
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional polyamides with gem-diazido units: synthesis and diversification†
Phillip
Biallas
,
Janina
Heider
and
Stefan F.
Kirsch
 *
*
Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany. E-mail: sfkirsch@uni-wuppertal.de
First published on 9th November 2018
Abstract
A diazidated malonate derivative was employed as a monomer for the reagent-free one-pot synthesis of several polyamides containing geminal diazido moieties within the polymer backbone. Using different diamines, azide-rich polyamides with diverse properties and sensitivities dependent on the structure of the diamine units were obtained. Postpolymerization CuAAC reactions of the geminal diazido units in the polymer backbone allowed for the introduction of additional functionalities, and the construction of densely functionalized polymeric structures.
Synthetic polyamides are a major polymer class with important uses ranging from plastics and fibers for commodities and automotives to biomedical applications.1,2 Polyamides combine high strength and stability with good biocompatibility and degradability.3 They are typically prepared through the condensation of diamines with dicarboxylic acid chlorides or with other activated dicarboxylic acid derivatives.4 Other methods involve the use of peptide coupling reagents or activating reagents.5 For the commercial synthesis of aliphatic polyamides, high temperature melt condensation is the standard method.1
Besides the development of new amide bond-forming methodologies,6 the introduction of functional groups into polyamides is a main issue since the properties of polymers can be altered significantly by functional groups. Conventional polycondensation methods provide mostly linear and barely functionalized polyamides, since the reactions do not allow the distinction between reactive groups within a monomer unit, and the formation of cross-linked polyamides is problematic. For the preparation of functionalized polyamides, a range of powerful strategies involving somewhat tedious protection-deprotection procedures the polycondensation of cyclic bis(anhydrides) or bis(thiolactones)with diamines, multicomponent reactions like the Passerini, or the Ritter reaction were developed.7
Recently, we became involved into the synthesis and reactivity of small molecules having geminal diazido groups, an uncommon structural motif mostly neglected in the literature.8 Since polymers with azide functional groups are highly attractive as cross-linking materials,9 energetic materials,10 and as tools for the preparation of new functional polymers,11 we were inspired to produce polymers containing geminal diazido units, a functional motif that was, to our knowledge, not attached onto polymer chains until now. To this end, we utilized a highly efficient amidation reaction where diazidated diethyl malonate 1 reacted with primary amines 2. The waste-free reaction does not require any additional reagents or activators, and amides 3 are produced in a straightforward way under neutral conditions, using THF as a solvent at room temperature (Scheme 1A). In this communication, we report the synthesis of several polyamides 5 through the simple amidation of diamines 4 and diazidated diethyl malonate 1 (Scheme 1B), thus producing macromolecules with a uniquely high grade of azido moieties. Importantly, the azidated polyamides are suitable for postmodification through CuAAC click chemistry with alkyne reagents,12–14 thus leading to the fully controlled attachement of additional functional units.
 | ||
| Scheme 1 Synthesis of polyamides with gem-diazides. | ||
Regarding our synthetic strategy for the formation of polyamides 5, diazidated diethyl malonate 1 was expected to directly react with diamines of different constitution. Since malonate 1 already possessed the two azide groups within its core, it appeared to be the ideal precursor molecule. The polycondensation generates no side products, apart from ethanol. This strategy was founded on our previous studies with diazidated compounds where we observed a strong activating effect of the geminal diazido group on neighboring carbonyl groups:15 We assumed that the enhanced electrophilicity of the carbonyls might facilitate a double amidation reaction with amines through activation of the adjacent ester groups; amide formation then proceeds through addition of the nucleophile followed by elimination of ethanol. In an early competition experiment with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diazide 1 and the analogous dimethyl malonate (6) having no diazido functionality, we observed an outstanding selectivity for the formation of the diazidated amide 3a under our amidation conditions (Scheme 2). According to 1H NMR of the crude mixture, the non-azidated malonate 6 remained fully unreacted when treated with benzylamine 2a in THF at room temperature for 12 h. Diazide 1, on the other hand, was rapidly consumed, smoothly generating the product of the double amidation. In the absence of diazidated malonate 1, malonate 6 also underwent slow conversion with benzylamine, albeit in an impractical manner.
:1 mixture of diazide 1 and the analogous dimethyl malonate (6) having no diazido functionality, we observed an outstanding selectivity for the formation of the diazidated amide 3a under our amidation conditions (Scheme 2). According to 1H NMR of the crude mixture, the non-azidated malonate 6 remained fully unreacted when treated with benzylamine 2a in THF at room temperature for 12 h. Diazide 1, on the other hand, was rapidly consumed, smoothly generating the product of the double amidation. In the absence of diazidated malonate 1, malonate 6 also underwent slow conversion with benzylamine, albeit in an impractical manner.
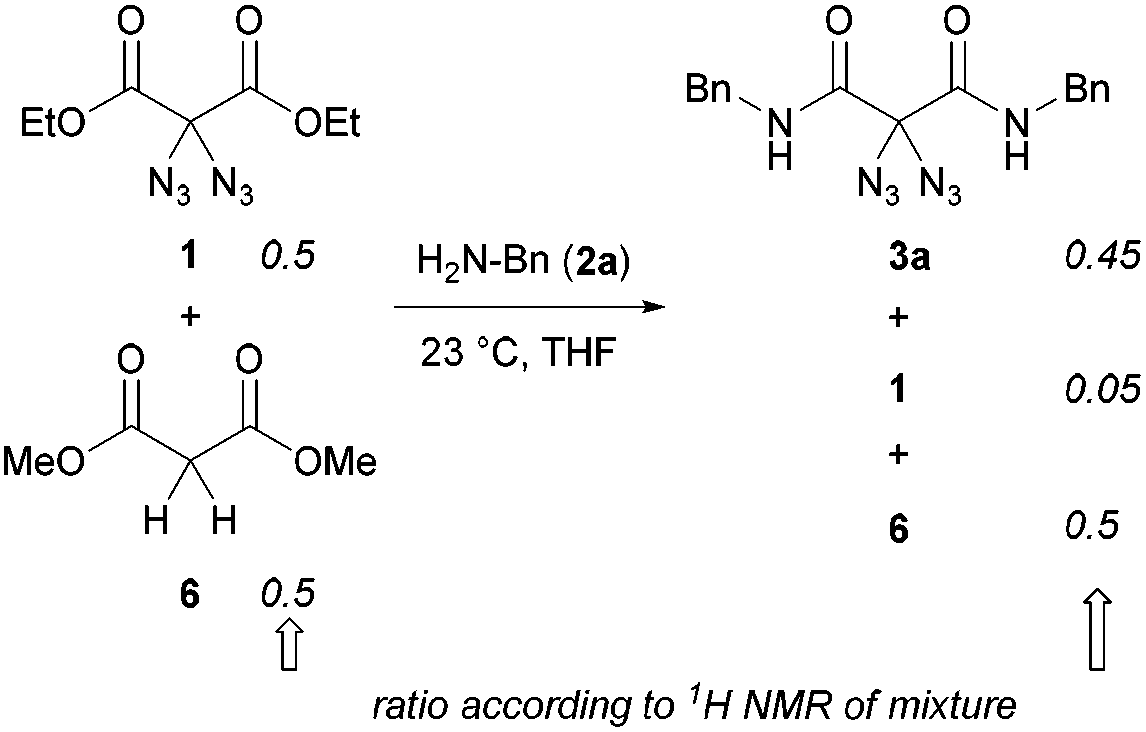 | ||
| Scheme 2 The direct diamidation of malonates at room temperature. | ||
Next, we began with the synthesis of polyamides 5 using diazidated malonate 1 and the five diamines 4a–e, depicted in Scheme 3.16 While malonates were frequently polymerized to yield polyesters for various applications, they happen to be rare monomer units of polyamides. Polymerisations leading to malonate-derived polyamides typically require high temperatures and/or additional reagents. Nevertheless, our model studies for the polyamidation between malonate 1 and diamines 4 were performed at room temperature, without any additives. The two monomer units were smoothly converted in a range of solvents, including THF, CH2Cl2, DMSO, DMF, NMP. The formation of side-products was never observed. However, we had severe problems with removing the polar solvents (DMSO, DMF and NMP) from the polymeric products, thus the proper characterization of polymers produced in polar solvents was not possible. The use of CH2Cl2 was also impractical due to the rapid precipitation of amide products during the reaction; however, it may become a useful solvent for other diamines having different solubility properties. The best choice, at least for our model substrates 4a–e, was THF: depending on the diamine, it was possible to run the polymerizations with a concentration of up to 8 M, without obvious precipitation during the reaction (e.g. with 4d and 4e). For less soluble polymers (e.g. with 4a and 4b), 1 M solutions in THF were chosen to prevent phase separation. Under typical conditions, the solutions of malonate 1 and diamine 4 were stirred for 72 h. In the case of diamines 4a–4c, the polyamides were precipitated through the addition of cold EtOH. Subsequent Soxhlet extraction with methanol resulted in the lowering of the PDI values, and the remaining residue was submitted to polymer analysis. When using 4d and 4e, the corresponding polymers were directly analyzed after removal of the solvent. Except of 5d, which is a yellowish and highly viscous oil, all polyamides were obtained as solids (5a–c: colorless; 5e: yellow foam) in yields from 15 to 97%.
 | ||
| Scheme 3 Diamines 4 used for the polymerizations with 1. | ||
The proposed structure of the five polyamides 5a–e was confirmed by NMR spectroscopy; in the case of 5e (and of 5c and 5b), it was possible to calculate the number of repeating monomer units from the integral values of the end groups in the 1H NMR spectrum: n(5e) ≈ 11, n(5c) ≈ 22, n(5b) ≈ 24.18
Table 1 gives an overview of the molecular weight characteristics of the polymers 5a–e obtained from the diamines 4a–e. Gel permeation chromatography (GPC) measurements were performed in hexafluoroisopropanol and calibrated with PMMA standards. The obtained molecular weights were ranging from small to moderate (from 4 to 9 kDa), with degrees of polymerization (DPs) ranging from 10 to 31, and depend somewhat on the amine used in the polycondensation. The lowest number-average molecular weight was obtained for diamine 4e. The highest number-average molecular weights were obtained with the structurally simple and highly flexible diamines 4a, 4b and 4d. The PDI values for 5a–5e are ranging from two to four, as one might expect for a polycondensation. We note that polymers 5d and 5e have the lowest molecular weight characteristics, despite better solubility during the polymerization process: this may be due to an increase of coiled structures with the long-chain diamines 4d and 4e that leads to a less effective polymerization.
| Entry | # | Yielda [%] |
M
nb [g mol−1] |
PDIc | Chemical formulae |
T
decomp.f [°C] |
ISg [J] |
|---|---|---|---|---|---|---|---|
| a Conditions: 1 (1.0 equiv.), 4 (1.0 equiv.), room temperature, THF; isolated yields after 72 h. b Number average molecular weight, determined by GPC. c Polydispersity index, determined by GPC. d 120 h. e Repeating unit. f Onset temperature, determined by TGA. g Impact sensitivity, determined by (BAM drophammer (1 of 6). h Addition of NEt3 (1.0 equiv.). | |||||||
| 1 | 5a | 40 | 9400 | 3.93 | C8H12N8O2 | 156 | 6 |
| 2d | 5b | 57 | 7900 | 2.97 | C9H14N8O2 | 160 | 17.5 |
| 3 | 5c | 52 | 4800 | 2.80 | C11H10N8O2 | 154 | 10 |
| 4 | 5d | 84 | 6200 | 2.85 | C9H14N8O4 | 150 | 18 |
| 5 | 5e | 97 | 3900 | 2.22 | C13H26N8O3Si2 | 153 | >20 |
| 6h | 5a | 44 | 9100 | 3.68 | C8H12N8O2 | — | — |
The thermal properties of the polyamides were studied by differential scanning calorimetric analysis (DSC) and thermogravimetric analysis (TGA) (see ESI†). TGA curves show that decomposition of all polymers starts similar to diazide 1 at around 120–140 °C, above which the polymers totally decompose.17 This leads to the conclusion that the thermal stability of the polyamides is mostly controlled by the geminal diazido functionality introduced by monomer 1; the diamine influence is only minor. According to the DSC profiles, melting occurs directly before the decomposition starts, i.e. in the cases of 5a, 5b, and 5c.
With regard to the stability of the polymers toward impact,19 the diamine monomer 4 is a major factor, as outlined in Table 1. Drophammer experiments showed that polyamide 5a with 1,5-pentanediamine units holds a high sensitivity (impact ca. 6 J). On the other hand, the use of a larger diamine monomer leads to significantly less sensitive materials. For example, 5b, 5d and 5e have a markedly reduced impact sensitivity of greater than 17 J. Thus, the highly azidated polyamides 5 show great potential for applications as energetic materials. Through simple fine-tuning of the diamine unit, it may become possible to acquire the desirable sensitivity.
The geminal diazido moieties deliver polyamides with a high level of energetic density, but they also are valuable starting points for the creation of functional density through post-polymerization functionalisation.20 We anticipated that the polymer analogous modification of the azide groups through the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) would alter the chemical nature of the polyamide structures by incorporating functional groups of choice. With the previously unused diazido moieties, we now provide new options for the creation of densely functionalized polyamides.
As summarized in Scheme 4, we attempted the CuAAC reaction of polymer 5a with several alkynes 7 using standard conditions with catalytic amounts of CuSO4 and sodium ascorbate at room temperature in DMF. It became easily possible to synthesize polyamides where, for example, aryls (i.e., 8a), chromophores (i.e., 8b) and carbohydrates (i.e., 8c) are attached at the main chain via triazoles. We note that the removal of the copper catalysts was demanding, and repeated washing with an aqueous solution of EDTA was required. On the other hand, polyamide 5a reacted under copper-free conditions with cyclooctyne, a prime example for a highly reactive strained alkyne, to give polymer 8d.
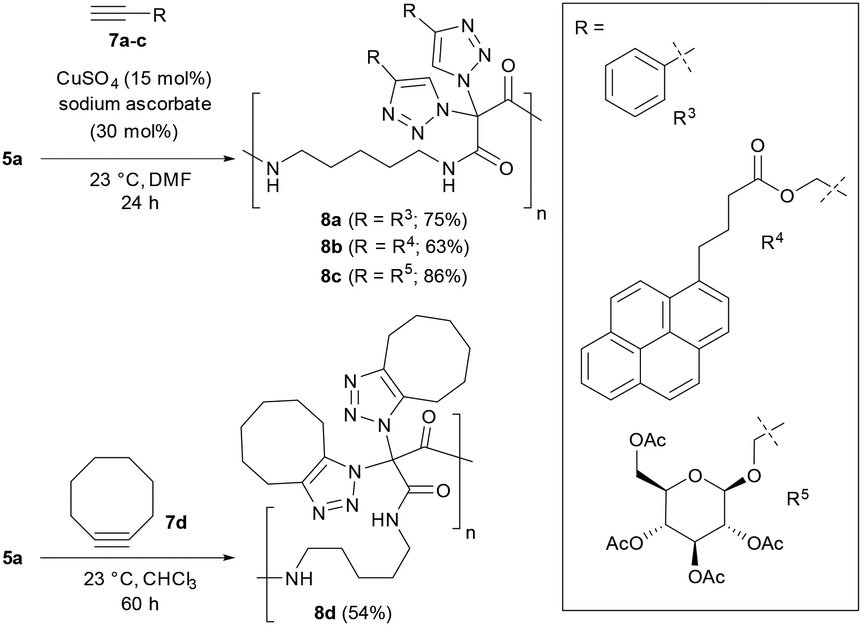 | ||
| Scheme 4 Postpolymerization modifications with alkynes. | ||
As exemplified for material 8b in Fig. 1, the post-functionalized polymers have UV/vis spectra that are dominated by the attached groups since the parent polymer 5a, bearing only azide groups, shows no absorption in the range of 250–400 nm (Fig. 1, green). The neat chromophore 7b, on the other hand, shows significant absorptions in the range of 260–285 nm and 305–355 nm, with an absorbtion maximum at 280 nm (Fig. 1, blue). Upon cycloaddition of polymer 5a with chromophore 7b, the functional polymer 8b (Fig. 1, red) shows the complete adaption of the absorption properties of the chromophore, thus resulting to very similar absorption spectra of polymer 8b compared to alkyne 7b.
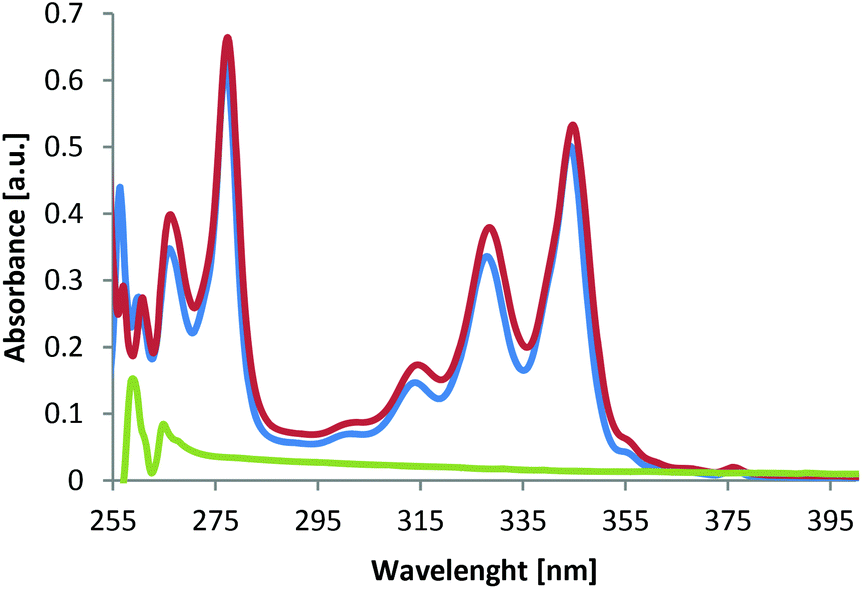 | ||
| Fig. 1 UV/vis spectra from 5a (green), 8b (red) and 7b (blue). | ||
We then realized that, when using a combination of two different alkynes for the postpolymerization modifications, two types of structures became accessible: (A) one-pot postpolymerization modification of azidated polymer 5a with different alkynes, using an equimolar mixture of 7a (R = R3) and 7c (R = R5), provided the polyamide 8e (Fig. 2A). The statistical copolymer contains three different bistriazole units: (i) with R3 and R3 residues, (ii) with R5 and R5 residues, and (iii) with R3 and R5 residues. (B) The sequential CuAAC reaction of 5a with one equivalent of 7a followed by the reaction with one equivalent of 7c gave the polyamide 8f consisting of two different bistriazole units, (i) and (ii) (Fig. 2B). Switching from one-pot to sequential postpolymerization CuAAC provides, therefore, a unique opportunity of how to create variations in molecular structures, despite the fact that the identical set of functional groups is introduced to the polymer precursors.
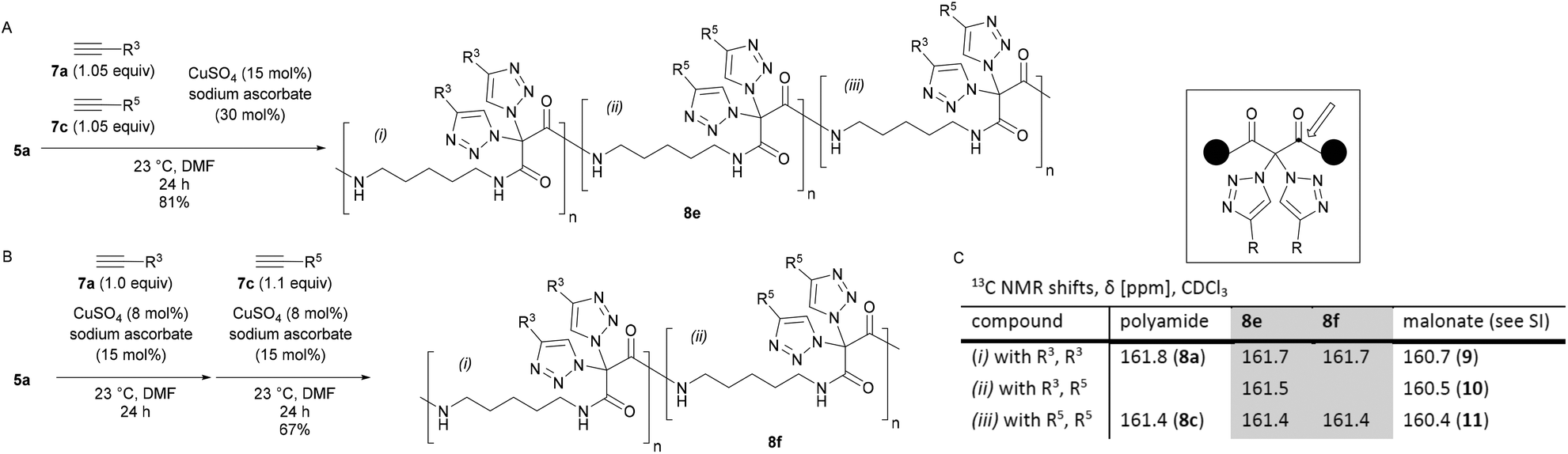 | ||
| Fig. 2 Postpolymerization modification of 5a with different alkynes. A: One-pot addition. B: Sequential addition. C: 13C NMR data of 8evs. 8f. | ||
The proposed structure of 8e and 8f was supported by 13C NMR spectroscopy (Fig. 2C): the chemical shift of the carbonyl peak was markedly different, depending on the exact nature of the geminal bistriazole units. For example, bistriazoles with two phenyl substituents (i.e., (i) with R3, R3) were found to have a signal at δ = 161.7 ppm in both polymers 8e and 8f, and the chemical shift was similar to the one obtained for polyamide 8a (δ = 161.8 ppm). A signal at δ = 161.4 ppm was assigned to bistriazole units of type (ii) with two R5 substituents, as observed for 8e, 8f (and 8c). In the case of polyamide 8e, an additional signal at δ = 161.5 ppm was clearly found for the bistriazole units of type (iii).
Finally, we attempted the reduction of the diazido functionalities of polyamide 5a.21 It was found that complete hydrogenolysis occurred when using hydrogen over palladium on charcoal in pure methanol at room temperature (Scheme 5). Polyamide 14 having free amino groups was accomplished in quantitative yield. We believe that this polymer is a potential precursor for the crosslinking of polyamide structures, a feature that will be studied in due course.
 | ||
| Scheme 5 Hydrogenolysis of 5e. | ||
Conclusions
In summary, we report here the first synthesis of polyamides containing geminal diazido moieties. The polymerization is a direct polyamidation between simple diamines and malonate-derived monomers with already installed diazido functionalities. This method avoids the use of activating reagents and provides a straightforward process at room temperature with high atomic economy. The new polyamides have highly reactive geminal dia-zido units and may become of interest as energetic materials with easily tuned stabilities and sensitivities. Post-polymerization modification of the azido groups through classical CuAAC or hydrogenolysis also offers the opportunity of installing a range of additional functionalities. Importantly, the polyamide design is based on difunctionalities in geminal position and, thus, allows for the installation functional groups in particularly close proximity to each other, a feature that may become attractive to researchers. Given the broad significance of functional polyamides, this polyamidation may find, in combination with the postpolymerization modification strategies, widespread utility in several technological and biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr Ullrich Scherf (BUW) for helpful discussions. Anke Helfer (BUW) is thanked for TGA-DSC measurements. We also thank Dr Björn Beele (BUW) for TGA-DSC measurements. We thank Thomas Fickinger (PSS Polymer Standards Service, Mainz) for SEC instrumentation assistance and helpful discussions. We thank Prof. Dr Roland Görtz (BUW) and Alexander Schaberg (BUW) for impact sensitivity measurements.Notes and references
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., Hoboken, NJ, 4th edn, 2004 Search PubMed.
- (a) K. Marchildon, Macromol. React. Eng., 2011, 5, 22 CrossRef CAS; (b) J. M. Garcia, F. C. Garcia, F. Serna and J. L. de la Pena, Prog. Polym. Sci., 2010, 35, 623 CrossRef CAS; (c) M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259 CrossRef CAS PubMed; (d) M. Pervaiz, M. Faruq, M. Jawaid and M. Sain, Curr. Org. Synth., 2017, 14, 146 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS PubMed.
- (a) C. L. R. Zaliz and O. Varela, Tetrahedron: Asymmetry, 2005, 16, 97 CrossRef; (b) Y. Fan, M. Kobayashi and H. Kise, Polym. J., 2000, 32, 817 CrossRef CAS; (c) Y.-T. Chern and W.-H. Chung, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 117 CrossRef CAS; (d) J. Thiem and F. Bachmann, Makromol. Chem., 1993, 194, 1035 CrossRef CAS.
- (a) S.-H. Hsiao and Y.-M. Chang, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4056 CrossRef CAS; (b) A. Okamura, T. Hirai, M. Tanihara and T. Yamaoka, Polymer, 2002, 43, 3549 CrossRef CAS.
- (a) J. Malineni, C. Merkens, H. Keul and M. Möller, Catal. Commun., 2013, 40, 80 CrossRef CAS; (b) H. Zeng and Z. Guan, J. Am. Chem. Soc., 2011, 133, 1159 CrossRef CAS; (c) B. Gnanaprakasam, E. Balaraman, C. Gunanathan and D. Milstein, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1755 CrossRef CAS; (d) T. J. Deming, Adv. Polym. Sci., 2006, 202, 1 CrossRef CAS.
- (a) E. M. O. Chiellini, R. Bizzarri, P. Bonaguidi, P. Talamelli and R. Solaro, J. Macromol. Sci., Part A: Pure Appl.Chem., 1999, 36, 901 CrossRef; (b) S. Mommer, H. Keul and M. Möller, Macromol. Rapid Commun., 2014, 35, 1986 CrossRef CAS; (c) Y.-Z. Wang, X.-X. Deng, L. Li, Z.-L. Li, F.-S. Du and Z.-C. Li, Polym. Chem., 2013, 4, 444 RSC; (d) A. Sehlinger, P.-K. Dannecker, O. Kreye and M. A. R. Meier, Macromolecules, 2014, 47, 2774 CrossRef CAS; (e) N. Ogata and K. Saito, J. Polym. Sci., 1975, 13, 2763 CAS.
- (a) A. P. Häring and S. F. Kirsch, Molecules, 2015, 20, 20042 CrossRef; (b) P. Klahn, H. Erhardt, A. Kotthaus and S. F. Kirsch, Angew. Chem., Int. Ed., 2014, 53, 7913 CrossRef CAS; (c) H. Erhardt, A. P. Häring, A. Kotthaus, M. Roggel, M. L. Tong, P. Biallas, M. Jübermann, F. Mohr and S. F. Kirsch, J. Org. Chem., 2015, 80, 12460 CrossRef CAS; (d) T. Harschneck, S. Hummel, S. F. Kirsch and P. Klahn, Chem. – Eur. J., 2012, 18, 1187 CrossRef CAS.
- C. J. Ruud, J. P. Jia and G. L. Baker, Macromolecules, 2000, 33, 8184 CrossRef CAS.
- (a) M. D. Ger, W. H. Hwu and C. C. Huang, Thermochim. Acta, 1993, 224, 127 CrossRef CAS; (b) T. Parr and D. Hanson-Parr, Combust. Flame, 2004, 137, 38 CrossRef CAS; (c) I. K. Varma, Macromol. Symp., 2004, 210, 121 CrossRef CAS.
- (a) D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369 RSC; (b) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952 CrossRef CAS; (c) P. Lundberg, C. J. Hawker, A. Hult and M. Malkoch, Macromol. Rapid Commun., 2008, 29, 998 CrossRef CAS; (d) G. Li, H. Zheng and R. Bai, Macromol. Rapid Commun., 2009, 30, 442 CrossRef CAS PubMed.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (c) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- (a) L. Billiet, D. Fournier and F. Du Prez, Polymer, 2009, 50, 3877 CrossRef CAS; (b) M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016 CrossRef CAS; (c) G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296 CrossRef CAS; (d) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef CAS.
- (a) B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404 CrossRef CAS; (b) X. Jiang, E. B. Vogel, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 1937 CrossRef CAS; (c) D. Fournier and F. Du Prez, Macromolecules, 2008, 41, 4622 CrossRef CAS; (d) O. Jazkewitsch, A. Mondrzyk, R. Staffel and H. Ritter, Macromolecules, 2011, 44, 1365 CrossRef CAS.
- A. P. Häring, P. Biallas and S. F. Kirsch, Eur. J. Org. Chem., 2017, 1526 CrossRef.
- The diazidated compound 1 is potentially hazardous and should be handled with great care. Although we never encountered any problems, we advise the use of protective gear.
- Full decomposition was not at an explosive rate: Instead of rapidly detonating, the material decomposed over a range of 60 °C (corresponding to 6 min), according to TGA measurements.
- End group analysis was not possible for 5a and 5d since end group signals were not unequivocally detected in the 1H NMR spectrum.
- Tests were conducted using BAM methods according to the UN Recommendations on the Transport of Dangerous Goods, Manual of Tests and Criteria, United Nations Publication, New York, 5th edn, 2009: Impact insensitive >40 J, less sensitive ≥35 J, sensitive ≥4 J, very sensitive ≤3 J Search PubMed.
- (a) N. ten Brummelhuis and M. Weck, ACS Macro Lett., 2012, 1, 1216 CrossRef CAS; (b) M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messerschmidt, B. Voit, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2005, 127, 14942 CrossRef CAS; (c) J. S. Oakdale, L. Kwisnek and V. V. Fokin, Macromolecules, 2016, 49, 4473 CrossRef CAS; (d) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS; (e) C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC; (f) S. Reinicke, P. Espeel, M. M. Stamenovic and F. E. Du Prez, ACS Macro Lett., 2013, 2, 539 CrossRef CAS; (g) A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC; (h) L. Billiet, X. K. D. Hillewaere and F. E. Du Prez, Eur. Polym. J., 2012, 48, 2085 CrossRef CAS; (i) M. Trigo-López, J. L. Barrio-Manso, F. Serna, F. C. García and J. M. García, Macromol. Chem. Phys., 2013, 214, 2223 Search PubMed; (j) X. Zhang, Z. Zhong and R. Zhuo, Macromolecules, 2011, 44, 1755 CrossRef CAS.
- P. Biallas and S. F. Kirsch, Tetrahedron Lett., 2017, 58, 4209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01087k |
| This journal is © The Royal Society of Chemistry 2019 |