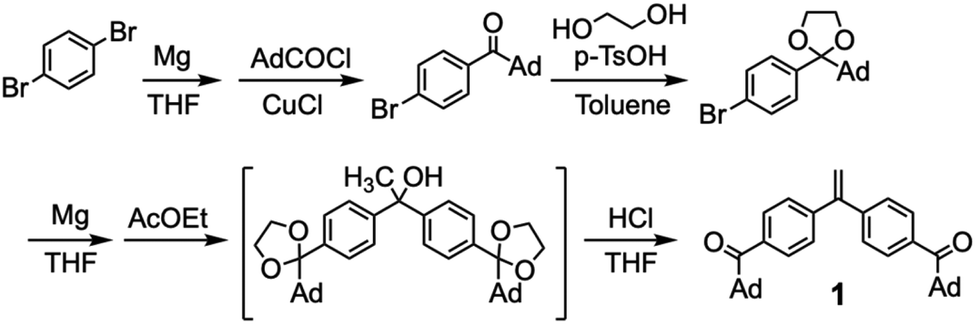 | ||
| Scheme 1 Synthesis of 1. | ||
Reaction of 1 and living anionic polymers
At first, a difunctional living PSt was synthesized by the reaction of St with potassium naphthalenide (K-Naph) at −78 °C in THF. To the dark red solution of living PSt, a THF solution of 1 was added at −78 °C, and the color of the reaction mixture immediately changed to dark blue.34 The blue coloration was maintained for 15 min and instantaneously disappeared by adding acetic acid (AcOH) to terminate the reaction, indicating the presence of the DPE carbanion derived from 1. After precipitation of the polymerization system in MeOH, the PSt was quantitatively obtained and characterized by GPC. The GPC curve showed a multimodal shape, and the molecular weight (Mn = 17 kg mol−1) was much higher than the calculated value (Mn = 5.5 kg mol−1) as shown in Fig. 1A. The elution volume of the lowest molecular weight fraction in the GPC curve corresponded to the theoretical molecular weight of the PSt. The main fraction in the higher molecular weight region indicated that a serious intermolecular side reaction occurred in the reaction of the difunctional living PSt and 1 even at −78 °C. The polystyryl anion might simultaneously attack not only the carbon–carbon double bond of 1 but also the carbonyl groups, as shown in Scheme 2. This resulted in the formation of the higher molecular weight fraction in the GPC curve (Fig. 1A). | ||
| Fig. 1 GPC curves of polymers obtained by the reaction of 1 with the polystyryl anion, Mw/Mn = 2.24 (A, run 1), and the reaction of 1 with the DPE-capped polystyryl anion, Mw/Mn = 1.05 (B, run 2). | ||
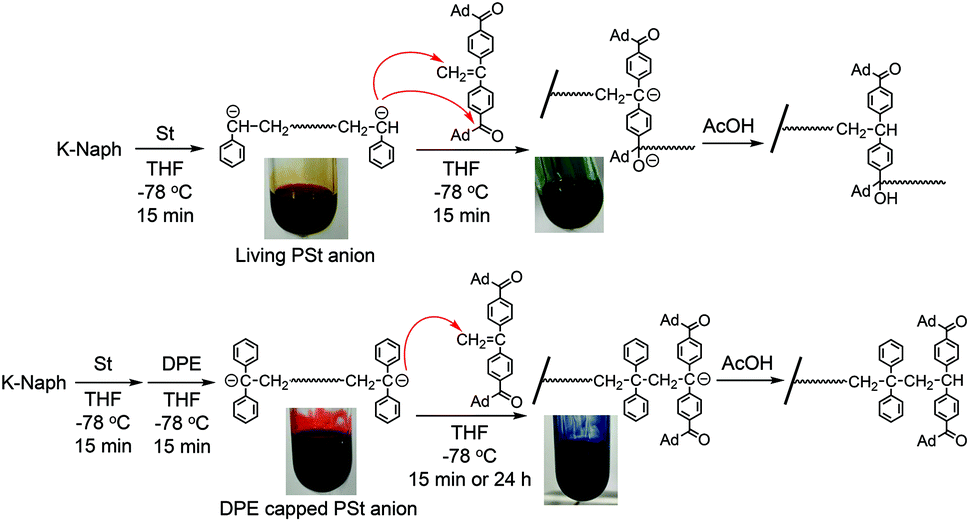 | ||
| Scheme 2 End-functionalization of 1 with the living anionic PSt and its DPE capped anion. | ||
Then, we reduced the nucleophilicity of the carbanion of the difunctional living PSt by reaction with DPE in order to prevent the side reaction to the carbonyl group, as shown in Scheme 2.35,36 The polystyryl anion was reacted with DPE at −78 °C for 15 min to form a red-colored DPE-capped anion. When 1 was added to the DPE capped living PSt at −78 °C, a rapid color change similarly occurred from red to dark blue. After 15 min, the reaction was again quenched with AcOH, and the blue coloration immediately disappeared. The GPC curve of the resulting PSt is unimodal and narrow (Mw/Mn = 1.05) (Fig. 1B). The resulting PSt had a predicted molecular weight based on the molar ratio of St and K-Naph along with the quantitative introduction of 1. In the 1H NMR spectrum of the end-functionalized polymer, typical signals corresponding to the adamantyl groups were observed between 1.76 and 1.99 ppm, while those signals overlapped with the signals of the polymer main chain (Fig. S1†). The signal of the carbonyl carbon of the ketone moiety was also observed at 209.3 ppm in the 13C NMR spectrum in addition to the four carbon signals derived from the adamantyl ring in the aliphatic region (Fig. S2†). In the IR spectrum, a strong adsorption of the CO group appeared at 1668 cm−1 in the end-functionalized polymer (Fig. S3†). Fig. 2 illustrates the MALDI-TOF-MS of the polymer after treating with 1. Only one series of signals derived from similar structures having different degrees of polymerization of St and the same terminal moieties was observed. In fact, a series of observed masses agreed with the calculated values of the St monomer unit multiplied by the degree of polymerization plus two DPE units and two 1 units plus a silver cation (Ag+). These results clearly indicate that the serious side reaction toward the CO groups is completely suppressed in the reaction of the DPE anion and 1 (Scheme 2). In other words, the π-conjugated and bulky DPE anion at the polymer terminals exclusively attacks the carbon–carbon double bond of 1 not its CO groups. It is noteworthy that even a less nucleophilic DPE anion quantitatively undergoes the 1:1 addition reaction with a similar DPE derivative, 1, to form the blue-colored DPE anion. It is believed that DPE derivatives cannot undergo homopolymerization to form the DPE–DPE sequence because of steric reasons or the low ceiling temperature.28,29 However, our results confirm that the crossover 1:1 reaction of the DPE anion and other DPE derivatives is possible to form the DPE–DPE sequence if the electrophilicity of the DPE derivative is significantly enhanced by the electron-withdrawing carbonyl groups. This also suggests that the low ceiling temperature is more important than the steric effect in the homopolymerization of the DPE derivatives. As shown in Table 1, a series of well-defined CO end-functionalized PSts with different molecular weights could be similarly obtained using the DPE-capped living PSt and 1 by changing the molar ratio between St and K-Naph (runs 2–4). These polymers possessed the predicted molecular weights, narrow molecular weight distributions (Mw/Mn < 1.09) and quantitative carbonyl end-functionalities.
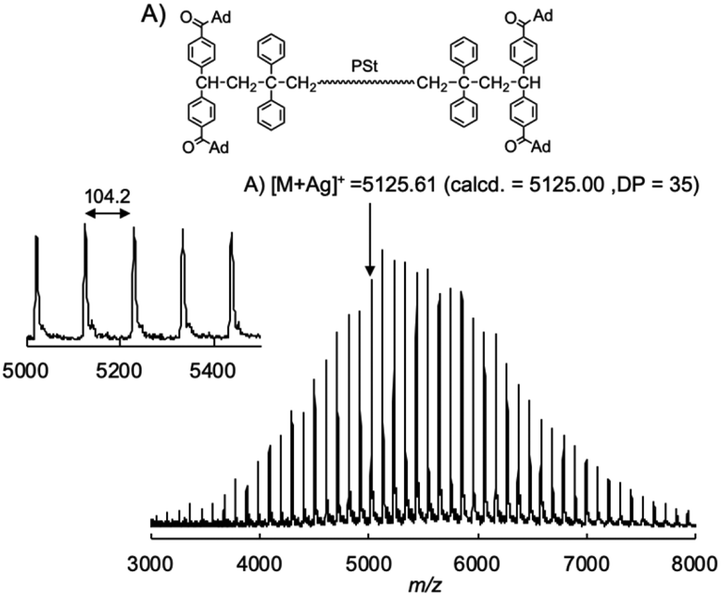 | ||
| Fig. 2 MALDI-TOF-MS of the chain end acyl-functionalized PSt. | ||
| Run | Initiator | Monomer | DPE | 1 | Timea | M n (kg mol−1) |
M
w/Mnd |
T
ge (°C) |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| mmol | mmol | mmol (eq.) | mmol (eq.) | calcdb | obsdc | ||||||
| All polymerization reactions were conducted in THF at −78 °C. St was polymerized for 15 min. Isp was polymerized for 4 h. tBMA was polymerized for 1 h. DPE was reacted for 15 min.a Reaction time of 1 with living anionic polymers.b [(MW of monomer) × (M/I) + (MW of DPE derivatives)] × 2.c By GPC with triple detectors using PSt standards.d By GPC using PSt standards.e Measured by DSC.f Potassium naphthalenide.g Lithium naphthalenide.h (MW of initiator) + (MW of monomer) × (M/I) + (MW of DPE derivatives).i Diphenylmethylpotassium. | |||||||||||
| 1 | K-Naphf | 0.170 | St | 2.56 | — | 0.244 (1.4) | 15 min | 5.3 | 17 | 2.24 | — |
| 2 | K-Naph | 0.183 | St | 2.54 | 0.295 (1.6) | 0.307 (1.7) | 15 min | 4.3 | 3.9 | 1.05 | 119 |
| 3 | K-Naph | 0.108 | St | 5.49 | 0.207 (1.9) | 0.234 (2.2) | 15 min | 12 | 12 | 1.05 | 110 |
| 4 | K-Naph | 0.137 | St | 3.19 | 0.232 (1.7) | 0.227 (1.7) | 24 h | 6.2 | 6.3 | 1.07 | 117 |
| 5 | Li-Naphg | 0.120 | St | 2.71 | — | 0.214 (1.8) | 15 min | 5.7 | 9.0 | 1.79 | — |
| 6 | sec-BuLi | 0.0751 | St | 2.39 | — | 0.214 (2.8) | 15 min | 3.8h | 4.0 | 1.20 | — |
| 7 | Li-Naph | 0.152 | St | 2.90 | 0.233 (1.6) | 0.306 (2.0) | 2 h | 5.4 | 5.8 | 1.08 | n.d. |
| 8 | sec-BuLi | 0.112 | St | 4.49 | 0.223 (2.0) | 0.220 (2.0) | 2 h | 4.8h | 3.5 | 1.01 | 99 |
| 9 | K-Naph | 0.147 | Isp | 6.25 | 0.265 (1.8) | 0.286 (1.9) | 2 h | 7.3 | 7.5 | 1.03 | 21 |
| 10 | Ph2CHKi | 0.114 | tBMA | 3.88 | — | 0.230 (2.0) | 24 h | 5.3h | 4.6 | 1.08 | 97 |
We next changed the countercation of the polystyryl anion from potassium to lithium to check the versatility of the end-functionalization of 1. We used lithium naphthalenide (Li-Naph) and sec-BuLi to initiate the polymerization of St in THF and masked the resulting polystyryllithiums with DPE to lower the nucleophilicity of the propagating chain ends. The end-functionalization of the polystyryllithium with 1 was also attempted at −78 °C in THF for 15 min to 2 h. Similar to the result of polystyryldipotassium, an α,ω-difunctional PSt with a tailored structure was obtained after addition of 1 to the DPE-capped difunctional living PSt synthesized with Li-Naph (run 7). In the case of sec-BuLi, a well-defined semitelechelic PSt possessing two carbonyl groups at the ω-terminal formed after the end-functionalization using 1 (run 8). On the other hand, the multimodal GPC curves of the polymers were observed when the end-functionalization was performed without DPE-capping of the living anionic polymers (runs 5 and 6). This also suggests that a serious side reaction occurred between the CO groups of 1 and the polystyryl anion carrying the lithium countercation along with the desirable nucleophilic addition to the CC bond.
We also succeeded in the end-functionalization of living anionic PIsp with 1. In this case, a difunctional living PIsp was first synthesized by the initiation of isoprene (Isp) with K-Naph in THF at −78 °C for 4 h, and it was reacted with DPE to reduce the nucleophilicity of the propagating polyisoprenyl anion. After reacting the DPE-capped polyisoprenyldipotassium with 1 for 2 h, a tailored CO end-functionalized PIsp was obtained (run 9). The introduction of CO moieties into the PIsp was confirmed by 13C NMR, IR, and MALDI-TOF-MS measurements.
We then attempted to use the living anionic polymer of tert-butyl methacrylate (tBMA) in the end-functionalization of 1. tBMA was first polymerized with diphenylmethylpotassium (Ph2CHK) in THF at −78 °C for 1 h. To living PtBMA, 1 was directly added to react in THF at −78 °C, as shown in Scheme 3. A color change of the reaction system instantaneously occurred from colorless to dark blue, indicating that the rapid nucleophilic addition of the enolate anion of PtBMA toward 1 took place (run 10). After termination with AcOH, the resulting PtBMA was characterized by 1H NMR, 13C NMR, IR, GPC, and MALDI-TOF MS measurements.37,38 All the analytical results supported that the quantitative end-functionalization of PtBMA was achieved by the 1:1 addition reaction of 1 and the propagating enolate anion of PtBMA. This means that the electrophilicity of 1 is markedly enhanced by two electron-withdrawing carbonyl substituents similar to the corresponding St derivative, AdK.33 Therefore, even the less nucleophilic enolate anion of PtBMA can quantitatively undergo the 1:1 addition reaction to 1 to form a DPE end-functionalized PtBMA. Thus, 1 acts as an effective electrophile in the anionic end-functionalization of PSt, PIsp, and PtBMA to introduce the electrophilic CO moieties quantitatively without protection of the carbonyl group.
 | ||
| Scheme 3 End-functionalization of the living PtBMA with 1. | ||
Synthesis of H-shaped polymers by grafting reaction
As described above, we successfully synthesized the telechelic PSt possessing four reactive CO functionalities in both terminals. This is a suitable starting material for synthesizing an H-shaped polymer via a grafting reaction to the four carbonyl groups. Then, 1.4 equivalents of the living anionic PSt prepared with sec-BuLi in THF at −78 °C were directly reacted with the CO groups of the end-functionalized PSt in THF at −78 °C for 12 h (Scheme 4). During the reaction, the orange coloration of the living PSt was maintained and disappeared on the addition of MeOH to quench the polymer reaction. After the reaction, a bimodal GPC curve of the crude reaction product was obtained as shown in Fig. 3B. The lower molecular weight side was corresponding to an excess fraction of the reacted PSt (Mn = 16000 g mol−1), and the higher molecular weight side should be a grafted H-shaped polymer. No trace of the starting CO end-functionalized PSt (Mn = 3900 g mol−1) was observed in the crude product, indicating the high efficiency of the grafting reaction. The higher molecular weight fraction of the reaction product could be isolated in 36% yield by the fractional precipitations in a mixed solvent of cyclohexane and hexane. Fig. 3 illustrates three GPC curves of the starting CO end-functionalized PSt, the crude product after the grafting reaction, and the isolated H-shaped PSt, respectively. The GPC curve of the isolated H-shaped PSt possessed narrow molecular weight distribution (Mw/Mn = 1.04). Furthermore, the molecular weight of the isolated H-shaped PSt (Mn = 69000 g mol−1) measured by RALLS-GPC agreed with the calculated value (Mn = 67000 g mol−1) of the expected structure. All these results obtained indicated that the quantitative grafting reactions on the four ketone moieties at both chain ends smoothly proceeded with the living polystyryllithium to give the well-defined H-shaped PSt.
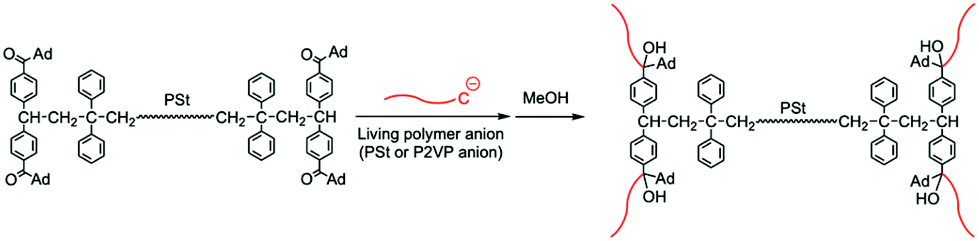 | ||
| Scheme 4 Synthesis of H-shaped polymers using the CO end-functionalized telechelic PSt and living anionic polymers of St and 2VP. | ||
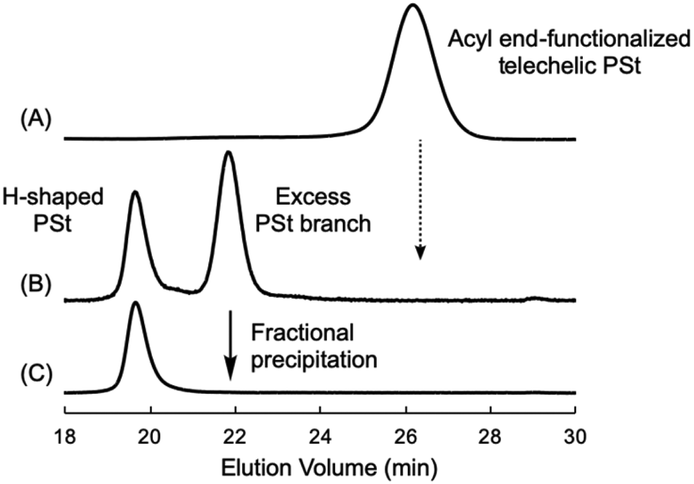 | ||
| Fig. 3 GPC curves of the starting acyl end-functionalized telechelic PSt (A), the crude product after the grafting reaction (B), and the isolated H-shaped PSt (C). | ||
Furthermore, a tailored H-shaped block copolymer possessing a PSt middle segment and four poly(2-vinylpyridine) (P2VP) terminal branch segments was successfully synthesized via similar synthetic strategy (Scheme 4). In this case, a living P2VP prepared with sec-BuLi in THF was reacted with the acyl end-functionalized PSt having four CO terminal groups in THF at −78 °C for 24 h. The observed composition and molecular weight of the isolated copolymer agreed with the calculated values along with a narrow molecular weight distribution (Mw/Mn = 1.02), indicating the formation of the well-defined H-shaped block copolymer. Thus, even the CO groups possessing a neighboring bulky adamantyl group still keep the sufficient electrophilicity toward the nucleophilic addition reactions using living polymeric anions of St and 2-vinylpyridine (2VP). In fact, this observed reactivity is in accordance with the observation in the end-functionalization reaction of 1 with the living PSt showing high nucleophilicity. In each case, the 1-adamantylcarbonyl group in 1 or the end-functionalized polymer was attacked by the non-DPE-capped polystyryl anion or P2VP anion to produce the addition products.
The glass transition temperature (Tg) of the CO end-functionalized polymers was finally analyzed by differential scanning calorimetry (DSC). In each measurement, a glass transition behavior was observed in the DSC profile before the thermal degradation. It should be noted that the Tgs of the polymers did not increase with the Mn values. Interestingly, the Tgs of the PSt samples having lower Mn values were rather higher than those of the higher Mn polymers. The movement of the polymer main chain might be restricted by the bulky adamantyl groups at the terminals to result in the high Tg, particularly observed in the lower Mn samples. In fact, the Tg value of the PSt reached 119 °C in the lowest Mn sample (Mn = 3900 g mol−1), and it was significantly higher than that of the standard PSt having a comparable Mn value (Mn = 3400 g mol−1, Tg = 75 °C). It is well known that the introduction of adamantyl groups into polymers usually induced the drastic increase of the Tg values.33,39–45 The observed results indicated that the remarkable effect of the adamantyl substituents on increasing the Tg was realized not only in the repeating units and the main chain but also at the terminals of the end-functionalized polymers.
Experimental
Materials
All reagents were purchased from Kanto Chemical Co., Inc., and used without purification unless otherwise stated. St was washed with aqueous solution of NaOH (10 wt%) and water and dried over MgSO4 (Aldrich). After filtration, it was distilled over CaH2 under reduced pressure. Finally, it was distilled over dibutylmagnesium (3 mol%) and dissolved in dry THF on a high vacuum line (10−6 mmHg). DPE was purified by vacuum distillation over CaH2 and finally distilled over n-BuLi on the vacuum line. tBMA was distilled over CaH2 under reduced pressure and finally distilled from trioctylaluminum on the vacuum line. Isp was dried over CaH2 and distilled from dibutylmagnesium (3 mol%) on the vacuum line. 2VP was stirred over KOH overnight. After removal of KOH by filtration, it was distilled over CaH2 under reduced pressure. Finally, it was distilled over trioctylaluminum on the vacuum line. LiCl was dried under heating for 72 h on the vacuum line. The regents were diluted with dry THF as 0.7–1.0 M solutions and were stored at −30 °C in glass ampules equipped with a break-seal until ready to use for the anionic polymerization. THF used as a polymerization solvent was refluxed over a sodium wire for 5 h, distilled over LiAlH4 under nitrogen, and finally distilled from the sodium naphthalenide solution on the vacuum line. n-Heptane was washed with concentrated H2SO4, water, and NaHCO3 aqueous solution, dried over anhydrous MgSO4, and then dried over P2O5 for 1 day under reflux. Finally, it was distilled from 1,1-diphenylhexyllithium solution on the vacuum line.Initiators
K-Naph and Li-Naph were prepared by the reaction of potassium or lithium and 1.5-fold naphthalene in THF at room temperature for 48 h under argon atmosphere. sec-BuLi (1.0 M in cyclohexane) was used without purification and diluted with dry n-heptane. Ph2CHK was synthesized by the reaction of K-Naph and 1.5-fold diphenylmethane in dry THF under argon at room temperature for 48 h. The concentrations of initiators were determined by colorimetric titration using standardized 1-octanol in THF in a sealed reactor under vacuum, as previously reported.46 These initiators were stored at −30 °C in ampules equipped with break-seals.Synthesis of 1-adamantyl-4-bromophenyl ketone
1,4-Dibromobenzene (26.0 g, 110 mmol) was reacted with Mg (3.15 g, 130 mmol) in dry THF (100 mL) at room temperature for 1 h under nitrogen. The resulting Grignard reagent in THF was added dropwise to a THF solution (50 mL) of 1-adamantanecarbonyl chloride33 (19.9 g, 100 mmol) and CuCl (0.50 g, 4.9 mmol) at 0 °C under nitrogen, and the mixture was stirred at room temperature for 12 h. The reaction was quenched with water (50 mL) and acidified with 2 M HCl (50 mL), and the solvent was evaporated under reduced pressure. The residue was extracted with CHCl3 (50 mL) three times, and the combined organic layer was washed three times with water (50 mL) and dried over MgSO4. After removal of MgSO4 by filtration, the solvent of the filtrate was evaporated, and a pale yellow solid was obtained. The resulting solid was purified by flash column chromatography (silica gel, hexane) followed by recrystallization using a mixture of CH2Cl2 and hexane to give 1-adamantyl-4-bromophenyl ketone (22.8 g, 71.2 mmol, 71%, mp = 108–109 °C) as a white solid.1H NMR (Fig. S17,† 300 MHz; CDCl3; ppm) δ = 1.68–2.10 (m, 15H, adamantyl), 7.45 (d, J = 8.5 Hz, 2H, aromatic), 7.51 (d, J = 8.5 Hz, 2H, aromatic).
13C NMR (Fig. S18,† 75 MHz; CDCl3; ppm) δ = 28.2 (adamantyl), 36.5 (adamantyl), 39.1 (adamantyl), 47.0 (adamantyl), 124.9 (Ar, C–Br), 129.0 (Ar), 131.3 (Ar), 138.2 (Ar–CO), 208.8 (CO).
Synthesis of 2-(1-adamantyl)-2-(4-bromophenyl)-1,3-dioxolane
A toluene (200 mL) solution of 1-adamantyl-4-bromophenyl ketone (11.3 g, 35.3 mmol), ethylene glycol (12.4 g, 200 mmol), and a catalytic amount of p-toluenesulfonic acid (10 mg) was refluxed with a Dean–Stark trap to remove water azeotropically for 5 h. The reaction was quenched with a saturated aqueous NaHCO3 solution (100 mL). The organic layer was washed three times with water (50 mL), dried over MgSO4, and filtered. After the solvent was evaporated in vacuo, a yellow solid was obtained. The resulting yellow solid was purified by flash column chromatography (silica gel, hexane) and by recrystallization using a mixture of CH2Cl2 and hexane to afford a white solid of 2-(1-adamantyl)-2-(4-bromophenyl)-1,3-dioxolane (5.89 g, 16 mmol, 45%, mp = 139–140 °C).1H NMR (Fig. S19,† 300 MHz; CDCl3; ppm) δ = 1.50–2.10 (m, 15H, adamantyl), 3.64 and 3.91 (2s, 4H, O–CH2–CH2–O), 7.27 (d, J = 8.3 Hz, 2H, aromatic), 7.41 (d, J = 8.3 Hz, 2H, aromatic).
13C NMR (Fig. S20,† 75 MHz; CDCl3; ppm) δ = 28.3 (adamantyl), 36.4 (adamantyl), 37.0 (adamantyl), 39.8 (adamantyl), 64.7 (–O–CH2–CH2–O–), 112.9 (Ar), 121.8 (Ar), 130.1 (Ar), 130.3 (Ar), 139.0 (O–C–O).
Synthesis of 1,1-bis(4-(1-adamantanecarbonyl)phenyl)ethylene (1)
2-(1-Adamantyl)-2-(4-bromophenyl)-1,3-dioxolane (4.97 g. 13.7 mmol) was reacted with Mg (0.67 g, 27.5 mmol) in dry THF (20 mL) at refluxing temperature for 7 h under nitrogen. A THF (20 mL) solution of ethyl acetate (0.48 g, 5.5 mmol) was added dropwise to the resulting Grignard regent at 0 °C, and the mixture was stirred at room temperature for 12 h. The reaction mixture was quenched with water (50 mL) and acidified with 2 M HCl (50 mL). After the solvent was evaporated, the aqueous layer was extracted three times with CHCl3 (50 mL). The organic layer was washed three times with water (50 mL), dried over MgSO4, and filtered. After the solvent was evaporated in vacuo, a pale yellow solid was obtained. Then, the resulting solid was dissolved in a mixture of THF (300 mL) and 2 M HCl (50 mL) and refluxed for 40 h. The reaction was quenched with a saturated aqueous NaHCO3 solution (100 mL), and the layers were separated. The aqueous layer was extracted three times with CHCl3 (50 mL), and the combined organic layer was washed three times with water (50 mL), dried over MgSO4, and filtered. The solvent was removed in vacuo to give a yellow liquid. The resulting yellow liquid was purified by flash column chromatography (silica gel, hexane:ethyl acetate = 98:2) followed by recrystallization using a mixture of CH2Cl2 and hexane to afford a white solid of 1 (0.57 g, 1.13 mmol, 21%, mp = 152–153 °C).
1H NMR (Fig. S21,† 300 MHz; CDCl3; ppm) δ = 1.75 (m, 12H, adamantyl), 2.03–2.10 (m, 18H, adamantyl), 5.56 (s, 2H, CCH2), 7.36 (d, J = 8.3 Hz, 2H, aromatic), 7.56 (d, J = 8.3 Hz, 2H, aromatic).
13C NMR (Fig. S22,† 75 MHz; CDCl3; ppm) δ = 28.1 (adamantyl), 36.6 (adamantyl), 39.0 (adamantyl), 47.0 (adamantyl), 116.4 (CH2), 127.6 (Ar), 127.9 (Ar), 138.9 (Ar–CO), 142.9 (Ar), 148.7 (–CCH2), 209.7 (Ad–CO).
IR (KBr; cm−1) 2909, 2881, 2847, 1654 (CO), 1601, 1267, 1234, 1181, 989, 856.
MALDI-TOF-MS [M + H]+ = 505.31 Da (calcd 505.31 Da).
After recrystallization, 1 was dried overnight on the vacuum line over P2O5. Then, 1 was diluted with dry THF as a 0.05 M solution and stored at −30 °C in a glass ampule equipped with a break-seal for the end-functionalization.
Synthesis of chain end acyl-functionalized PSt
All the anionic polymerization and end-functionalization reactions were carried out under high vacuum conditions (10−6 mmHg) in all-glass apparatus equipped with break-seals.46 In a typical procedure (Table 1, run 2), a difunctional living PSt was prepared by the anionic polymerization of St (0.26 g, 2.54 mmol) with K-Naph (0.183 mmol) as an initiator in THF at −78 °C for 15 min. Then, DPE (0.295 mmol, 1.6 equivalents) in THF (2 mL) was added and reacted at −78 °C for 15 min to reduce the nucleophilicity of the living anionic PSt. A THF solution (6 mL) of 1 (0.307 mmol, 1.7 equivalents) was added to the solution of DPE-capped living PSt and allowed to stand for 15 min at −78 °C. Finally, the polymerization reaction was terminated with degassed AcOH. A polymer (0.38 g, 98%) was quantitatively obtained by pouring the reaction solution to MeOH. The obtained polymer was purified by reprecipitation from THF to hexane and the subsequent freeze-drying from the benzene solution.1H NMR (400 MHz; CDCl3; ppm) δ = 0.90–2.07 (br, main chain CH2CH), 1.76 (br, 24H, adamantyl group), 1.99–2.04 (br, 36H, adamantyl group), 3.49 (s, 2H, terminal CH), 6.47–7.24 (br, aromatic).
13C NMR (100 MHz; CDCl3; ppm) δ = 28.3 (adamantyl), 36.7 (adamantyl), 39.2 (adamantyl), 40.4–44.1 (main chain), 47.9 (adamantyl), 125.8–128.4 (Ar), 145.4–147.7 (Ar), 209.3 (CO).
IR (KBr; cm−1) 3060, 3026, 2921, 2849, 1668 (CO), 1602 1493, 1452, 1028, 756, 700, 540.
Synthesis of chain end acyl-functionalized PtBMA
A living anionic PtBMA was obtained by the anionic polymerization of tBMA (0.55 g, 3.88 mmol) using Ph2CHK (0.114 mol) as an initiator in THF at −78 °C for 1 h (Table 1, run 10). Then, a THF solution (5 mL) of 1 (0.230 mmol, 2.0 equivalents) was added to the living PtBMA anion and reacted for 24 h at −78 °C. The end-functionalization reaction was terminated with AcOH. A polymer (0.59 g, 94%) was obtained by pouring the reaction solution to a mixture of MeOH and H2O (MeOH:H2O = 5:1). The obtained PtBMA was purified by reprecipitation from its THF solution to MeOH/H2O and by freeze-drying from the benzene solution.
1H NMR (500 MHz; CDCl3; ppm) δ = 1.42 (s, –C–(CH3)3), 0.74–2.04 (br, –CH2–C(CH3)–), 1.72 (br, 12H, adamantyl), 1.97–2.04 (br, 18H, adamantyl), 3.90 (s, 1H, terminal CH), 4.20 (s, 1H, terminal CH), 7.11–7.53 (br, aromatic).
13C NMR (125 MHz; CDCl3, ppm) δ = 18.5 (α-CH3), 27.9–28.2 (–C–(CH3)3), 29.8 (adamantyl), 36.7 (adamantyl), 39.2 (adamantyl), 46.3–53.0 (–CH2–C–), 46.5 (adamantyl), 80.6–80.7 (–O–C(CH3)3), 127.6–128.6 (Ar), 176.3–177.0 (CO ester), 209.2 (CO ketone).
IR (KBr; cm−1) 2978, 2932, 1725 (CO ester), 1668 (CO ketone), 1476, 1458, 1392, 1367, 1271, 1252, 1138, 849.
Synthesis of H-shaped PSt
A polymer reaction was also carried out under high vacuum conditions (10−6 mmHg) in all-glass apparatus equipped with break-seals. A living polystyryl anion was firstly prepared by the polymerization of St (1.27 g, 12.2 mmol) with sec-BuLi (0.101 mmol) in THF at −78 °C. Then, to the solution of the polystyryl anion, a THF solution of chain end acyl tetra-functionalized PSt (Mn = 3.9 kg mol−1, 0.0180 mmol, 0.072 mol of the CO functionalities) was added and reacted at −78 °C for 12 h. During the reaction, the orange coloration of the living PSt was maintained. Finally, the reaction was terminated with MeOH. A polymer of white powder was obtained by pouring the reaction solution to MeOH. A bimodal GPC curve of the reaction system was obtained, which was corresponding to the objective H-shaped polymer and the excess amount of PSt branch. The H-shaped PSt was isolated in 36% yield by repeating fractional precipitations (cyclohexane/hexane) and purified by freeze-drying from the benzene solution.
1H NMR (400 MHz; CDCl3; ppm) δ = 0.58–0.72 (m, 24H, sec-Bu), 0.98–2.19 (br, main chain and adamantyl group), 3.75 (s, 2H, terminal CH), 6.15–7.25 (br, aromatic).
IR (KBr; cm−1) 3059, 3025, 2922, 2848, 1492, 1452, 755, 698, 539.
Measurements
Both 1H and 13C NMR spectra were measured on a Bruker DPX300, a JEOL JMN-AL400, or a Bruker Ascend 500 in CDCl3. Chemical shifts were recorded in ppm downfield relative to CHCl3 (δ 7.26) and CDCl3 (δ 77.1) for 1H and 13C NMR as a standard, respectively. IR spectra were recorded on a JASCO FT/IR-4100 instrument using either an attenuated total reflectance (ATR) attachment or a KBr disk. SEC curves for determination of Mw/Mn were obtained in THF at 40 °C at a flow rate of 1.0 mL min−1 with a Viscotek TDA305 equipped with three PSt gel columns (TSKgelG5000HHR + G4000HHR + G3000HHR). Three detectors including a viscometer, right angle laser light scattering detection (RALLS), and refractive index (RI) were applied for the online SEC system in order to determine the absolute molecular weights of the polymer. The melting point of 1 and the Tg of chain end acyl-functionalized polymers were measured by differential scanning calorimetry (DSC) using a Seiko instrument DSC6220 apparatus under nitrogen flow. The sample was heated to 200 °C, cooled to 30 °C, and then scanned at a rate of 1 °C min−1 (for melting point) or 5 °C min−1 (for Tg). MALDI-TOF-MS spectra were recorded on a SHIMADZU AXIMA-CFR MASS spectrometer by using a nitrogen laser (λ = 337 nm) and with the operating conditions at an accelerating potential of 20 kV in a linear-positive ion mode with pulsed ion extraction. A sample solution was prepared by mixing a THF solution containing a polymer specimen (10 μL, 5 mg mL−1), a THF solution of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB, Tokyo Chemical Industry Co., Ltd) or dithranol (10 μL, 20 mg mL−1), and a THF solution of silver trifluoroacetate or potassium trifluoroacetate (10 μL, 1 mg mL−1). Mass values were calibrated by the four-point method using peaks of peptide plus Na+ at m/z = 3613.44, peptide plus Na+ at m/z = 4793.90, peptide plus Na+ at m/z = 5975.30, and peptide plus Na+ at m/z = 7241.87. The calibration sample of MALDI-TOF-MS was purchased from Polymer Factory.Conclusions
In conclusion, we have succeeded in the synthesis of chain end 1-adamantylcarbonyl-functionalized polymers by the 1:1 addition of the acyl-substituted DPE derivative 1 and a series of living anionic polymers derived from St, Isp, and tBMA. Undesired side reactions between the CO bond of 1 and the living anionic polymers often take place when the nucleophilicity of the propagating anion is too strong. On the other hand, the direct end-functionalization of 1 quantitatively proceeds without side reactions when the less nucleophilic DPE-capped anion or PtBMA enolate anion is employed in the reaction. The high electrophilicity of 1 derived from the electron-withdrawing acyl groups is essential for the quantitative end-functionalization using the propagating carbanions showing low nucleophilicity.
The CO groups in 1 during the course of the 1:1 addition functionalization are intact, indicating the coexistence of the DPE anion derived from 1 and the CO substituents. On the other hand, the high reactivity of the CO groups in the resulting telechelic polymers is demonstrated by the quantitative reaction using the living anionic PSt or P2VP. It is thus confirmed that the acyl end-functionalized polymers are versatile precursors for the synthesis of H-shaped polymers with well-defined chain structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in Aid (No. 17K19149) from the Ministry of Education, Science, Sports, and Culture, Japan.Notes and references
- A. Bertrand, F. Lortie and J. Bernard, Macromol. Rapid. Commun., 2012, 33, 2062–2091 CrossRef CAS PubMed.
- H. Durmaz, A. Sanyal, G. Hizal and U. Tunca, Polym. Chem., 2012, 3, 825–835 RSC.
- X. Wang, J. Xia, J. He, F. Yu, A. Li, J. Xu, H. Lu and Y. Yang, Macromolecules, 2006, 39, 6898–6904 CrossRef CAS.
- N. Hadjichristidis and A. Hirao, Anionic Polymerization: Principles, Practice, Strength, Consequences and Applications, Springer, 2015 Search PubMed.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- A. Hanisch, H. Schmalz and A. H. E. Müller, Macromolecules, 2012, 45, 8300–8309 CrossRef CAS.
- R. Goseki, S. Ito, Y. Matsuo, T. Higashihara and A. Hirao, Polymers, 2017, 9, 470 CrossRef PubMed.
- R. P. Quirk, J. Yin and L. J. Fetters, Macromolecules, 1989, 22, 85–90 CrossRef CAS.
- R. P. Quirk and J.-J. Ma, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2031–2037 CrossRef CAS.
- R. P. Quirk and J. Kim, Macromolecules, 1991, 24, 4515–4522 CrossRef CAS.
- M. O. Hunt Jr., A. M. Belu, R. W. Linton and J. M. DeSimone, Macromolecules, 1993, 26, 4854–4859 CrossRef.
- M. A. Peters, A. M. Belu, R. W. Linton, L. Dupray, T. J. Meyer and J. M. DeSimone, J. Am. Chem. Soc., 1995, 117, 3380–3388 CrossRef CAS.
- Y. Tezuka and A. Araki, Polymer, 1993, 34, 5180–5185 CrossRef CAS.
- A. Hirao, I. Hattori, T. Sasagawa, K. Yamaguchi, S. Nakahama and N. Yamazaki, Makromol. Chem., Rapid Commun., 1982, 3, 59–63 CrossRef CAS.
- K. Ueda, A. Hirao and S. Nakahama, Macromolecules, 1990, 23, 939–945 CrossRef CAS.
- A. Hirao, H. Nagahama, T. Ishizone and S. Nakahama, Macromolecules, 1993, 26, 2145–2150 CrossRef CAS.
- M. Tohyama, A. Hirao, S. Nakahama and K. Takenaka, Macromol. Chem. Phys., 1996, 197, 3135–3148 CrossRef CAS.
- R. P. Quirk and L. Zhu, Makromol. Chem., 1989, 190, 487–493 CrossRef CAS.
- R. P. Quirk and L. Zhu, Br. Polym. J., 1990, 23, 47–54 CAS.
- R. P. Quirk and Y. Wang, Polym. Int., 1993, 31, 51–59 CrossRef CAS.
- R. P. Quirk and T. Lynch, Macromolecules, 1993, 26, 1206–1212 CrossRef CAS.
- G. J. Summers and R. P. Quirk, Polym. Int., 1996, 40, 79–83 CrossRef CAS.
- J. Kim, S. Kwak, K. U. Kim, K. H. Kim, J. C. Cho, W. H. Jo, D. Lim and D. Kim, Macromol. Chem. Phys., 1998, 199, 2185–2191 CrossRef CAS.
- A. Hirao and M. Hayashi, Macromolecules, 1999, 32, 6450–6460 CrossRef CAS.
- A. Hirao, S. Loykulnant and T. Ishizone, Prog. Polym. Sci., 2002, 27, 1399–1471 CrossRef CAS.
- W.-B. Zhang, B. Sun, H. Li, X. Ren, J. Janoski, S. Sahoo, D. E. Dabney, C. Wesdemiotis, R. P. Quirk and S. Z. D. Cheng, Macromolecules, 2009, 42, 7258–7262 CrossRef CAS.
- A. G. Evans and D. B. George, J. Chem. Soc., 1961, 4653–4659 RSC.
- R. P. Quirk, C. Garcés, S. Collins, D. Dabney, C. Wesdemiotis and V. Dudipala, Polymer, 2012, 53, 2162–2167 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- A. Hirao, K. Kato and S. Nakahama, Macromolecules, 1992, 25, 535–540 CrossRef CAS.
- The electrophilic acetyl group of 4-acylstyrene was converted to the electron-rich silyl enol ether, and the acidic α-hydrogen of the CH3CO group was simultaneously eliminated to prevent the side reactions with the nucleophilic and basic anionic initiator and propagating carbanion.
- D. Matsuoka, R. Goseki, S. Uchida and T. Ishizone, Macromol. Chem. Phys., 2017, 218, 1700015 CrossRef.
- When we terminated the reaction with MeOH, the blue coloration did not disappear at −78 °C within 30 min. Then, we employed more acidic AcOH to quench the carbanion derived from 1 immediately.
- A similar modification of the nucleophilicity was often used in the sequential copolymerization of St and methyl methacrylate (MMA) to prevent the nucleophilic substitution to the ester carbonyl group of MMA. Prior to the addition of MMA, a propagating polystyryl anion was masked with DPE to reduce its nucleophilicity.
- See ref. 33. It should be noted that the living polymer derived from AdK was stable in the presence of the ketone moieties of the repeating units because of the stabilization of the propagating benzylic carbanion by the electron-withdrawing carbonyl group.
- See ref. 38. Two series of the mass spectrum signals were observed in the MALDI-TOF-MS of the resulting PtBMA after the end-functionalization. The major series were corresponding to the expected chemical structures of an end-functionalized PtBMA possessing a diphenylmethyl initiator residue and a terminal DPE unit carrying two 1-adamantylcarbonyl groups.
- M. Kubo, T. Nishigawa, T. Uno, T. Itoh and H. Sato, Macromolecules, 2003, 36, 9264–9266 CrossRef CAS.
- T. Ishizone and R. Goseki, Polym. J., 2018, 50, 805–819 CrossRef CAS.
- S. Inomata, S. Matsuoka, S. Sakai, H. Tajima and T. Ishizone, Macromolecules, 2012, 45, 4184–4195 CrossRef CAS.
- A. J. van Reenen, L. J. Mathias and L. Coetzee, Polymer, 2004, 45, 799–804 CrossRef CAS.
- T. Otsu, A. Matsumoto, A. Horie and S. Tanaka, Chem. Lett., 1991, 20, 1145–1148 CrossRef.
- W. Lu, C. Huang, K. Hong, N.-G. Kang and J. W. Mays, Macromolecules, 2016, 49, 9406–9414 CrossRef CAS.
- S. Kobayashi, T. Matsuzawa, S. Matsuoka, H. Tajima and T. Ishizone, Macromolecules, 2006, 39, 5979–5986 CrossRef CAS.
- S. Kobayashi, H. Kataoka, R. Goseki and T. Ishizone, Macromol. Chem. Phys., 2018, 219, 1700450 CrossRef.
- A. Hirao, K. Takenaka, S. Packirisamy, K. Yamaguchi and S. Nakahama, Makromol. Chem., 1985, 186, 1157–1166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py00642g |
| This journal is © The Royal Society of Chemistry 2019 |