
Synthesis of peptide–vinyl polymer multiblock hybrids by nitroxide-mediated polymerization: breaking the limitations of monomer compatibility†
Shin-nosuke
Nishimura
 ,
Nobuyuki
Higashi
* and
Tomoyuki
Koga
*
,
Nobuyuki
Higashi
* and
Tomoyuki
Koga
*
Department of Molecular Chemistry & Biochemistry, Faculty of Science & Engineering, Doshisha University, Kyotanabe, Kyoto, 610-0321, Japan. E-mail: tkoga@mail.doshisha.ac.jp; nhigashi@mail.doshisha.ac.jp
First published on 9th November 2018
Abstract
Significant efforts have been dedicated toward designing conjugated sequence-controlled peptides and synthetic vinyl polymers as a new class of polymeric materials with ordered structures and specific biofunctions. The creation of novel synthetic strategies that enables the precise incorporation of multiple tailored peptides along a vinyl polymer backbone remains highly challenging. Herein, we report a useful method for preparing multiblock architectures composed of alternately aligned sequential peptides and various vinyl polymers through nitroxide-mediated polymerization (NMP). A new cyclic oligopeptide was developed containing a 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (TIPNO)-derived alkoxyamine bond in the framework. Controlled radical polymerization in a homogeneous liquid phase using this peptide initiator successfully provided a well-defined multiblock hybrid polymer in one step, in which chain extension and multimerization proceeded simultaneously. This method was applied to the polymerizations of a wide range of vinyl monomers, including styrene, acrylamides, acrylates and acrylonitrile. Expanding the compatibility of monomers with this approach is valuable for constructing complex multiblock architectures with structural and functional diversity.
Introduction
Controlling the synthesis of multiblock copolymers with well-regulated structures, such as uniform block lengths and good monodispersities, based on chemically distinct polymer backbones remains a significant challenge in polymer chemistry. Multiblock architectures have enabled the spatial arrangement of specific blocks in a single polymer chain,1 which allows the incorporation of multiple and/or amplified functions. Such architectures are important for the design of novel polymeric materials with potential applications in various fields, such as self-assembling, folding, and degradable materials.2–8 Artificial peptides are an attractive block segment because they can provide enormous structural and functional diversity. Their chemical and physiological properties, including their self-assembling capabilities, cell adhesion, and antimicrobial activities, can be precisely designed based on the primary amino acid sequence.9–15 Recent significant advances in controlled radical polymerization techniques16–19 offer opportunities for synthesizing complex vinyl polymer architectures possessing multiblock and sequence-controlled structures20–23 as well as sequential peptide–vinyl polymer hybrids24–29 by incorporating solid phase peptide synthesis (SPPS) approaches. Although simple diblock, triblock, or graft copolymers can be well synthesized using these hybrid approaches, few studies have examined alternating peptide–vinyl polymer multiblock architectures30–32 due to a lack of efficient synthetic methodologies.One successful approach to constructing multiblock architectures involves a combination of click chemistry and living radical polymerizations.30,31 This approach, however, requires precise transformations of the chain ends on each block to provide appropriate reactive groups (i.e., azide/alkyne). Chromatographic purification is needed for isolating the objective polymer after the click reaction. Recently, we reported that the nitroxide-mediated polymerization (NMP) of styrene and its derivatives by cyclic peptide initiators having a 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO)-derived alkoxyamine bond in the framework successfully yielded multiblock hybrids composed of alternately aligned peptides and well-defined polystyrenes with low dispersities in one step.32 Significantly, this method permitted easy manipulation of the peptide sequence in the multiblock hybrids,32 although it was unfortunately only applicable to aromatic vinyl monomers due to the nature of the TEMPO group, which featured a relatively low dissociation rate constant (kd) of the C–ON bond.33 A new cyclic peptide initiator that provides more flexibility for polymer design is strongly required to produce functional multiblock architectures with structural diversity.
To overcome the limitations of monomer compatibility, we designed a novel hetero-bifunctional 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (TIPNO)-derived alkoxyamine (Fmoc-NH-TIPNO-COOH) for the synthesis of a versatile peptide initiator. A significant structural feature of TIPNO was the presence of a hydrogen atom attached to one of the α-carbons bonded to the nitrogen. This proton changed the kd value of the alkoxyamine bond and consequently increased the range of vinyl monomers that could be polymerized in a well-controlled living manner.33,34
Herein, we describe the preparation of a novel TIPNO-based cyclic peptide and its usefulness as an NMP initiator for the one-step production of a wide variety of multiblock copolymers composed of alternately aligned sequential peptides and various vinyl polymers such as polyacrylamides, polyacrylates, polyacrylonitrile and polystyrene.
Results and discussion
First, the synthesis of the bifunctional TIPNO-containing alkoxyamine, Fmoc-NH-TIPNO-COOH, was carried out, as outlined in Scheme 1. Mono Fmoc-protected ethylenediamine hydrochloride (Fmoc-EDA·HCl, 1) was synthesized by treating commercially available mono Boc-protected ethylenediamine with (i) Fmoc-OSu and (ii) hydrogen chloride. Bromine-terminated Fmoc-EDA (Fmoc-EDA-Br, 2) was then synthesized using a conventional condensation reaction between 1 and 2-bromo-2-methylpropanoyl bromide. N-tert-Butyl-α-isopropylnitrone (3) was obtained by reductive condensation of 2-methyl-2-nitropropane and isobutylaldehyde, reported by Hawker et al.34 for the preparation of α-hydrogen-bearing nitroxide in a single high-yield step. tert-Butyl 4-iodobenzoate (4) was obtained from the reaction of potassium tert-butoxide with the 4-iodobenzoic acid-derived acyl chloride. In general, Grignard reagents can only be prepared from simple halides without sensitive functional groups; however, Knochel et al. reported the successful synthesis of Grignard reagents containing ester and nitrile groups via a metal–halogen exchange reaction between the corresponding aryl iodides and iPrMgX under mild conditions at low temperatures.35 According to this procedure, the metal–halogen exchange reaction of 4 was carried out with iPrMgBr in THF at −30 °C over an hour. The resultant Grignard reagent was reacted with nitroxide 3 and then oxidized using Cu(II) as a catalyst under an ambient atmosphere to give the objective nitroxide radical (TIPNO-COOtBu, 5). Subsequently, Fmoc-NH-TIPNO-COOtBu (6) was synthesized by an atom transfer radical coupling reaction36 between 2 and 5 in the presence of Cu(0), Cu(II), and 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (Me4Cyclam). The desired Fmoc-TIPNO-COOH (7) was finally obtained by deprotection of the tert-butyl group using 2,2,2-trifluoroacetic acid (TFA). The obtained compounds 1–7, except for 5, which could not be detected by NMR spectroscopy due to the influence of the electron spin of the nitroxide radical, were characterized by 1H NMR analysis (Fig. S1–S5, S7 and S8†). The molecular weight and nitroxide radical of 5 were checked by DART-MS and electron spin resonance (ESR) spectroscopy, respectively (Fig. S6A and B†).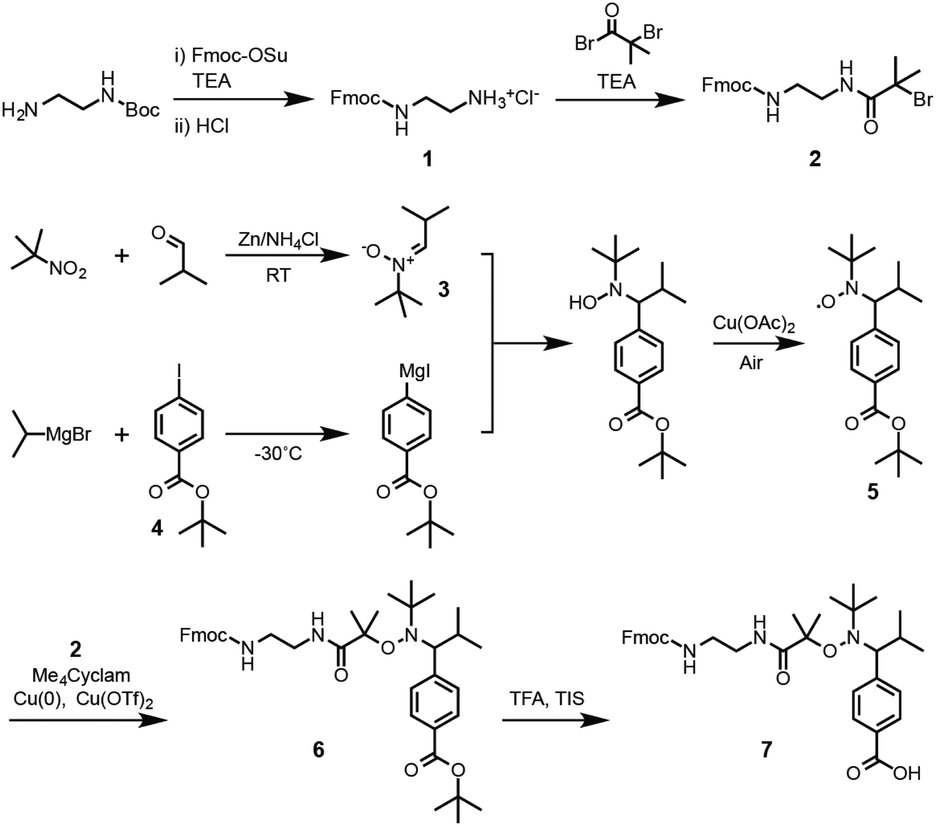 | ||
| Scheme 1 Synthetic route to the hetero-bifunctional TIPNO-derived alkoxyamine. | ||
We next prepared a new TIPNO-based cyclic peptide as an NMP initiator. In this system, the thermally promoted homolysis of the alkoxyamine bond generates a reactive carbon radical and a nitroxide radical simultaneously at the C and N termini of the peptide, respectively. In the presence of vinyl monomers, chain polymerization proceeds only from the carbon radical at the peptide terminus, not from the stable nitroxide radical; however, the nitroxide radical can trap the intermediate growing polymer radical reversibly, which provides a multiblock architecture during polymerization.
First, the peptide segment was constructed on an Fmoc-NH-SAL MBHA resin by SPPS using Fmoc-derivatives. We selected a tetraleucine sequence as an example because of its structural simplicity and easy characterization. TIPNO-derived alkoxyamine was next introduced at the N-terminus of the peptide using 7. The allyl group of the Asp residue and Fmoc group were removed by treating with palladium(0) and piperidine, respectively. Subsequently, intramolecular cyclization of the peptide was carried out on the resin through condensation between the N-terminus and the Asp side chain. Finally, the resin was treated with TFA to give the objective TIPNO-based cyclic peptide initiator. The molecular structure was verified by MALDI-TOF MS and 1H NMR analyses (Fig. 1). The observed molecular weight ([M + H]+obsd. = 1233.68) agreed well with the theoretical value ([M + H]+calcd. = 1233.57). 1H NMR analysis also gave satisfactory results.
 | ||
| Fig. 1 A. MALDI-TOF MS spectrum of the TIPNO-based cyclic peptide initiator. Matrix: DHBA. B. 1H NMR spectrum of the peptide in DMSO-d6 at 25 °C. | ||
The versatility of this new type of peptide initiator was demonstrated by conducting the polymerization using a wide range of vinyl monomers, including acrylamides, acrylates, acrylonitrile, and styrene (Scheme 2). Initially, NMPs of the acrylamides were tested. Polymerization of N-isopropylacrylamide (NIPAM) was performed in DMF at 120 °C, and the molecular weights and polydispersity indexes (Đ) were evaluated by size-exclusion chromatography (SEC). Table 1 summarizes the resulting products obtained after polymerization for 1 (P1), 3 (P2), 6 (P3), 12 (P4), and 24 h (P5). The SEC traces of all polymers were unimodal and showed a clear shift toward high molecular weights with conversion (Fig. 2); however, the number average molecular weights (Mn) of these polymers deviated remarkably from the calculated theoretical values (Mn,Theor) based on the monomer conversion, assuming one peptide segment per chain (for P1 (Mn = 7000 g mol−1, Mn,Theor = 2820 g mol−1), P2 (Mn = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1, Mn,Theor = 4850 g mol−1), P3 (Mn = 19700 g mol−1, Mn,Theor = 6400 g mol−1), P4 (Mn = 24500 g mol−1, Mn,Theor = 9100 g mol−1), P5 (Mn = 38600 g mol−1, Mn,Theor = 11400 g mol−1)). The polydispersities were relatively broad (Đ ≈ 1.8). These polymers were soluble in water and showed lower critical solution temperature (LCST) behaviors, due to the PNIPAM block. On the other hand, circular dichroism (CD) analysis clearly indicated the presence of a peptide segment in the polymer chain (e.g., for P2; Fig. 3B (blue line)). The CD spectrum showed a mixed pattern of β-sheet and random coil structures with a negative maximum at 208 nm and a shoulder at 218 nm. Moreover, the 1H NMR spectrum (Fig. 3C, blue line) supported the presence of both peptide and PNIPAM segments. These results indicated the production of multiblock structures through the ring-opening of the peptide initiator and subsequent NMP. Note that the observed LCST for the multiblock hybrid was distinctly shifted to the lower temperature side compared with that for the PNIPAM homopolymer even in the case of the hybrid with a long PNIPAM block length (Fig. S9†). This can be attributed to the introduction of hydrophobic tetraleucine blocks.
200 g mol−1, Mn,Theor = 4850 g mol−1), P3 (Mn = 19700 g mol−1, Mn,Theor = 6400 g mol−1), P4 (Mn = 24500 g mol−1, Mn,Theor = 9100 g mol−1), P5 (Mn = 38600 g mol−1, Mn,Theor = 11400 g mol−1)). The polydispersities were relatively broad (Đ ≈ 1.8). These polymers were soluble in water and showed lower critical solution temperature (LCST) behaviors, due to the PNIPAM block. On the other hand, circular dichroism (CD) analysis clearly indicated the presence of a peptide segment in the polymer chain (e.g., for P2; Fig. 3B (blue line)). The CD spectrum showed a mixed pattern of β-sheet and random coil structures with a negative maximum at 208 nm and a shoulder at 218 nm. Moreover, the 1H NMR spectrum (Fig. 3C, blue line) supported the presence of both peptide and PNIPAM segments. These results indicated the production of multiblock structures through the ring-opening of the peptide initiator and subsequent NMP. Note that the observed LCST for the multiblock hybrid was distinctly shifted to the lower temperature side compared with that for the PNIPAM homopolymer even in the case of the hybrid with a long PNIPAM block length (Fig. S9†). This can be attributed to the introduction of hydrophobic tetraleucine blocks.
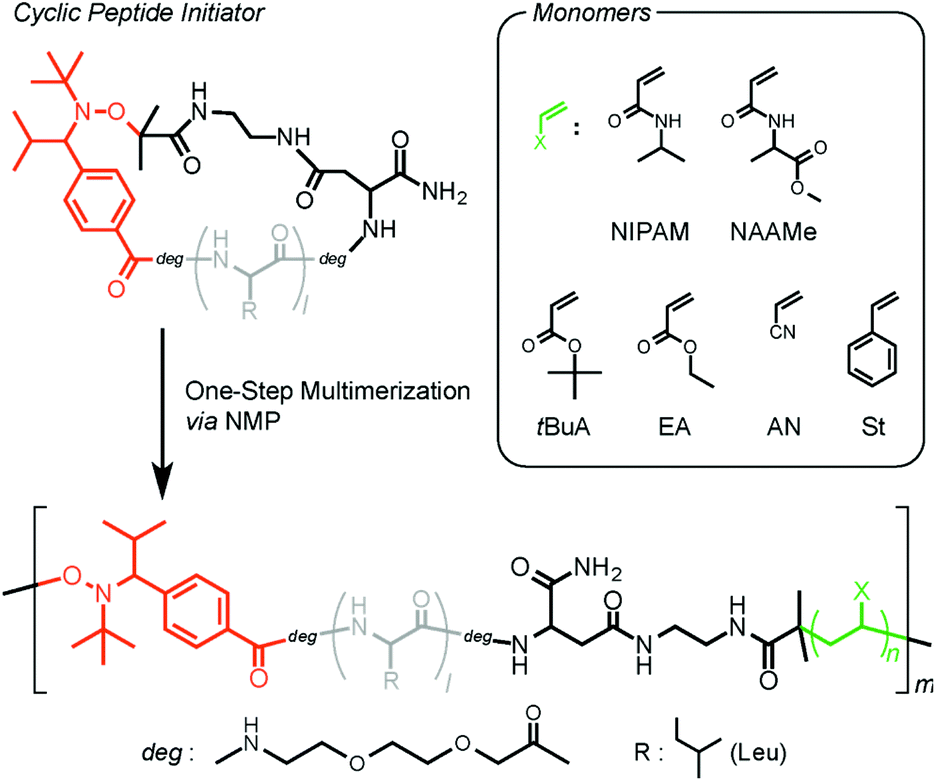 | ||
| Scheme 2 Schematic illustration of the one-step synthesis of versatile multiblock peptide–polymer hybrids via NMP utilizing the TIPNO-based cyclic peptide initiator. | ||
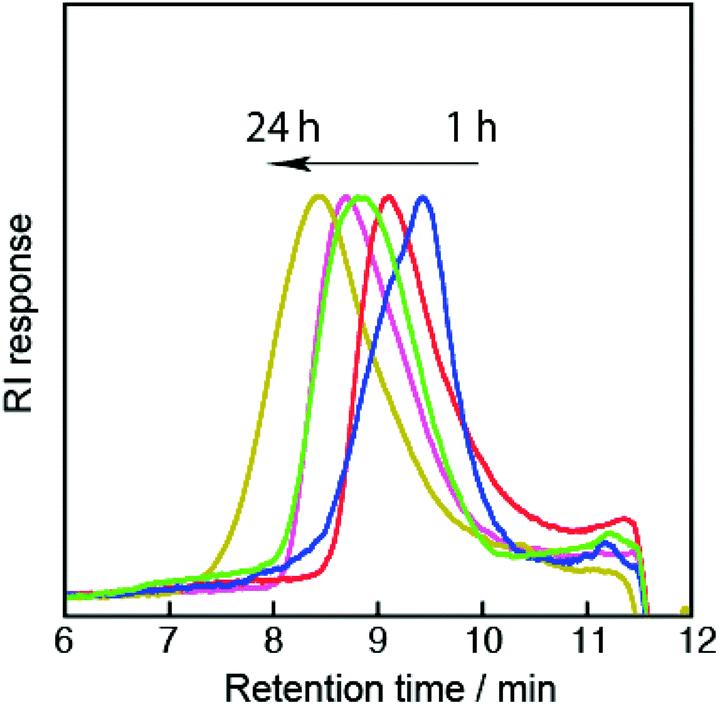 | ||
| Fig. 2 SEC traces (THF, 40 °C) of the multiblcok copolymers (P1–P5) obtained after polymerization for 1 (blue), 3 (red), 6 (green), 12 (pink) and 24 h (yellow). | ||
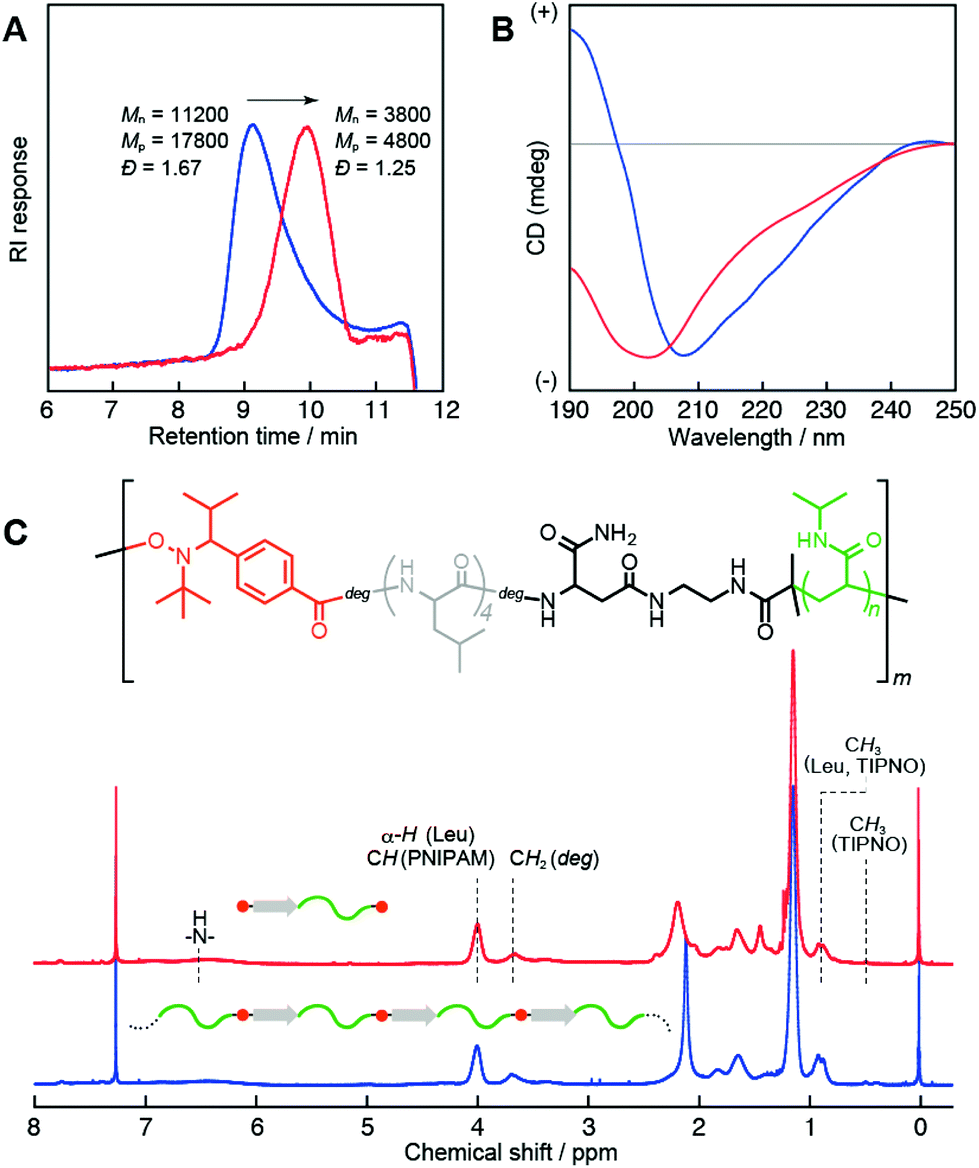 | ||
| Fig. 3 A. SEC traces (THF, 40 °C) of the multiblock P2 obtained before (blue) and after (red) fragmentation. B. CD spectra of P2 (blue) and fragmented P2 (red) in water at 25 °C. [polymer] = 0.01 wt%. C. 1H NMR spectra of P2 (blue) and fragmented P2 (red) in CDCl3 at 25 °C. | ||
| Polymer | Monomer | Polymn. time (h) | Polymn. temp. (°C) | Conv. (%) |
M
n, Theora (g mol−1) |
Before fragmentation | After fragmentation | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M n (g mol−1) |
M
pb (g mol−1) |
Đ (Mw/Mn) | M n (g mol−1) |
M
pb (g mol−1) |
Đ (Mw/Mn) | ||||||
| a Theoretical number-average molecular weight of the fragmented polymer calculated by using the following equation: Mn,Theor. = [monomer]/[initiator] × conversion × molecular weight of monomer + molecular weight of the initiator. b Molecular weight at the top of the peak in the SEC curve. c Number-average molecular weight calculated by SEC analysis (PSt standard) in THF at 40 °C. d Number-average molecular weight calculated by SEC analysis (PMMA standard) in THF at 40 °C. e Number-average molecular weight calculated by SEC analysis (PMMA standard) in DMF (containing 10 mM LiBr) at 40 °C. | |||||||||||
| P1 | NIPAM | 1 | 120 | 14.0 | 2820 | 7000d | 10300 |
1.77 | 2400d | 3400 | 1.20 |
| P2 | NIPAM | 3 | 120 | 32.1 | 4850 | 11200d |
17800 |
1.67 | 3800d | 4800 | 1.25 |
| P3 | NIPAM | 6 | 120 | 45.6 | 6400 | 19700d |
30600 |
1.87 | 7500d | 9600 | 1.19 |
| P4 | NIPAM | 12 | 120 | 69.8 | 9100 | 24500d |
36100 |
1.67 | 8300d | 12900 |
1.21 |
| P5 | NIPAM | 24 | 120 | 89.9 | 11400 |
38600d |
64300 |
2.12 | 12400d |
21800 |
1.28 |
| P6 | NAAMe | 18 | 120 | 87.6 | 15000 |
32300d |
53000 |
1.46 | 12700d |
19800 |
1.23 |
| P7 | tBuA | 17 | 120 | 79.4 | 11400 |
26300d |
42800 |
1.89 | 9500d | 13100 |
1.18 |
| P8 | EA | 25 | 120 | 42.6 | 5500 | 9500d | 18400 |
1.63 | 3700d | 6200 | 1.22 |
| P9 | AN | 16 | 120 | 97.7 | 6400 | 34200e |
43500 |
1.54 | 13400e |
15200 |
1.38 |
| P10 | St | 72 | 110 | 76.5 | 9200 | 38800c |
55000 |
1.77 | 8400c | 9100 | 1.10 |
These structures were characterized by conducting fragmentation experiments using a radical crossover reaction.37 Assuming a multiblock structure, the obtained polymer must include thermally labile alkoxyamine bonds in the main chain at multiple points, reflective of the block number. Fragmentation reactions of P1–P5 were performed in the presence of a large excess of TIPNO (>70 equiv.) in N-methyl-2-pyrrolidone at 120 °C over 12 h, and the resultant polymers were isolated using a reprecipitation method. As a representative example, SEC analysis of P2 (Fig. 3A) showed a marked decrease in both the molecular weight and polydispersity index after treatment, from Mn = 11200 g mol−1 (Đ = 1.67) to Mn = 3800 g mol−1 (Đ = 1.25). Other polymers showed similar fragmentation behaviors (Fig. S11A–S14A†). On the other hand, the 1H NMR (Fig. 3C, S11C–S14C†) and FTIR spectra before and after fragmentation (Fig. S10, S11B–S14B†) did not change significantly, demonstrating cleavage of the polymer chain without altering the compositions of the PNIPAM/peptide blocks, namely, fragmentation from a multi- to a diblock structure. Note that fragmentation caused a conformational change in the peptide block. The CD spectrum of the fragmented P2 (diblock form) showed a pattern typical of a random coil peptide, with a negative maximum at 200 nm (Fig. 3B, red line). Thus, the unique multiblock structure with alternately aligned peptides and PNIPAM blocks increased the local peptide concentration. As a result, the hydrophobic tetraleucine blocks self-organized into β-sheets in water. In fact, increasing the chain lengths of the PNIPAM blocks tended to suppress high-order structuring of the peptide blocks (Fig. S11D–14D†).
The most important feature of this method is that the PNIPAM block length was precisely controlled during conversion. In other words, a specific spatial arrangement of peptide blocks could be designed in a single polymer chain. The SEC charts of the fragmented diblock polymers obtained from P1–P5 were symmetrically unimodal and showed a clear shift toward high molecular weights as the polymerization time increased (Fig. 4A). Fig. 4B plots Mn and Đ of the fragmented polymers as a function of the monomer conversion. In this figure, the calculated values of the theoretical Mn,Theor are included as a solid line. The Đ values fell below 1.3 throughout the polymerization reaction, indicating that nearly monodisperse polymers were formed. The linearity of the Mnversus conversion plot, which was comparable to the Mn,Theor plot, clearly showed that NMP proceeded in a well-controlled living manner. These results also indicated that the initiation efficiency of this cyclic peptide initiator was sufficiently high. The ratio of the molecular weight of the original polymer to the corresponding fragmented one (Table 1) suggested that the multiblock copolymers [(Leu)4-b-PNIPAM]m contained approximately three repeats (m ≈ 3) of the (Leu)4-b-PNIPAM diblock unit (i.e. total hexablock). This TIPNO-based cyclic peptide initiator could be applied to other acrylamides derived from the amino acid, N-acryloyl-L-alanine O-methyl ester (NAAMe).
 | ||
| Fig. 4 A. SEC traces (THF, 40 °C) of the fragmented block copolymers obtained from P1–P5, demonstrating excellent controlled polymerization. B. Plots of Mn (blue circles) and Đ (red triangles) as a function of the conversion rate. The solid line represents the theoretical Mn (see the footnote (a) in Table 1). | ||
Amino acid-based vinyl polymers have enormous potential as smart biomaterials because they show tunable thermo-responsive behaviors in water and excellent biocompatibilities.38–41 For example, we reported that the LCST/UCST behaviors of amino acid-based vinyl polymers depended on the type of amino acid and the chemical modification of the pendant group.39 The objective multiblock hybrid (P6) was successfully obtained by NMP in one step and was characterized by SEC, 1H NMR, and FTIR analyses in the same manner as described above (Table 1, Fig. S15†). This polymer was soluble in water (below the LCST of the PNAAMe block, 18 °C (ref. 39)) and cleavable by a thermal treatment (under excess TIPNO). The fragmented P6 showed an Mn value close to the Mn,Theor as well as a relatively low dispersity (Đ = 1.23). The hybrid P6 assumed an unprecedented and unique multiblock architecture composed of sequential peptides and amino acid-based polymers; therefore, it is highly attractive as both a stimuli-responsive and protein-mimetic biomaterial.
We next polymerized two acrylates: tert-butyl acrylate (tBuA) and ethyl acrylate (EA). Acrylate polymers are useful as general-purpose plastics over large areas. The one-step syntheses of the multiblock hybrids using TIPNO-based cyclic peptides were successful in both cases (P7 (Mn ≈ 26300) and P8 (Mn ≈ 9500)) (Table 1, Fig. S16–S18†). The fragmentation experiments showed that the dispersities of the PtBuA and PEA blocks remained satisfactorily low (Đ < 1.25), and both polymers featured three repeats (m ≈ 3) of the peptide-b-polyacrylate diblock unit. It should be noted that the PtBuA blocks in P7 could be converted easily into poly(acrylic acid) (PAA) blocks by removing the tBu groups (Fig. S17†). The resultant fully deprotected hybrid was soluble in water and exhibited pH-responsiveness.
In a previous study,32 we reported that the copolymerization of acrylonitrile (AN) and styrene (St) proceeded via a TEMPO-based cyclic peptide initiator only when St was the major component; however, the TIPNO-based cyclic peptide initiator allowed homopolymerization of AN to proceed in DMF at 120 °C and yielded a multiblock peptide/PAN hybrid of Mn ≈ 34200 (P9) (Table 1, Fig. S19†). FTIR and 1H NMR analyses verified the formation of PAN blocks (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching bond at ν = 2242 cm−1 and 1H signal at δ ≈ 3.2 ppm (–CH(CN)–), although the observed dispersity (Đ = 1.38) was slightly larger than that of the other vinyl monomers. Of course, this new cyclic initiator, as well as the previously reported TEMPO-based initiator, is applicable to the polymerization of aromatic St and gives well-regulated multiblock hybrids (P10, Table 1, Fig. S20†). Notably, an enhanced polymerization rate was observed for the TIPNO-based cyclic peptide system (conversion 77% at 72 h) compared to the rate obtained using the TEMPO-based initiator (34% at 70 h)32 under the same reaction conditions, reflecting the difference between the dissociation rate constants of the two alkoxyamines.
N stretching bond at ν = 2242 cm−1 and 1H signal at δ ≈ 3.2 ppm (–CH(CN)–), although the observed dispersity (Đ = 1.38) was slightly larger than that of the other vinyl monomers. Of course, this new cyclic initiator, as well as the previously reported TEMPO-based initiator, is applicable to the polymerization of aromatic St and gives well-regulated multiblock hybrids (P10, Table 1, Fig. S20†). Notably, an enhanced polymerization rate was observed for the TIPNO-based cyclic peptide system (conversion 77% at 72 h) compared to the rate obtained using the TEMPO-based initiator (34% at 70 h)32 under the same reaction conditions, reflecting the difference between the dissociation rate constants of the two alkoxyamines.
Conclusions
In summary, we successfully developed a new type of TIPNO-derived alkoxyamine-containing cyclic peptide as an NMP initiator. Importantly, this unique polymerization method is applicable to a wide variety of vinyl monomers for the one-step production of multiblock architectures composed of sequential peptides and well-defined vinyl polymers. The block length of the vinyl polymer was precisely controlled by conversion, enabling a spatial arrangement of peptide blocks in a single polymer chain. We believe that this robust synthetic strategy will enable the creation of complex and functional macromolecular architectures with unlimited potential in the nanotechnology, biomedical, and industrial fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by Grants-in-Aid for Scientific Research (KAKENHI) (No. 17K04994 and 16K05800) from the Japan Society for the Promotion of Science (JSPS), and a MEXT-Supported Program for the Strategic Research Foundation at Private University.References
- F. S. Bates, M. A. Hilmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS.
- D. J. Pochan, Z. Chen, H. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS.
- T.-B. Yu, J. Z. Bai and Z. Guan, Angew. Chem., Int. Ed., 2009, 48, 1097–1101 CrossRef CAS.
- M. T. Krejchi, E. D. Atkins, A. J. Waddon, M. J. Fournier, T. L. Mason and D. A. Tirell, Science, 1994, 265, 1427–1432 CrossRef CAS.
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924 CrossRef CAS.
- B. Schmidt, N. Fechler, J. Falkenhagen and J. F. Lutz, Nat. Chem., 2011, 3, 234–238 CrossRef CAS.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS.
- Y. Takahashi, A. Ueno and H. Mihara, Chem. – Eur. J., 1998, 4, 2475–2484 CrossRef CAS.
- H. A. Lashuel, S. R. LaBrenz, L. Woo, L. C. Serpell and J. W. Kelly, J. Am. Chem. Soc., 2000, 122, 5262–5277 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- T. Koga, M. Higuchi, T. Kinoshita and N. Higashi, Chem. – Eur. J., 2006, 12, 1360–1367 CrossRef CAS.
- H. T. More, K. S. Zhang, N. Srivastava, J. A. Frezzo and J. K. Montclare, Biomacromolecules, 2015, 16, 1219–1217 CrossRef.
- M. D. Pierschbacher and E. Ruoslahti, Nature, 1984, 309, 30–33 CrossRef CAS.
- T. Ganz, M. E. Selsted, D. Szklarek, S. S. Harwig, K. Daher, D. F. Bainton and R. I. Lehrer, J. Clin. Invest., 1985, 76, 1427–1435 CrossRef CAS.
- M. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- A. H. Soeriyadi, C. Boyer, F. Nystrom, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS.
- Y. Mei, K. L. Beers, H. C. MichellesByrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS.
- J. Hentschel and H. G. Börner, J. Am. Chem. Soc., 2006, 128, 14142–14149 CrossRef CAS PubMed.
- T. Koga, S. Kamiwatari and N. Higashi, Langmuir, 2013, 29, 15477–15484 CrossRef CAS.
- N. Higashi, Y. Yasufuku, M. Matsuo, T. Matsumoto and T. Koga, Colloid Interface Sci. Commun., 2014, 1, 50–53 CrossRef CAS.
- T. Koga, E. Aso and N. Higashi, Langmuir, 2016, 32, 12378–12386 CrossRef CAS PubMed.
- S. Nishimura, A. Hirata, Y. Taki, Y. Morita, N. Higashi and T. Koga, Chem. Lett., 2018, 47, 555–558 CrossRef CAS.
- K. Luo, J. Yang, P. Kopečková and J. Kopeček, Macromolecules, 2011, 44, 2481–2488 CrossRef CAS.
- S. E. Grieshaber, B. A. Paik, S. Bai, K. Kiick and X. Jia, Soft Matter, 2013, 9, 1589–1599 RSC.
- S. Nishimura, N. Higashi and T. Koga, Chem. – Eur. J., 2017, 23, 15050–15058 CrossRef CAS.
- S. Marque, C. L. Mercier, P. Tord and H. Fischer, Macromolecules, 2000, 33, 4403–4410 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- A. E. Jensen, W. Dohle, I. Sapountzis, D. M. Lindsay, V. A. Vu and P. Knochel, Synthesis, 2002, 565–569 CrossRef CAS.
- K. Matyjaszewski, B. E. Woodworth, X. Zhang, S. G. Gaynor and Z. Metzner, Macromolecules, 1998, 31, 5955–5957 CrossRef CAS.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, Chem. Commun., 2002, 2838–2839 RSC.
- H. Mori, H. Iwaya, A. Nagai and T. Endo, Chem. Commun., 2005, 4872–4874 RSC.
- N. Higashi, R. Sonoda and T. Koga, RSC Adv., 2015, 5, 67652–67657 RSC.
- N. Higashi, A. Hirata, S. Nishimura and T. Koga, Colloids Surf., B, 2017, 159, 39–46 CrossRef CAS.
- N. Higashi, D. Sekine and T. Koga, J. Colloid Interface Sci., 2017, 500, 341–348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplemental figures. See DOI: 10.1039/c8py01330f |
| This journal is © The Royal Society of Chemistry 2019 |