
A fast semi-continuous anionic process for fluorene-functionalized homo and block oligomers with uniform molecular weights†
Xinming
Pu
and
Junpo
He
 *
*
The State Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai, 200433, China. E-mail: jphe@fudan.edu.cn
First published on 22nd November 2018
Abstract
A semi-continuous strategy based on anionic reactions was developed for the step-by-step synthesis of oligomers with uniform molecular weights. With the homemade monomers, the strategy allows the one-pot synthesis of oligomers up to tetramers without intermediate separation owing to the highly efficient stoichiometric anionic reaction. The combination of the continuous process with the coupling reaction affords homo-oligomers and triblock oligomers of large molecular weights with a precisely inserted fluorenyl functionality. Aggregation-induced emission was observed in the suspensions of the coupled homo-oligomers and triblock oligomers in the mixed solvents of THF and water. The current approach provides a new method in the preparation of oligomers and block oligomers with uniform molecular weights.
Natural biopolymers such as proteins, DNA and polysaccharides are highly defined macromolecules with uniform molecular weights, precise sequences and stereo-regularities that accomplish a great diversity of biochemical and biophysical functions.1,2 Synthetic polymers possess statistically distributed structures that lack the precision as biopolymers, although several living or controlled polymerization methods (e.g., living anionic, cationic and controlled radical polymerizations) provide control on the molecular weights, end-functionality and topological architecture of polymers.3–5 Studies on precision polymers can help us understand the relationship between the structure and property of polymers. Therefore, although it is challenging, the preparation of polymers with precise structures, such as a uniform molecular weight, has been reported recently.6,7
Up to now, a number of strategies have been developed for the synthesis of monodisperse macromolecules both in liquid and solid phases.8–10 Almost all of these synthetic strategies were established on iterative processes which involved the successive steps of monomer coupling and functional group transformation reactions. The molecular weight increase was accomplished through either linear or exponential mode. In the former, one monomer unit was added to the polymer chain, while in the latter, two or three n-mers were coupled in one step. For instance, single unit monomer insertions (SUMI) were achieved through the radical addition process mediated by the reversible addition–fragmentation chain transfer (RAFT) reaction11–14 and metal-catalyzed atom transfer radical addition (ATRA).15,16 In these studies, oligomeric products were purified via preparative column chromatography which was very laborious. A faster mode for molecular weight increase is through iterative exponential growth (IEG), where orthogonal terminal activations/transformations and couplings of one kind of doubly-protected monomer were performed in a repetitive way. IEG has been employed to prepare various kinds of polymers with uniform molecular weights, such as polyethylene, poly(ethylene glycol), and oligo(ε-caprolactone).17–21 Very recently, the IEG approach was extended to the IEG plus side-chain functionalization (IEG+) strategy to synthesize monodisperse polymers with a defined sequence and block copolymers with uniform structures.22–24 The scalability of the synthesis of the uniform macromolecules was achieved through a semi-automated multi-step flow system.
In both linear and exponential modes, highly efficient orthogonal coupling and transformation reactions are key elements since the reactions only take place at the chain ends. Among others, Suzuki coupling reactions,25–29 copper catalyzed azide–alkyne cycloaddition (CuAAC),30,31 thiol–ene coupling reactions,32,33 and multicomponent reactions34,35 are the widely used reactions in the synthesis of uniform macromolecules.
Carbanions are highly reactive organic species and in many cases allow complete conversion of the substrates within minutes. A particularly efficient reaction is the monoaddition of organolithium to 1,1-diphenylethylene (DPE) and its derivatives, which are widely used in the synthesis of functional and topological macromolecules.36–45 The reaction between n-butyllithium and DPE was found to be stoichiometric and quantitative,46,47 although in a recent study Quirk and coworkers found that oligomers formed when using excess DPE in a hydrocarbon solvent.48 The reason for the stoichiometry is due to the strong stabilization effect of the two phenyl rings and the steric hindrance of the molecule preventing homopolymerization. Nevertheless, the monoaddition reaction has never been used for the synthesis of macromolecules with uniform molecular weights and precision functionalities. We report here a fast semi-continuous synthetic strategy based on the stoichiometric reaction between butyllithium and the DPE moiety for the synthesis of monodisperse homo-oligomers and triblock oligomers with a fluorenyl functionality at the core of the molecules.
Four DPE-based monomers, as shown in Scheme 1, were prepared through a three-step procedure involving the Wittig reaction and Grignard reaction/coupling with a chlorosilane followed by a hydrosilylation reaction. The details of monomer synthesis are given in the Experimental section. The overall yields ranged from 28% to 53%. The structures of the products were confirmed by NMR and mass spectrometry (ESI, Fig. S1–10†).
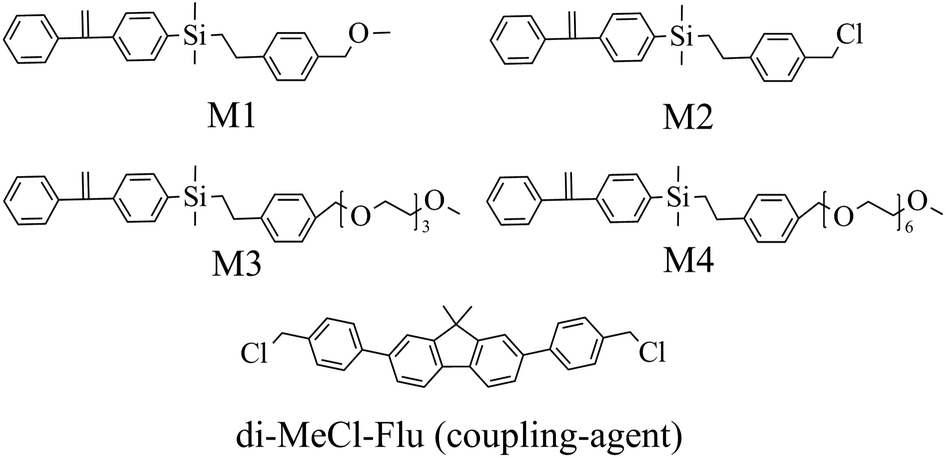 | ||
| Scheme 1 DPE-based monomers (M1, M2, M3, M4) and fluorene-based coupling agent (di-MeCl-Flu) used in the present work. | ||
First, we prepared homo-oligomers using M1 and M2 as monomers. The synthetic route is outlined in Scheme 2A. The synthesis started from the quantitative addition of sec-BuLi to the DPE moiety of M1 in THF. The feed ratio of Li to DPE moiety was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The reaction was completed within 15 min at −80 °C (ethyl acetate/liquid nitrogen). The resulting dark red colored solution was used directly to react with chloromethyl of M2. Upon addition of M2, the solution immediately became pale yellow within 1 min, which was kept at −80 °C for 5 min and at room temperature for another 10 min. Samples were taken from the system with a needle syringe for analysis where no traces of M1 or M2 were observed by size exclusion chromatography (SEC) analysis The above reaction produced a dimer with the DPE moiety and the methoxy group at a yield of 99% based on the integration of the SEC chromatogram (Fig. S11(A)†). The DPE moiety of the dimer was activated in situ by the quantitative reaction with sec-BuLi, again resulting in a dark red colored solution of the living anions. The activation reaction was followed by the coupling reaction between living anionic species and chloromethyl of M2, leading to the formation of a trimer with a yield of 90%. Thus, starting from M1, the alternating addition of sec-BuLi and M2 to the reaction system led to the formation of a dimer, trimer and tetramer within 90 min. The reaction was performed in a continuous mode since there was no need for the repetitive protecting/deprotecting reactions.
:1. The reaction was completed within 15 min at −80 °C (ethyl acetate/liquid nitrogen). The resulting dark red colored solution was used directly to react with chloromethyl of M2. Upon addition of M2, the solution immediately became pale yellow within 1 min, which was kept at −80 °C for 5 min and at room temperature for another 10 min. Samples were taken from the system with a needle syringe for analysis where no traces of M1 or M2 were observed by size exclusion chromatography (SEC) analysis The above reaction produced a dimer with the DPE moiety and the methoxy group at a yield of 99% based on the integration of the SEC chromatogram (Fig. S11(A)†). The DPE moiety of the dimer was activated in situ by the quantitative reaction with sec-BuLi, again resulting in a dark red colored solution of the living anions. The activation reaction was followed by the coupling reaction between living anionic species and chloromethyl of M2, leading to the formation of a trimer with a yield of 90%. Thus, starting from M1, the alternating addition of sec-BuLi and M2 to the reaction system led to the formation of a dimer, trimer and tetramer within 90 min. The reaction was performed in a continuous mode since there was no need for the repetitive protecting/deprotecting reactions.
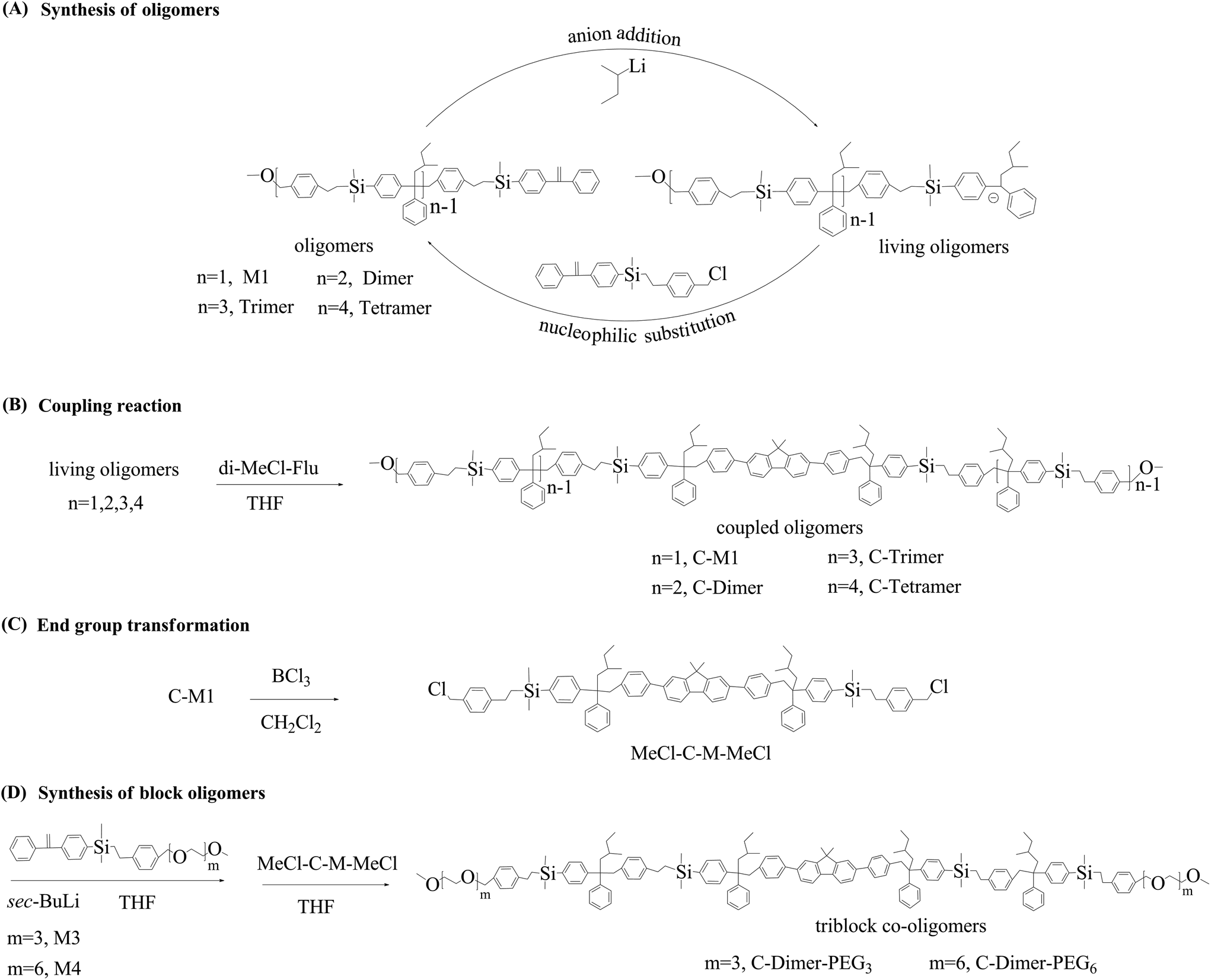 | ||
| Scheme 2 Synthetic route for the oligomers (A), coupled oligomers (B), end-functional oligomers (C) and triblock oligomers (D). | ||
As shown in Fig. 1, all SEC traces of the monomer to tetramer show narrow distributions with discrete incremental shifts to the larger molecular weight region. Although the crude products of the trimer and tetramer showed broader profiles (ESI, Fig. S11†), these products were easily obtained after precipitation in methanol and subsequent separation with preparative SEC.
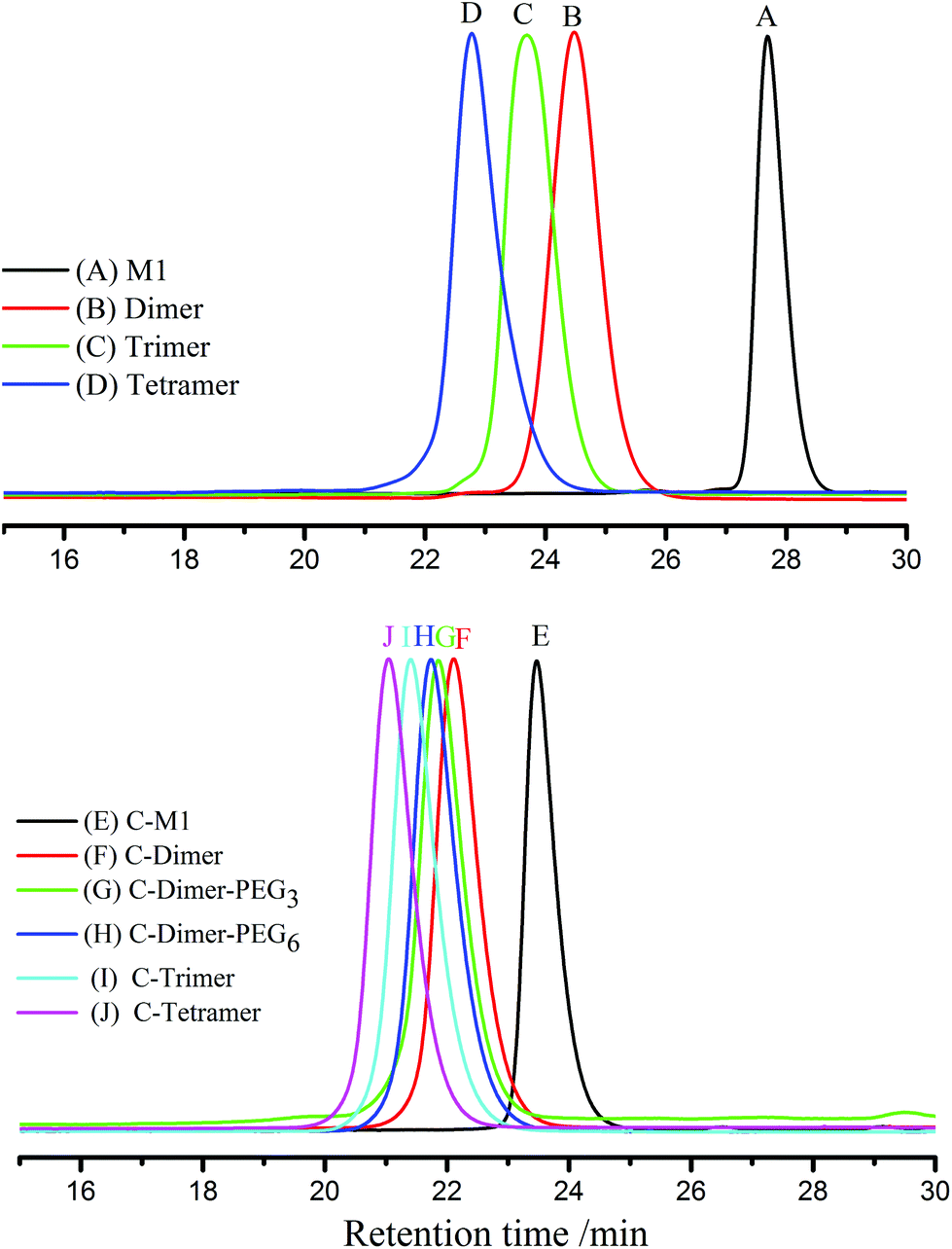 | ||
| Fig. 1 SEC diagrams of the products of different steps before (upper part) and after (lower part) the coupling reaction with sample names indicated in the figure. Triblock oligomers were also shown as curves G and H in the lower part. | ||
The product of each step was also characterized by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS). As shown in Fig. 2, all of the products show a single peak, indicating the uniform molecular weight. The interval between the peaks is equal to ca. 412, which is corresponding to the molecular weight of the repeat unit. Furthermore, the observed isotopic pattern of each peak agrees well with the theoretical one, as exemplified by those of the tetramer in the inset in Fig. 2A.
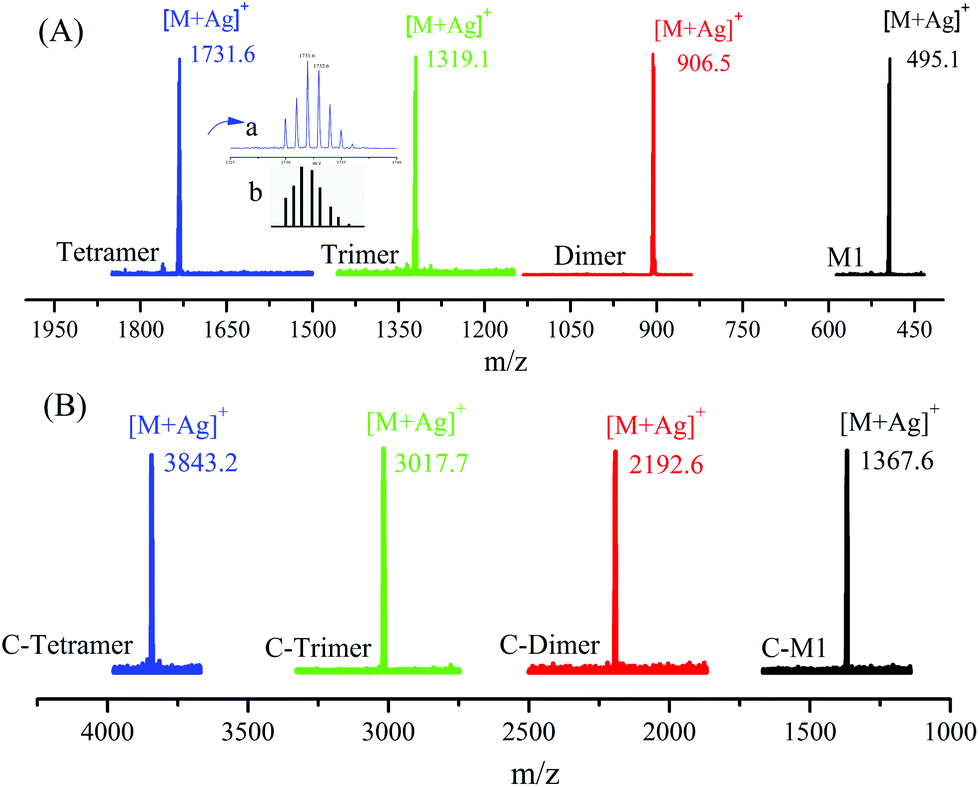 | ||
| Fig. 2 MALDI-TOF MS spectra of the oligomers of different steps with sample names indicated in the figure. The insets in A are measured (upper) and calculated (lower) isotopic patterns. | ||
The molecular structures of homo-oligomers were further confirmed from the 1H NMR results. As an example, the spectrum in Fig. S14† shows all the characteristic proton signals of the trimer. The signals of the vinyl protons appear as double doublets at a chemical shift of around 5.5 ppm, while that of the methoxy group appears as a singlet at 3.4 ppm. The ratio of the integration is 2/3, which agrees well with the theoretical value.
Thus a step-by-step molecular weight growth, in which one monomer was added in a controlled way to the oligomer chain end in each step, was achieved by operating the highly efficient reactions of living carbanions in a continuous mode. Nevertheless, the efficiency became lower when the degree of polymerization was larger than the tetramer because of a number of reasons such as the anion-quenching impurities might be introduced by the repetitive operation procedure, the accumulation of LiCl as the side product, and the entrapping of the end-groups in the globular coil as the molecular weight increased. Actually, we had tried to synthesize a pentamer from the purified tetramer. However, the separation of the pentamer and tetramer from the reaction mixture turned out to be difficult due to close overlapping of the two species on SEC.
In the following, we give the results of coupling reactions (Scheme 2B) of the above products with a fluorene derivative, 2,7-bis(4-(chloromethyl)phenyl)-9,9-dimethyl-9H-fluorene (di-MeCl-Flu) (Scheme 1), synthesized according to the pertinent literature.49,50Sec-BuLi was added respectively to equal moles of monomer (M1), dimer, trimer and tetramer (Scheme 2A & B). The resulting living anionic addition products were subsequently coupled using di-MeCl-Flu as the coupling agent in THF for total 25 min. The collected white powders were purified using preparative SEC to obtain the corresponding coupled products. The resulting coupled products, C-M1, C-dimer, C-trimer and C-tetramer, are monodisperse oligomers with precise functionality used for the study of photophysical properties (Table 1).
| Oligomers | M n,NMR (Da) | M n,MS (Da) | M n,SEC (Da) | PDISECc |
|---|---|---|---|---|
| a Calculated from the 1H NMR spectra. For homo-oligomers Mn,NMR was obtained using the integration ratio of the signal intensity of the benzylic protons in the repeat units to the terminal vinylic protons; for block oligomers Mn,NMR was obtained using the integration ratio of the signals of the benzylic protons to the OEG protons. b Measured by MALDI-TOF MS with dithranol as the matrix and silver trifluoroacetate as the ionization agent. c Determined by SEC calibrated using narrow disperse PS standards. | ||||
| M1 | 387 | 387.1 | — | — |
| Dimer | 799 | 798.5 | 810 | 1.001 |
| Trimer | 1210 | 1211.1 | 1200 | 1.003 |
| Tetramer | 1630 | 1623.6 | 1630 | 1.023 |
| C-M1 | 1260 | 1259.6 | 1220 | 1.002 |
| C-Dimer | 2090 | 2084.6 | 2000 | 1.014 |
| C-Trimer | 2910 | 2909.7 | 2890 | 1.010 |
| C-Tetramer | 3760 | 3735.2 | 3650 | 1.020 |
| C-Dimer-PEG3 | 2490 | — | 2340 | 1.060 |
| C-Dimer-PEG6 | 2700 | — | 2610 | 1.060 |
The SEC results of the coupled products show a series of narrowly disperse profiles with an incremental decrease of elution volume along with the molecular weight growth (Fig. 1B). The MALDI-TOF MS in Fig. 2B also shows a single peak for each sample with expected values of m/z, i.e., 1367, 2192, 3017, and 3843 corresponding to the product derived from C-M1 to C-tetramer, respectively. The NMR results in the ESI (Fig. S22–29†) further confirm the successful coupling reactions.
Triblock co-oligomers were prepared using PEG bearing monomers M3 and M4 with different lengths of PEG segments (Scheme 2D). First, the methoxy groups of the C-monomer were converted into chloromethyl groups by reaction with boron trichloride in dichloromethane (Scheme 2C). The resulting product was used to couple the living adduct of M3 or M4. The above reactions went smoothly, leading to the formation of triblock co-polymers containing a fluorenyl core, four monomer units and out segments of PEG with six or twelve units of ethylene oxide. SEC traces are narrow (Fig. 1) single peaks although MALDI-TOF mass spectrometry did not give reasonable signals. The NMR spectra of the triblock co-polymers are shown in ESI Fig. S32 and 33† with respective peak assignments. Furthermore, the molecular weights were calculated to be 2490 and 2700 Da from these NMR spectra, respectively, which were consistent with the theoretical values.
The coupled products exhibited the behavior of aggregation- induced emission (AIE) or enhanced emission (AIEE). AIE and AIEE were reported in the seminal studies by Tang and then by Park, respectively.51,52 As shown in Fig. 3, while a weak fluorescence was observed in THF solution, the fluorescence intensities of the coupled oligomers increased remarkably to ca. 7–9 fold in a mixed solvent of THF/H2O with the water content increasing up to 70% (vol%) in which the oligomers formed a suspension. This phenomenon was a typical AIE effect and was explained by the restricted intramolecular rotation of the chromophore. Blue shift or bathochromic shift emission was reported in the literature,49,53 but was not found in this study.
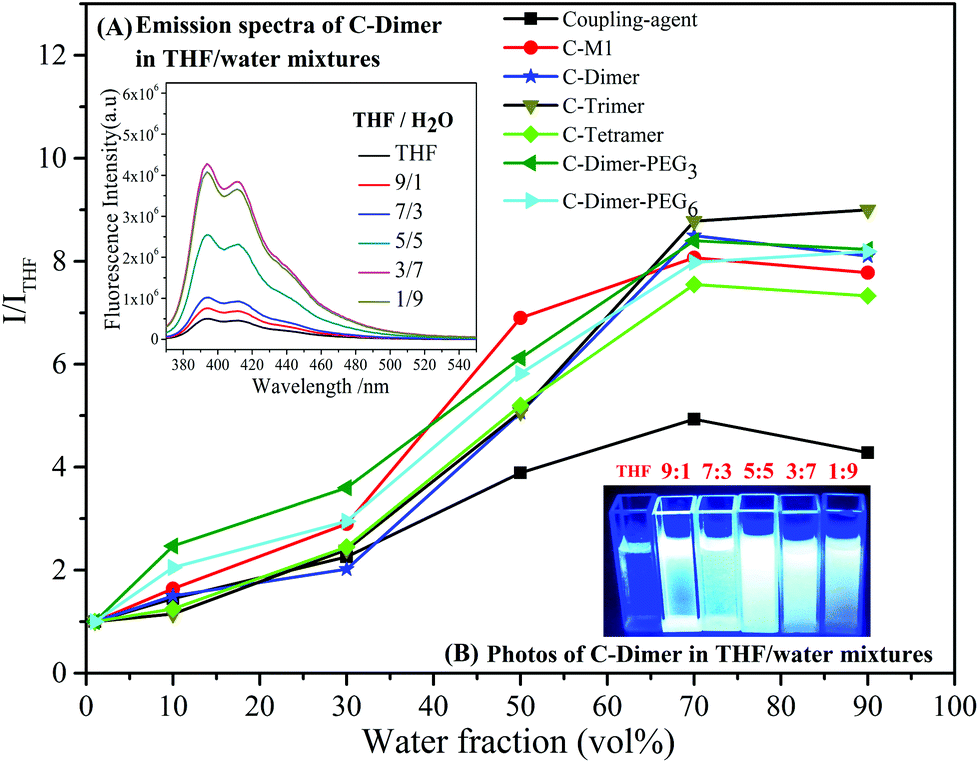 | ||
| Fig. 3 Dependence of the PL peak intensity ratio (I/ITHF) of the oligomers in THF/water mixtures on water contents at a concentration of 5 × 10−7 M, excitation wavelength: 360 nm. Insets: (A) emission spectra of C-Dimer in THF/water mixtures; (B) photos of C-Dimer in THF/water mixtures under a 365 nm UV lamp. | ||
It is interesting to note in Fig. 3 that the coupling agent itself showed a much weaker AIE behavior in comparison with the coupled oligomers. This clearly indicates that the intermolecular stacking between fluorenyl moieties, which usually leads to the aggregation-caused quenching (ACQ) effect, was suppressed by the attached oligomers.
The AIE effect was also observed in vesicular aggregates formed by triblock oligomers, i.e., C-Dimer-PEG3 and C-Dimer-PEG6. Fig. 4 shows the snapshots of laser scanning confocal microscopy (LSCM) of the two samples. The average sizes of the vesicles are 5 μm and 3 μm for C-Dimer-PEG3 and C-Dimer-PEG6 in THF/water (7/3, vol/vol), respectively. The intensity of the fluorescence increased by 3 fold from THF solution to the vesicular solution due to the more compact stacking of the chromophore in the vesicle walls. However, it is difficult to judge the number of layers that form the walls from the snapshots of LSCM.
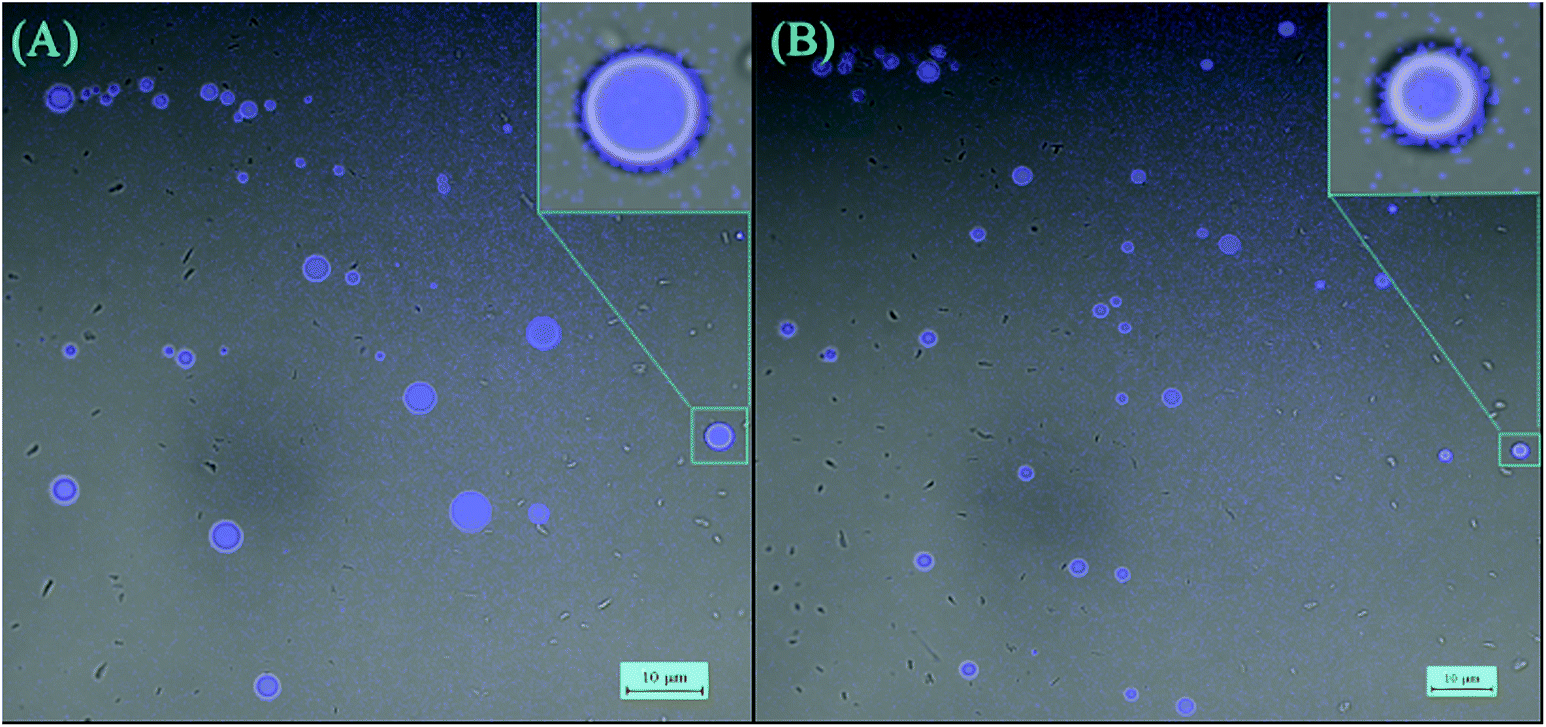 | ||
| Fig. 4 (A) LSCM images of C-Dimer-PEG3 in THF/water (7/3 vol/vol). (B) LSCM images of C-Dimer-PEG6 in THF/water (7/3 vol/vol). | ||
Conclusions
In conclusion, we have developed a semi-continuous strategy based on anionic reactions for the step-by-step synthesis of oligomers with uniform molecular weights. Owing to the high efficiency and the stoichiometry of the reactions, there was no need for the separation of the intermediates. Starting from M1, a tetramer was obtained within 2 h where the coupling reaction of the purified tetramer was completed within 20 min. Triblock co-oligomers of atomic precision with PEG segments on both sides were also prepared. Since the coupling agent was a fluorene derivative, the coupled products exhibited the AIE effect in the suspensions of the homo-oligomers and vesicular solutions of the triblock analogues in mixed solvents of THF and water.Considering the livingness of the anionic species, it is envisaged that the present method may serve as a unique tool, in combination with other strategies such as monomer design, IEG, etc., for the synthesis of uniform macromolecules with higher molecular weights, sequence control, specific functionalities, and various architectures. The development of this synthesis is currently underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support by the National Natural Science Foundation of China (NSFC) (Grant No. 21474016).Notes and references
- A. G. Murzin, S. E. Brenner, T. Hubbard and C. Chothia, J. Mol. Biol., 1995, 247, 536–540 CAS.
- G. M. Church, Y. Gao and S. Kosuri, Science, 2012, 337, 1628 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Chem. – Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. Meier, Macromol. Rapid Commun., 2017, 1600711, 1–45 Search PubMed.
- S. Binauld, D. Damiron, L. A. Connal, C. J. Hawker and E. Drockenmuller, Macromol. Rapid Commun., 2011, 32, 147–168 CrossRef CAS PubMed.
- K. Hatada, T. Kitayama, K. Ute and T. Nishiura, Macromol. Rapid Commun., 2010, 25, 1447–1477 CrossRef.
- T. T. Trinh, L. Oswald, D. Chan-Seng and J. F. Lutz, Macromol. Rapid Commun., 2014, 35, 141–145 CrossRef CAS.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS PubMed.
- J. Vandenbergh, G. Reekmans, P. Adriaensens and T. Junkers, Chem. Commun., 2013, 49, 10358–10360 RSC.
- S. Houshyar, D. J. Keddie, G. Moad, R. J. Mulder, S. Saubern and J. Tsanaktsidis, Polym. Chem., 2012, 3, 1879–1889 RSC.
- J. Xu, S. Shanmugam, C. Fu, K. F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376–8383 CrossRef CAS PubMed.
- J. Vandenbergh, G. Reekmans, P. Adriaensens and T. Junkers, Chem. Sci., 2015, 6, 5753–5761 RSC.
- D. Oh, M. Ouchi, T. Nakanishi, H. Ono and M. Sawamoto, ACS Macro Lett., 2016, 5, 745–749 CrossRef CAS.
- O. I. Paynter, D. J. Simmonds and M. C. Whiting, J. Chem. Soc., Chem. Commun., 1982, 14, 1165–1166 RSC.
- I. Bidd, D. W. Holdup and M. C. Whiting, ChemInform, 1988, 19, 2455–2463 CrossRef.
- A. C. French, A. L. Thompson and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 1248–1252 CrossRef CAS.
- F. A. Loiseau, K. K. Hii and A. M. Hill, J. Org. Chem., 2004, 69, 639–647 CrossRef CAS PubMed.
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718–1726 CrossRef CAS PubMed.
- F. A. Leibfarth, J. A. Johnson and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10617–10622 CrossRef CAS PubMed.
- Y. Jiang, M. R. Golder, H. V. T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369–9372 CrossRef CAS.
- J. C. Barnes, D. J. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS.
- N. Iwadate and M. Suginome, Org. Lett., 2009, 11, 1899–1902 CrossRef CAS PubMed.
- Y. Geng, S. W. Culligan, A. Trajkovska, J. U. Wallace and S. H. Chen, Chem. Mater., 2003, 15, 542–549 CrossRef CAS.
- Y. Geng, A. Trajkovska, D. Katsis, J. J. Ou, S. W. Culligan and S. H. Chen, J. Am. Chem. Soc., 2002, 124, 8337–8347 CrossRef CAS.
- Q. Wang, Q. Yao, H. Tian, Y. Geng and F. Wang, Macromolecules, 2011, 44, 1256–1260 CrossRef CAS.
- X. Zhang, H. Tian, Q. Liu, L. Wang, Y. Geng and F. Wang, Org. Chem., 2006, 71, 4332–4335 CrossRef CAS.
- F. Amir, Z. Jia and M. J. Monteiro, J. Am. Chem. Soc., 2016, 138, 16600–16603 CrossRef CAS PubMed.
- S. Binauld, C. J. Hawker, E. Fleury and E. Drockenmuller, Angew. Chem., Int. Ed., 2009, 48, 6654–6658 CrossRef CAS PubMed.
- X. Jiang, J. Lu, F. Zhou, Z. Zhang, X. Pan, W. Zhang, Y. Wang, N. Zhou and X. Zhu, Polym. Chem., 2016, 7, 2645–2651 RSC.
- Z. Huang, J. Zhao, Z. Wang, F. Meng, K. Ding, X. Pan, P. N. Zhou, P. X. Li, P. Z. Zhang and P. X. Zhu, Angew. Chem., Int. Ed., 2017, 56, 13612–13617 CrossRef CAS PubMed.
- S. C. Solleder, D. Zengel, K. S. Wetzel and M. A. R. Meier, Angew. Chem., Int. Ed., 2016, 55, 1204–1207 CrossRef CAS PubMed.
- Y. Wu, J. Zhang, F. Du and Z. Li, ACS Macro Lett., 2017, 6, 1398–1403 CrossRef CAS.
- R. N. Young, R. P. Quirk and L. J. Fetters, Adv. Polym. Sci., 1984, 56, 1–90 CrossRef CAS.
- R. P. Quirk, T. Yoo, Y. Lee, J. Kim and B. Lee, Adv. Polym. Sci., 2000, 153, 67–162 CrossRef CAS.
- A. Hirao, M. Tohoyama and S. Nakahama, Macromolecules, 1997, 30, 3484–3489 CrossRef CAS.
- S. Ito, R. Goseki, S. Senda and A. Hirao, Macromolecules, 2012, 45, 4997–5011 CrossRef CAS.
- W. Sang, H. Ma, Q. Wang, X. Hao, Y. Zheng, Y. Wang and Y. Li, Polym. Chem., 2015, 7, 219–234 RSC.
- Q. Wang, H. Ma, W. Sang, L. Han, P. Liu, H. Shen, W. Huang, X. Gong, L. Yang and Y. Wang, Polym. Chem., 2016, 7, 3090–3099 RSC.
- L. Hong, S. Yang and J. He, Eur. Polym. J., 2015, 65, 171–190 CrossRef CAS.
- W. Sun, J. He, X. Wang, C. Zhang, H. Zhang and Y. Yang, Macromolecules, 2009, 42, 7309–7317 CrossRef CAS.
- N. Hadjichristidis and A. Hirao, Anionic polymerization: principles, practice, strength, consequences and applications, Springer, Japan, 2015 Search PubMed.
- A. Mavroudis and N. Hadjichristidis, Macromolecules, 2006, 39, 535–540 CrossRef CAS.
- K. Ziegler, F. Crossmann, H. Kleiner and O. Schafer, Liebigs Ann. Chem., 1929, 473, 300–306 CrossRef.
- G. Köbrich and I. Stöber, Chem. Ber., 1970, 103, 2744–2753 CrossRef.
- R. P. Quirk, C. Garcés, S. Collins, D. Dabney, C. Wesdemiotis and V. Dudipala, Polymer, 2012, 53, 2162–2167 CrossRef CAS.
- F. Zhang, R. Zhang, X. Liang, K. Guo, Z. Han, X. Lu, J. Xie, J. Li, D. Li and X. Tian, Dyes Pigm., 2018, 155, 225–232 CrossRef CAS.
- R. Chien, C. Lai and J. Hong, J. Phys. Chem. C, 2011, 115, 12358–12366 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Tang, Chem. Commun., 2001, 18, 1740–1741 RSC.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS.
- M. P. Aldred, C. Li, G. Zhang, W. Gong, A. D. Q. Li, Y. Dai, D. Ma and M. Zhu, J. Mater. Chem., 2012, 22, 7515–7528 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, MALDI-TOF MS spectra and FL spectra charts. See DOI: 10.1039/c8py01340c |
| This journal is © The Royal Society of Chemistry 2019 |