
Polymerization induced self-assembly: an opportunity toward the self-assembly of polysaccharide-containing copolymers into high-order morphologies
Jean-Luc
Six
 and
Khalid
Ferji
*
and
Khalid
Ferji
*
Université de Lorraine, CNRS, LCPM, F-5400 Nancy, France. E-mail: khalid.ferji@univ-lorraine.fr
First published on 3rd October 2018
Abstract
Glyco-nanostructures, nano-objects obtained from the self-assembly of amphiphilic copolymers containing natural polysaccharides called glycopolymers, are promising nanocarriers that have attracted great attention for many decades owing to their interesting biodegradability and biocompatibility. However, the conventional self-assembly processes used currently lead to the production of primitive assemblies. This mini-review focuses on the recent efforts in the self-assembly of amphiphilic glycopolymers and highlights the advantages of Polymerization Induced Self-Assembly (PISA) to offer them a new breakthrough in biomedical applications, as their synthetic copolymer counterparts.
 Jean-Luc Six | Jean-Luc Six is a professor at Lorraine University (France). He received his Ph.D. degree in polymer chemistry in 1996 from the University of Bordeaux. Then, he joined CERM (University of Liege, Belgium) as a postdoctoral fellow in the group of Pr. Robert Jérôme. His current research interests include the controlled synthesis of biodegradable/biocompatible amphiphilic (gly)copolymers and the formulation of glycopolymer-based submicronic biomaterials, which can be used as nanocarriers for drug delivery or as microcarriers for cell culture. |
 Khalid Ferji | Khalid Ferji is an associate professor at Lorraine University. He received his Ph.D. degree in polymer chemistry in 2013 from the University of Lorraine. In 2014, he joined the IMMM (University of Maine) as a postdoctoral fellow and started working on colloid science and light scattering techniques. Then, he moved to the LCPO (University of Bordeaux) as a postdoctoral fellow where he worked on the self-assembly of copolymers/lipids to produce biomimetic vesicles for drug/gene delivery. His research interests are currently focused on the polymer chemistry and colloidal assemblies of amphiphilic copolymers using photo-induced PISA for biomedical applications. |
1. Introduction
Soft nanostructures, including solid nanoparticles, spherical/worm-like micelles and vesicles, are the result of a spontaneous organization of amphiphilic species, i.e. phospholipids and/or amphiphilic copolymers (ACPs), in aqueous solution to reduce energetically unfavorable hydrophobic/water interactions.1 Such fascinating morphologies have found applications in many fields ranging from cosmetics to the food industries. Their use as drug delivery systems (DDS) has been one of their greatest attractive applications.2 Indeed, several drugs cannot be administered as they exhibit poor water solubility and/or suffer from limited stability in the human body. Encapsulation of drugs into the hydrophobic core of micelles or solid nanoparticles has been firstly used to overcome these issues.3 However, such types of DDS lead to the loading of mostly hydrophobic drugs, which limits the choice of drugs. DDS with advanced morphologies such as vesicles, including liposomes, polymersomes and hybrid polymer/lipid vesicles,4,5 are under intense scrutiny given their interesting multi-compartmental morphology (Fig. 1). In this morphology, the hydrophilic cavity can serve as a natural carrier environment for hydrophilic species (drugs or imaging agents) and the hydrophobic bilayer membrane can incorporate hydrophobic ones, while a hydrophilic shell allows stabilization of such nano-objects in aqueous solution. Formulations based on liposomes, which are vesicles resulting from the self-organization of phospholipids, are already marketed for some therapies (e.g. DaunoXome®, Doxil®/Caelyx®). However, liposomes exhibit some important drawbacks: (i) lack of mechanical stability in the high shear rate of bloodstream circulation through tiny vessels and (ii) the leakage phenomenon known as “burst release”, which compromises the release efficiency of encapsulated drugs at the target biological site.4 Polymersomes, polymeric counterparts of liposomes formed by the self-assembly of ACPs, exhibit higher mechanical stability and lower membrane permeability.6 This may provide an ideal solution to maintain the drug within a desired therapeutic range after a single dose.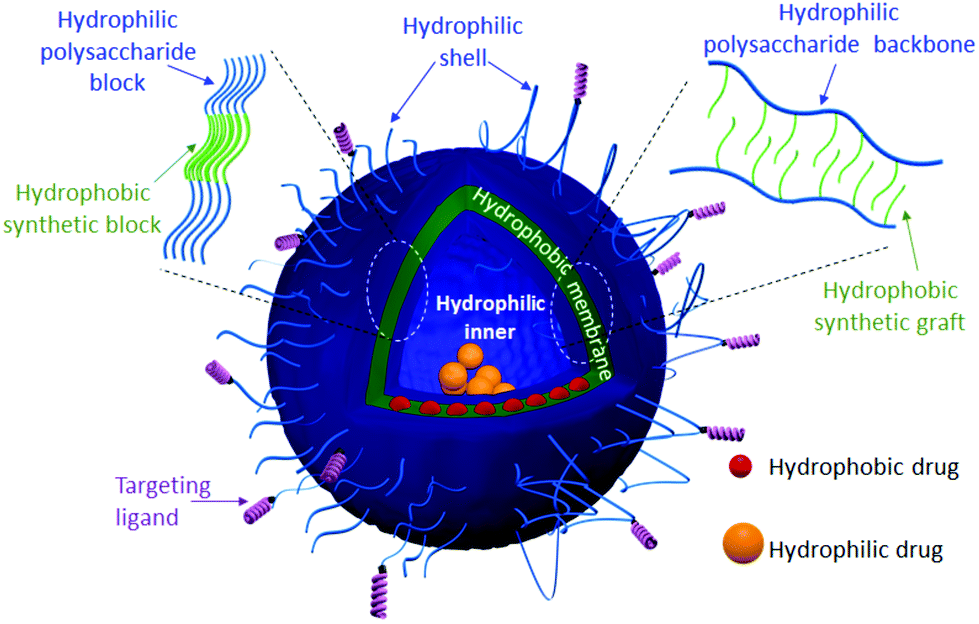 | ||
| Fig. 1 Representative structure of the expected glyco-polymersome for targeted drug delivery. Schematic description of the vesicle membrane prepared from diblock (left) and grafted (right) glycopolymers. | ||
For many decades, poly(ethylene oxide) (PEO)7 has been widely used in academia and industry, especially in biomedicine,8 to ensure the steric stabilization of the nanostructures and to enable them to escape the mononuclear phagocyte system, which prevents their preliminary elimination from the bloodstream.9
However, the low number of reactive functional groups in the PEO chain (only one hydroxyl function at its extremity) may limit its further chemical modification with specific ligands to reach specific cells for targeting and/or for specific cell uptake. Owing to their high hydrophilicity, non-toxicity and biocompatibility, neutral polysaccharides including dextran and pullulan have been reported as promising alternatives to PEO.10,11 In addition, such polysaccharides can enable an efficient surface functionalization with “bioactive” molecules, thanks to the numerous functional groups present on their backbone. Despite such enormous advantages, reports on the self-assembly of ACPs containing hydrophilic polysaccharides, called amphiphilic glycopolymers (AGPs), into vesicular morphology12,13 are less abundant compared to those based on synthetic ACPs.14 The term glycopolymers is still not clearly defined. According to the literature,15,16 glycopolymers may include chemically modified natural polysaccharides with synthetic polymers, as well as synthetic polymers containing sugar moieties as pendant groups. In this review, only modified natural polysaccharides will be considered.
This mini-review highlights the recent progress in the self-assembly of amphiphilic polysaccharide-based copolymers in aqueous medium. Here, we briefly discuss the synthesis strategies of AGPs and the conventional processes currently used to induce their self-assembly. The unsuitability of such processes for AGPs will be demonstrated through a comparison of nano-assemblies prepared using dextran and PEO-based amphiphilic copolymers. Finally, we introduce the Polymerization Induced Self-Assembly (PISA) method and its incontestable benefits for the AGP self-assembly.
2. Amphiphilic glycopolymer synthesis
Today, great achievements17 have been made regarding the synthesis of well-defined AGPs containing hydrophilic polysaccharides. Note that copolymers with pendant sugar moieties18,19 along their chains will not be considered in the present mini-review. Grafted/diblock AGPs were prepared from polysaccharides using both grafting from20,21 and grafting onto22–24 strategies (Fig. 2) combined with advanced controlled radical polymerization techniques including Atom Transfer Radical Polymerization (ATRP),21,24 Reversible Addition–Fragmentation Chain Transfer (RAFT) polymerization,25,26 Nitroxide Mediated Polymerization (NMP)27 and Single Electron Transfer Living Radical Polymerization (SET-LRP).23 The grafting from route (Fig. 2a) consists of polymerizing a synthetic hydrophobic monomer from a functionalized hydrophilic polysaccharide with reactive functions (initiators or the RAFT agent) (Fig. 2a) whereas the grafting onto strategy (Fig. 2b) involves the coupling of a preformed hydrophobic polymer chain to a functionalized hydrophilic polysaccharide using antagonist functions (Fig. 2b). In both strategies, a reversible partial protection of lateral hydroxyl groups of the polysaccharide is often required to make the latter soluble in the reaction organic medium. For more detailed information, the reader can refer to some excellent reviews dealing with the AGP synthesis.28–30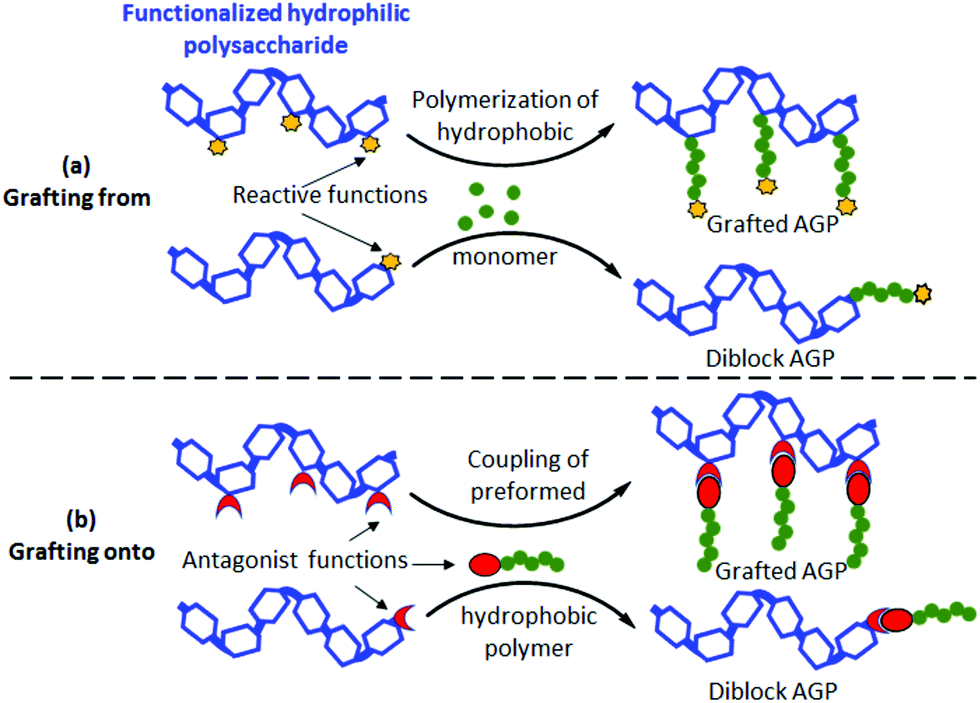 | ||
| Fig. 2 Synthesis strategies of grafted/diblock amphiphilic glycopolymers (AGPs) using (a) grafting from and (b) grafting onto routes. | ||
3. Conventional self-assembly process
Despite the development and the control of their synthesis, more work is still required to improve the self-assembly and nanostructure formulation of AGPs. Currently, while a broad set of nano-object morphologies have been reported with synthetic ACPs,1,14 spherical micelles17,29 and core/shell solid nanoparticles22,31,33 have been frequently reported for AGPs. Reports on the advanced morphologies of glycopolymer-based nanostructures (termed glyco-nanostructures, GNS) including vesicles12,13 are scarce as illustrated in Fig. 3. Table 1 shows several nanostructures generated by PEO-based ACPs and dextran-based AGPs as the hydrophilic part and different hydrophobic polymers (PLA, PCL and PS).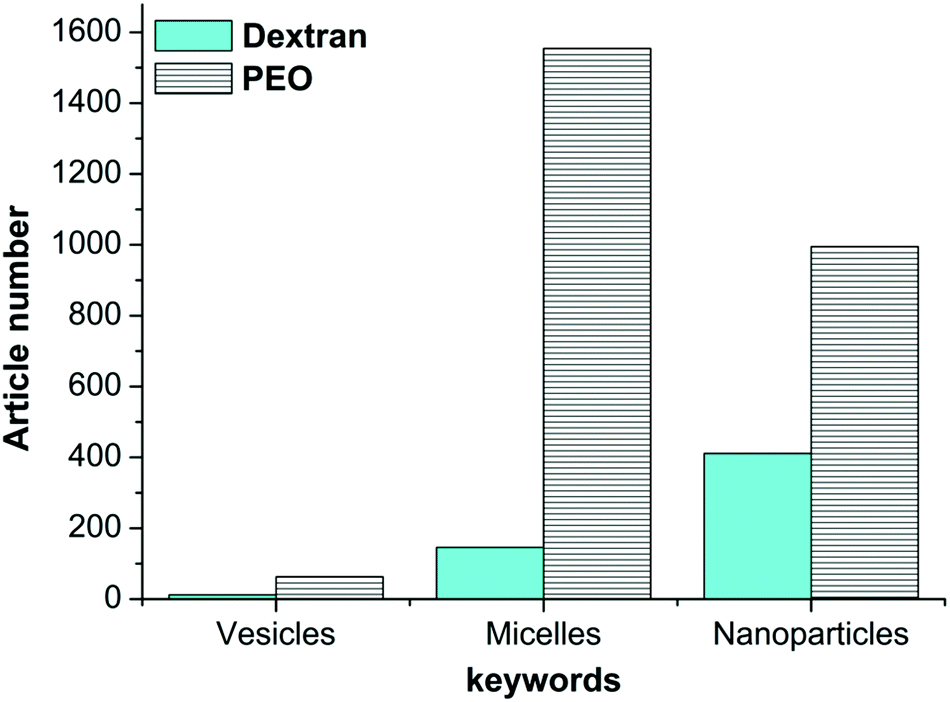 | ||
| Fig. 3 Number of publications dealing with dextran or PEO-based nanostructures. Statistics were made in May 2018 using the Web of Science database. | ||
| Hydrophobic part | Amphiphilic (gly)copolymer | f hydrophilic | Assembly morphology | Method (organic solvent) |
|---|---|---|---|---|
| Poly(D,L-lactide) (PLA) | Dex1.5k-b-PLA10k,20k,50k | 0.02 to 0.50 | Core/shell nanoparticles35,36 | Nanoprecipitation (DMSO) |
| Dex6k-b-PLA10k,20k,50k | ||||
| Dex10k-b-PLA10k,20k,50k | ||||
| Dex6k-b-PLA2k,4K | 0.75 | Micelles37 | ||
| 0.60 | ||||
| Dex29K-g14-PLA0.2K | 0.85 | Core/shell nanoparticles38 | Emulsion/solvent elimination (DCM) | |
| Dex33K-g36-PLA2.3K | 0.17 | |||
| Dex21K-g45-PLA3.1K | 0.10 | Core/shell nanoparticles32 | Nanoprecipitation (acetone or THF) | |
| Dex20k-g23-PLA16k | 0.05 | Microspheres39 | Emulsion/solvent elimination (DCM/DMSO) | |
| Dex20k-g12-PLA8k | 0.18 | |||
| Dex20k-g17-PLA3.7k | 0.25 | |||
| PEO4.2k-b-PLA1.9k | 0.68 | Micelles3 | Film rehydration (acetonitrile) | |
| PEO5k-b-PLA22k | 0.18 | Micelles, worm-like micelles40 | Emulsion/solvent elimination (THF) | |
| PEO5.8k-b-PLA17k,24k,48k | 0.25 to 0.10 | Vesicles 41 | Emulsion/solvent elimination (chloroform) | |
| Poly(ε-caprolactone) (PCL) | Dex6k-b-PCL2k,4k,6k | 0.75 to 0.50 | Micelles42 | Nanoprecipitation (DMSO) |
| Dex6k-b-PCL3.1k | 0.66 | Micelles43 | Nanoprecipitation (DMF/LiCl) | |
| PEO2k-b-PCL1k | 0.66 | Micelles, worm-like micelles44 | Film rehydration (DMF) | |
| PEO2k-b-PCL2k | 0.50 | |||
| PEO2k-b-PCL5.5k,7.5k,11.7k | 0.27 to 0.15 | Spherical nanoparticles45 | ||
| PEO2k-b-PCL17.4k | 0.10 | Vesicles 45 | ||
| Polystyrene (PS) | Dex6.6k-b-PS0.5k,1.6k,15k | 0.92 to 0.30 | Micelles46 | Direct dissolution or nanoprecipitation depending on the hydrophobic block size (DMSO/THF) |
| Dex6.6k-b-PS28k | 0.20 | Vesicles 47 | ||
| Dex6.6k-b-PS80k | 0.07 | |||
| PEO2k,3.5K-b-PS25k | 0.12 to 0.07 | Rod-like micelles | Nanoprecipitation (DMF) | |
| PEO0.6K-b-PS25k | 0.02 | Vesicles 48 | ||
According to the literature,1,34 the morphology of the nanostructures generated by the self-assembly of ACPs in water depends on two main factors: (i) the ACP compositions including the nature and the size of each block and (ii) the experimental conditions including the final ACP concentration in the medium, the self-assembly process and the common solvent used to solubilize the ACP before adding water. Concerning the first parameter, for an identical hydrophobic block in the ACP, decreasing the weight fraction of the hydrophilic block (fhydrophilic) should lead to a transition of the morphology from spherical micelles to vesicles via rod or worm-like micelles.6 While Table 1 shows that this empirical rule was proved with many PEO-based diblock ACPs, unfortunately it was rarely applicable for dextran-based AGPs, since simple spherical micelles or core/shell solid nanoparticles were mainly observed. It is important to note that the same primary morphologies were also observed in the case of AGP grafts despite the variation of the number and the size of hydrophobic grafts as shown in Table 1.
Such a low-order morphology of self-assembly observed for the AGP may be attributed to the classical self-assembly process used, from which we expect to have a strong influence on the morphologies of amphiphile assemblies in water. Two main processes are currently used as illustrated in Fig. 4: (i) the solvent displacement method (Fig. 4a), which consists of dissolving the amphiphiles in a common organic solvent, followed by either the addition of this mixture to water by nanoprecipitation or by the addition of water to make an emulsion. Finally, the nanostructures are formed after removing the organic solvent by dialysis or by vacuum. (ii) The film rehydration method (Fig. 4b), which also consists of dissolving the amphiphiles in a common organic solvent, followed by a slow removal of the solvent under reduced pressure to form a thin film. Then, the film is rehydrated with an aqueous solution to induce the assembly. However, for both methods, the solubilization of the AGP in a common organic solvent of the two parts (hydrophilic polysaccharide and the hydrophobic synthetic polymer) is a requirement that cannot be achieved for most of the AGPs as the neutral polysaccharides are soluble only in a few organic solvents including DMSO or DMF/LiCl. The non-compliance with the principal conditions of such a conventional method favors the formation of kinetically frozen aggregates and therefore limits assembly into primitive morphologies. Consequently, only simple descriptions of nanostructure morphologies have been generally reported in the literature for AGPs, without establishing clear relationships between their structural parameters (graft number and length for grafted glycopolymers, fhydrophilic) and the nanostructure morphologies formed. Thus, it is obvious that there is considerable interest in developing a versatile strategy to easily design well-defined AGPs, as well as to carry out their self-assembly using a straightforward process.
 | ||
| Fig. 4 Conventional self-assembly process used to prepare polymeric nano-objects. | ||
4. Polymerization induced self-assembly process
Recently, an emerging one-pot technique named Polymerization-Induced Self-Assembly (PISA) was developed to produce polymeric nanostructures in aqueous media, without the use of an organic solvent.49–53 It should be noted that PISA was also performed in non-aqueous media.54,55 Nevertheless, given the water solubility of polysaccharides, only aqueous PISA will be considered in this mini-review. Compared to the traditional self-assembly techniques, which are usually performed under dilute conditions (<0.5 wt%), PISA allows the production of nano-objects at high concentrations (up to 30 wt%). This is a significant benefit for a potential scale up. In PISA, the aqueous radical polymerization of a suitable monomer, which forms a water-insoluble polymer, is carried out using a water-soluble macroinitiator or macromolecular Chain Transfer Agent (macro-CTA). ACPs are progressively obtained when the hydrophobic block or grafts reach a critical size. At this time, an in situ phase separation occurs to finally generate stable colloidal nanostructures (Fig. 5). Depending on the nature of the monomer, PISA could be performed either under emulsion conditions50 when a hydrophobic monomer is used, or under dispersion conditions51 with a hydrophilic one.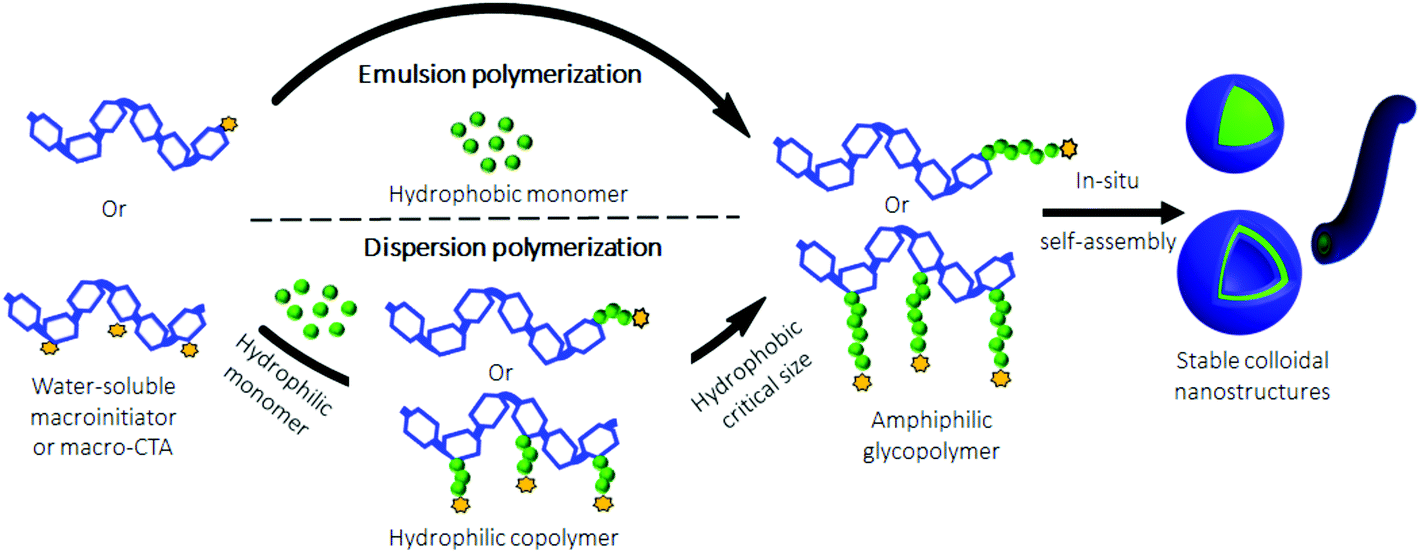 | ||
| Fig. 5 Schematic representation of PISA pathways under emulsion and dispersion conditions. | ||
The morphology of the assemblies is strongly influenced by two parameters: (i) the degree of polymerization of the hydrophobic block/grafts, and (ii) the solids concentration (the final copolymer concentration in water). Usually, polymersomes and worm-like morphologies are achieved either with a high hydrophobic block/graft length and/or at a high solid concentration. It should be noted that the non-spherical nanostructure morphologies have been usually achieved using PISA under aqueous dispersion conditions, whereas spherical micelles have been mostly obtained using aqueous emulsion PISA even with a high hydrophobic block size.56,57 Such a low-order morphology in emulsion PISA may be attributed either to the insufficient steric stabilization of some hydrophilic macroinitiators or macro-CTA used, or to the low reactivity of the hydrophobic monomers. To overcome the lack of stability, conventional stabilizers such as sodium dodecyl sulphate (SDS) are necessary to maintain the emulsion stability until the formation of ACPs.58
During the last few years, great efforts have been devoted to developing the PISA approach using various controlled radical polymerization techniques including ATRP,59 NMP,60 SET-LRP57 and Photoinduced Electron Transfer-Reversible Addition–Fragmentation Chain Transfer polymerization (PET-RAFT).61 Owing to its tolerance to water, and its ability to produce polymer chains with controlled parameters in a clean environment, RAFT62,63 is currently the most promising technique for PISA.25,51,64–67 Thermal initiation is currently mostly used for aqueous RAFT polymerization (Fig. 6).55 However, this process is relatively slow, requiring sometimes several hours of polymerization to reach high monomer conversions.
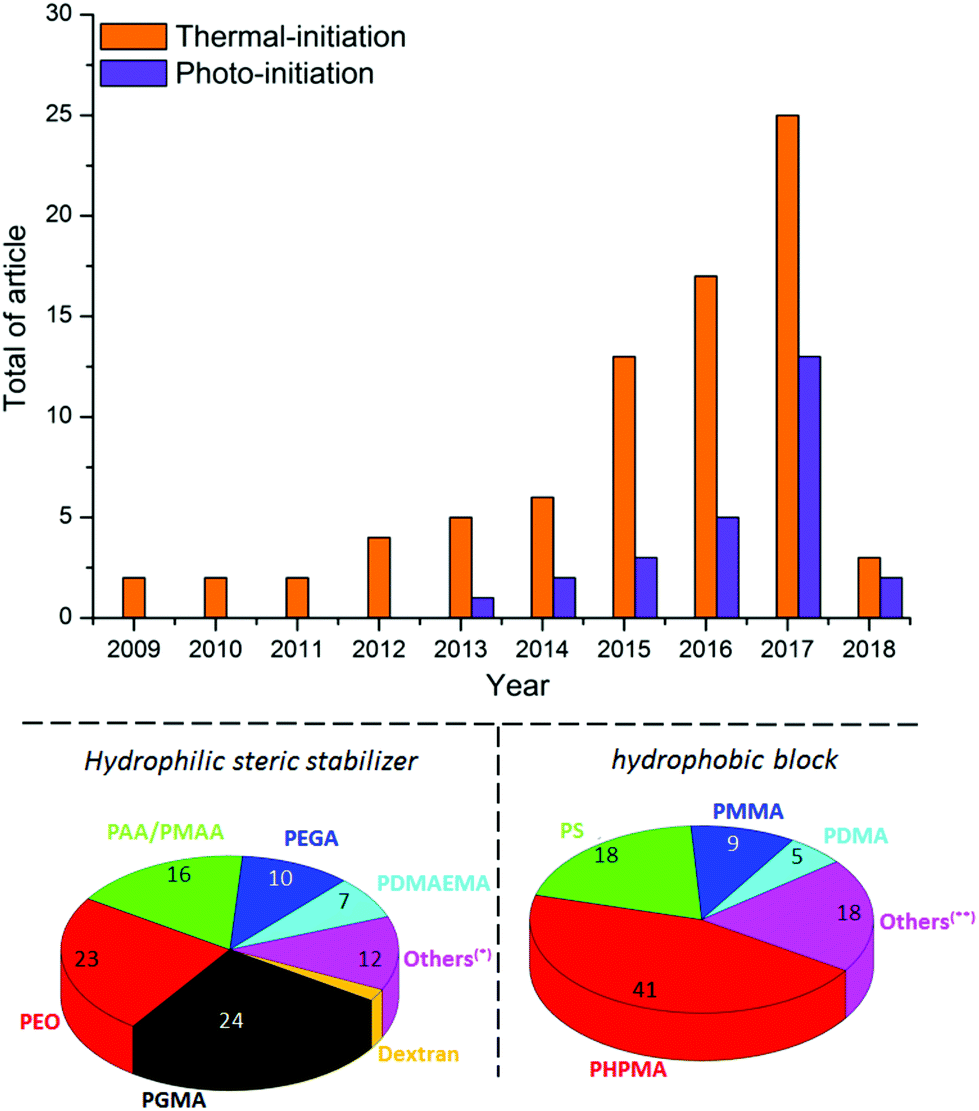 | ||
| Fig. 6 Statistical diagrams comparing the published items per year dealing with aqueous thermal and photo-initiated PISA, using hydrophilic steric stabilizers including PGMA: poly(glycerol monomethacrylate), PEO: poly(ethylene oxide), PAA: poly(acrylic acid), PMAA: poly(methacrylic acid), PEGA: poly(ethylene glycol acrylate), PDMAEMA: poly(2-(dimethylamino)ethyl methacrylate), dextran and others (*): POEGMA: poly(oligo(ethylene glycol)methyl ether methacrylate), PGalSMA: poly(galactose methacrylate), PTFEMA: poly(2,2,2-trifluoroethyl methacrylate), PHEMA: poly(hydroxyethyl methacrylate), PHEAA: poly(hydroxy-ethyl acrylamide) and PSBMA: poly((2-(methacryloyloxy)ethyl) dimethyl(3-sulfopropyl)ammonium hydroxide). (ii) Hydrophobic polymers including PHPMA: poly(2-hydroxypropyl methacrylate), PS: polystyrene, PMMA: poly(methyl methacrylate), PDMA: poly(N,N-dimethylacrylamide) and others (**): PVAc: poly(vinyl acetate), P4VP: poly(4-vinylpyridine), PDEAAm: poly(n,n-diethylacrylamide), PVCL: poly(n-vinylcaprolactam), PBA: poly(butyl acrylate), PVDC: poly(vinylidene chloride), PNIPAAm: poly(N-isopropylacrylamide), PVBA: poly(vinyl-benzaldehyde) and PBzMA: poly(benzyl methacrylate). Statistics were made in May 2018 using the Web of Science database. | ||
In addition, some recent studies55,68,69 have reported that an evolution of the nanostructure morphology was observed at high temperature (60 °C–70 °C, which is the range of temperature required for azo-initiator decomposition) after cooling to room temperature. This constitutes an additional drawback of thermal PISA, which may preclude experimental reproducibility. Photo-PISA25,49,67,70 using RAFT polymerization initiated by UV-irradiation has recently emerged as a versatile alternative (Fig. 6) due to its easiness, rapidity (99% conversion within less than 1 hour) and ability to produce a broad range of nanostructure morphologies at room temperature. This approach can also afford temporal control of polymerization by switching the light on or off, which facilitates the tracking of the polymerization progress and the isolation of nanostructure morphologies upon defined conversion. In addition, owing to its mild experimental conditions, the photo-PISA should ensure a good reproducibility of the nanostructure morphologies even with thermosensitive monomers, in contrast to the thermally-initiated PISA. Although research on PISA is growing rapidly, it has been mainly conducted with synthetic ACPs (Fig. 6), whereas its incontestable potential could be generalized to AGPs to offer a new breakthrough in terms of self-assembly.29 Currently, a hydrophilic steric stabilizer has been prepared either by RAFT polymerization of vinyl monomers, mainly acrylic acid/methacrylic acid (AA/MAA)71 for PISA in emulsion and glycerol monomethacrylate (GMA)72 for PISA in dispersion, or by end-functionalization of a commercially available polymer (i.e. PEO).67 The hydrophobic polymeric block has been derived from various vinyl monomers,66 usually polystyrene (PS)27,73,74 for PISA in emulsion and 2-hydroxypropyl methacrylate (HPMA)25,59,65,69,70 for PISA under dispersion conditions.
According to the literature, few studies25,26,57,75 reported on the use of polysaccharides as steric hydrophilic stabilizers for the formulation of nano-objects via the PISA process in water. Bernard et al.26 reported the first investigation in this field. In this seminal work, the authors described the preparation of stable submicronic latex particles by using aqueous RAFT polymerization of vinyl acetate (VAc) from xanthate-terminated dextran at 65 °C in the presence of a 2,2′-azobis(2-amidinopropane) dihydrochloride (V50) initiator to produce dextran-b-PVAc amphiphilic diblock glycopolymers (Fig. 7). Light scattering experiments have been performed for various colloidal suspensions to prove the stability of the latex produced. However, the authors did not analyze them by using imaging techniques to check the morphology of the nano-objects obtained.
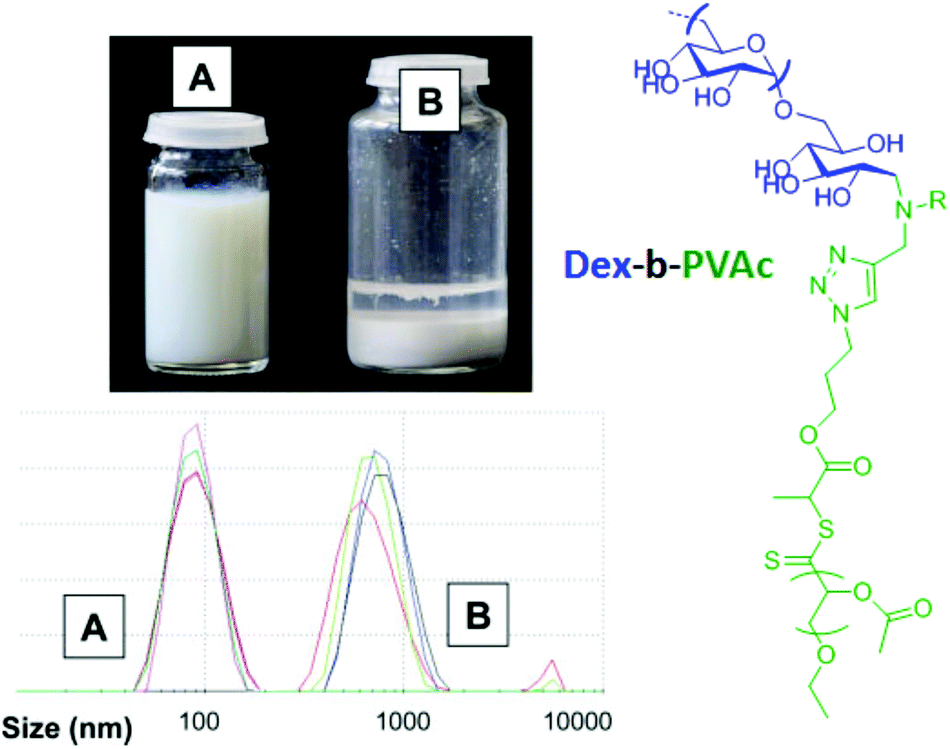 | ||
| Fig. 7 Comparison of the latex stability and particle size distribution for the colloidal suspension of emulsion polymerization of VAc in the presence of xanthate-terminated dextran (A) and native dextran (B). Reproduced from ref. 26 with permission from the Royal Society of Chemistry. | ||
Using a similar strategy, Hatton et al.75 reported the design of PMMA-based latex particles decorated and sterically stabilized by a noncharged hydrophilic xyloglucan (XG). Such a preparation has been achieved through a chain extension of a xyloglucan macroRAFT agent with PMMA in a RAFT-mediated surfactant-free emulsion polymerization. However, no higher-order morphologies other than spheres have been observed despite varying the degree of polymerization 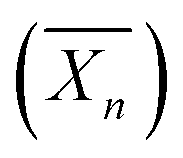 of the PMMA hydrophobic block (Fig. 8).
of the PMMA hydrophobic block (Fig. 8).
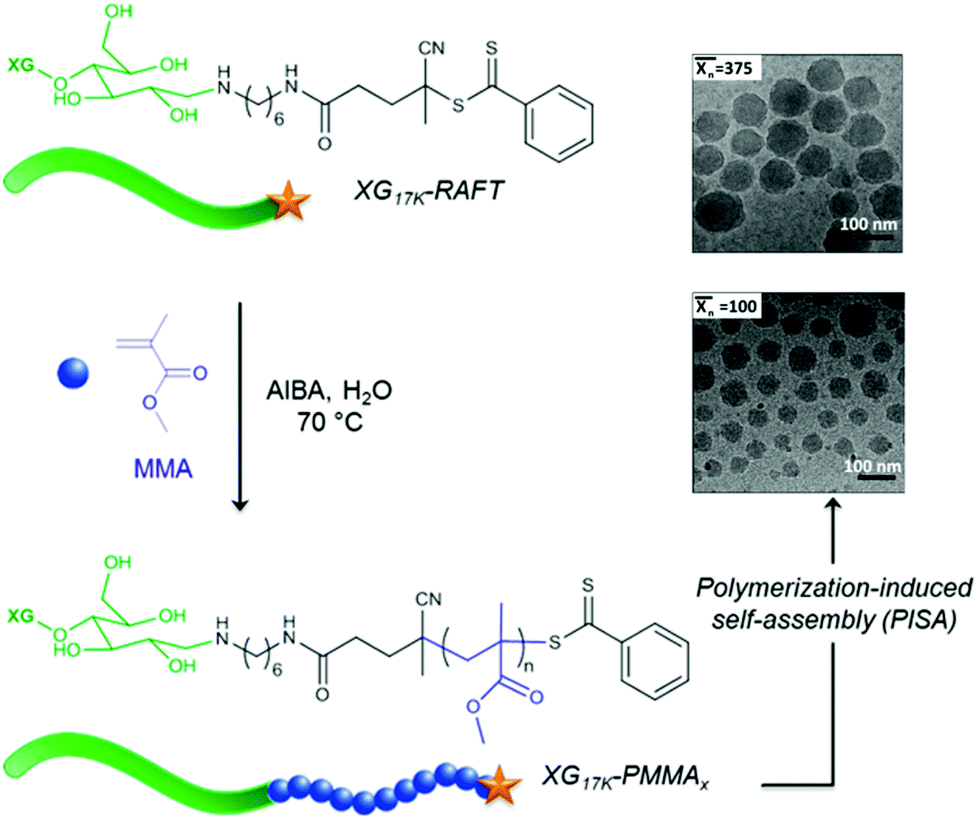 | ||
Fig. 8 Schematic representation of the formation of spherical XG17K-PMMAx latex particles via the PISA process under emulsion conditions. x is the number-average degree of polymerization 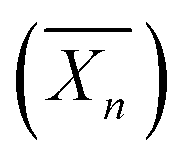 of the PMMA block. Reproduced from ref. 75 with permission from the American Chemical Society. of the PMMA block. Reproduced from ref. 75 with permission from the American Chemical Society. | ||
Kapishon et al.57 reported the preparation of spherical micelles based on alginate-g-PMMA via PISA under emulsion conditions by polymerization of a MMA monomer from an alginate macroinitiator in a water/methanol mixture using SET-LRP polymerization (Fig. 9). The authors assume that the lower order morphology observed for alginate-g-PMMA may be explained by the use of methanol, which prolongs the solvation of the grafts and most likely disturbs the self-assembly process.
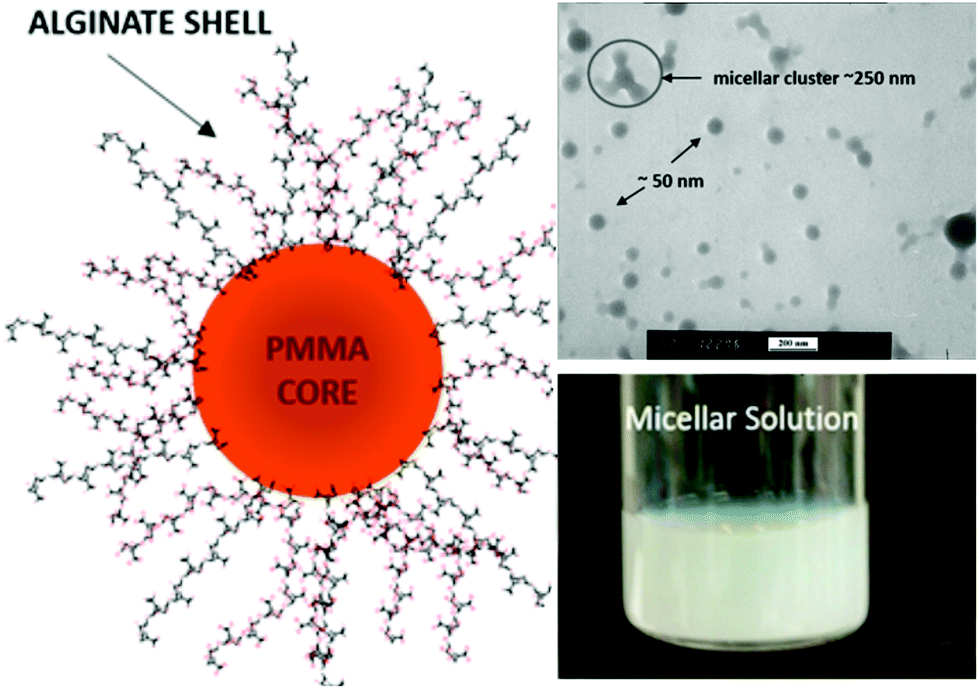 | ||
| Fig. 9 Synthesis of spherical micelles based on alginate-g-PMMA graft glycopolymers using aqueous PISA under emulsion conditions. Reproduced from ref. 57 with permission from the American Chemical Society. | ||
Recently, Ferji et al.25 reported the first example of non-spherical morphologies including worm-like micelles and vesicles using polysaccharides as steric stabilizers via an aqueous PISA under dispersion conditions (Fig. 9). In this study, the HPMA monomer was polymerized using UV irradiation (365 nm) from a photosensitive macro-CTA derived from dextran to produce grafted glycopolymers called Dex-g5-PHPMAy, where 5 is the number-average of PHPMA grafts per dextran chain and y is the number-average degree of polymerization 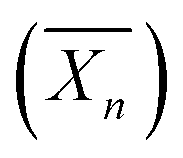 of each graft. Fast kinetics of RAFT polymerization in water was observed (10 min to 60 min) depending on the solids content (20% to 5% respectively). A broad set of advanced glyco-nanostructure morphologies was observed using light scattering analysis and Transmission Electron Microscopy (TEM). An evolution of morphology from spherical micelles to vesicles was observed depending on both the glycopolymer concentration and the length of the PHPMA grafts (Fig. 10).
of each graft. Fast kinetics of RAFT polymerization in water was observed (10 min to 60 min) depending on the solids content (20% to 5% respectively). A broad set of advanced glyco-nanostructure morphologies was observed using light scattering analysis and Transmission Electron Microscopy (TEM). An evolution of morphology from spherical micelles to vesicles was observed depending on both the glycopolymer concentration and the length of the PHPMA grafts (Fig. 10).
 | ||
Fig. 10 Synthesis of nanostructures based on graft glycopolymers Dex-gN-PHPMAY using aqueous Photo-PISA under dispersion conditions. N is the number average of PHPMA grafts per dextran chain. y is the number-average degree of polymerization 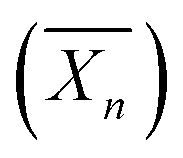 of each PHPMA graft. Reproduced from ref. 25 with permission from the Royal Society of Chemistry. of each PHPMA graft. Reproduced from ref. 25 with permission from the Royal Society of Chemistry. | ||
5. Conclusions
Amphiphilic glycopolymers certainly have many advantages and several interesting biocompatible and biodegradable properties to be promising biomaterials for biomedical applications and the food industry for instance. However, many challenges in terms of their self-assembly into high order morphologies as a vesicular one (polymersome) remain difficult to overcome using the classical self-assembly process. As described above, most of the studies dealing with amphiphilic glycopolymers reported the formulation of primitive morphologies, including spherical-micelles or core/shell nanoparticles, while those reporting the formulation of high-ordered morphologies such as vesicles are very limited. In this respect, we are convinced that the PISA process which is an exciting approach that has demonstrated a strong ability to produce synthetic nanostructure-based amphiphilic copolymers with amazing morphologies is the best way to overcome the limitation of amphiphilic glycopolymers in terms of self-assembly. The proof of concept was demonstrated using some polysaccharides (dextran and alginate), but more effort needs to be made with other polysaccharides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Agence Nationale de la Recherche of France (ANR JCJC, GlyNanEP N° 18-CE06-0002).Notes and references
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- S. Vrignaud, J. P. Benoit and P. Saulnier, Biomaterials, 2011, 32, 8593–8604 CrossRef CAS.
- H. C. Shin, A. W. G. Alani, D. A. Rao, N. C. Rockich and G. S. Kwon, J. Controlled Release, 2009, 140, 294–300 CrossRef CAS.
- T. P. T. Dao, F. Fernandes, E. Ibarboure, K. Ferji, M. Prieto, O. Sandre and J. F. Le Meins, Soft Matter, 2017, 13, 627–637 RSC.
- T. P. T. Dao, A. Brulet, F. Fernandes, M. Er-Rafik, K. Ferji, R. Schweins, J. P. Chapel, F. M. Schmutz, M. Prieto, O. Sandre and J. F. Le Meins, Langmuir, 2017, 33, 1705–1715 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- K. Ferji, I. Hamouda, C. Chassenieux, B. Nadal, B. Dubertret, C. Gaillard and E. Nicol, J. Colloid Interface Sci., 2016, 476, 222–229 CrossRef CAS.
- E. M. Pelegri-O'Day, E.-W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. Vanes and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS.
- M. A. Hussain, K. Abbas, I. Jantan and S. N. A. Bukhari, Int. Mater. Rev., 2017, 62, 78–98 CrossRef CAS.
- K. Ferji, C. Nouvel, J. Babin, M.-H. Li, C. Gaillard, E. Nicol, C. Chassenieux and J.-L. Six, ACS Macro Lett., 2015, 4, 1119–1122 CrossRef CAS.
- C. Schatz, S. Louguet, J.-F. Le Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. Okada, Prog. Polym. Sci., 2001, 26, 67–104 CrossRef CAS.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS.
- S. R. S. Ting, E. H. Min, P. B. Zetterlund and M. H. Stenzel, Macromolecules, 2010, 43, 5211–5221 CrossRef CAS.
- L. Dupayage, C. Nouvel and J. L. Six, J. Polym. Sci., Polym. Chem., 2011, 49, 35–46 CrossRef CAS.
- K. Ferji, C. Nouvel, J. Babin, P.-A. Albouy, M.-H. Li and J.-L. Six, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3829–3839 CrossRef CAS.
- M. Laville, J. Babin, I. Londono, M. Legros, C. Nouvel, A. Durand, R. Vanderesse, M. Leonard and J. L. Six, Carbohydr. Polym., 2013, 93, 537–546 CrossRef CAS.
- S. M. A. Soliman, L. Colombeau, C. Nouvel, J. Babin and J.-L. Six, Carbohydr. Polym., 2016, 136, 598–608 CrossRef CAS.
- F. Reyes-Ortega, F. J. Parra-Ruiz, S. E. Averick, G. Rodriguez, M. R. Aguilar, K. Matyjaszewski and J. S. Roman, Polym. Chem., 2013, 4, 2800–2814 RSC.
- K. Ferji, P. Venturini, F. Cleymand, C. Chassenieux and J.-L. Six, Polym. Chem., 2018, 9, 2868–2872 RSC.
- J. Bernard, M. Save, B. Arathoon and B. Charleux, J. Polym. Sci., Polym. Chem., 2008, 46, 2845–2857 CrossRef CAS.
- S. B. Chen, M. H. Alves, M. Save and L. Billon, Polym. Chem., 2014, 5, 5310–5319 RSC.
- F. Seidi, H. Salimi, A. A. Shamsabadi and M. Shabanian, Prog. Polym. Sci., 2018, 76, 1–39 CrossRef CAS.
- L. I. Atanase, J. Desbrieres and G. Riess, Prog. Polym. Sci., 2017, 73, 32–60 CrossRef CAS.
- O. Garcia-Valdez, P. Champagne and M. F. Cunningham, Prog. Polym. Sci., 2018, 76, 151–173 CrossRef CAS.
- M. El Founi, S. M. A. Soliman, R. Vanderesse, S. Acherar, E. Guedon, I. Chevalot, J. Babin and J. L. Six, J. Colloid Interface Sci., 2018, 514, 289–298 CrossRef CAS.
- C. Gavory, A. Durand, J. L. Six, C. Nouvel, E. Marie and M. Leonard, Carbohydr. Polym., 2011, 84, 133–140 CrossRef CAS.
- L. M. Ramirez, J. Babin, A. Durand, J. L. Six and C. Nouvel, Colloids Surf., A, 2015, 486, 60–68 CrossRef CAS.
- Y. Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. S. Verma and F. X. Gu, Carbohydr. Polym., 2012, 87, 2740–2744 CrossRef CAS.
- M. S. Verma, S. Liu, Y. Y. Chen, A. Meerasa and F. X. Gu, Nano Res., 2012, 5, 49–61 CrossRef CAS.
- Z. Zhao, Z. Zhang, L. Chen, Y. Cao, C. He and X. Chen, Langmuir, 2013, 29, 13072–13080 CrossRef CAS.
- C. Nouvel, J. Raynaud, E. Marie, E. Dellacherie, J. L. Six and A. Durand, J. Colloid Interface Sci., 2009, 330, 337–343 CrossRef CAS.
- T. Ouchi, T. Saito, T. Kontani and Y. Ohya, Macromol. Biosci., 2004, 4, 458–463 CrossRef CAS.
- N. V. Salim, N. Hameed, T. L. Hanley, L. J. Waddington, P. G. Hartley and Q. P. Guo, Langmuir, 2013, 29, 9240–9248 CrossRef CAS.
- F. H. Meng, C. Hiemstra, G. H. M. Engbers and J. Feijen, Macromolecules, 2003, 36, 3004–3006 CrossRef CAS.
- Z. Zhang, X. Chen, X. Gao, X. Yao, L. Chen, C. He and X. Chen, RSC Adv., 2015, 5, 18593–18600 RSC.
- H. L. Sun, B. N. Guo, X. Q. Li, R. Cheng, F. H. Meng, H. Y. Liu and Z. Y. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS.
- A. S. Mikhail and C. Allen, Biomacromolecules, 2010, 11, 1273–1280 CrossRef CAS PubMed.
- E. V. Konishcheva, U. E. Zhumaev and W. P. Meier, Macromolecules, 2017, 50, 1512–1520 CrossRef CAS.
- C. Houga, J. F. Le Meins, R. Borsali, D. Taton and Y. Gnanou, Chem. Commun., 2007, 3063–3065 RSC.
- C. Houga, J. Giermanska, S. Lecommandoux, R. Borsali, D. Taton, Y. Gnanou and J. F. Le Meins, Biomacromolecules, 2009, 10, 32–40 CrossRef CAS.
- K. Yu and A. Eisenberg, Macromolecules, 1998, 31, 3509–3518 CrossRef CAS.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- Y. Pei, A. B. Lowe and P. J. Roth, Macromol. Rapid Commun., 2017, 38, 1600528 CrossRef PubMed.
- F. H. Sobotta, F. Hausig, D. O. Harz, S. Hoeppener, U. S. Schubert and J. C. Brendel, Polym. Chem., 2018, 9, 1593–1602 RSC.
- V. Kapishon, R. A. Whitney, P. Champagne, M. F. Cunningham and R. J. Neufeld, Biomacromolecules, 2015, 16, 2040–2048 CrossRef CAS.
- I. Schreur-Piet and J. P. A. Heuts, Polym. Chem., 2017, 8, 6654–6664 RSC.
- K. Wang, Y. X. Wang and W. Q. Zhang, Polym. Chem., 2017, 8, 6407–6415 RSC.
- X. G. Qiao, M. Lansalot, E. Bourgeat-Lami and B. Charleux, Macromolecules, 2013, 46, 4285–4295 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2014, 47, 4930–4942 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS.
- J. B. Tan, D. D. Liu, Y. H. Bai, C. D. Huang, X. L. Li, J. He, Q. Xu, X. C. Zhang and L. Zhang, Polym. Chem., 2017, 8, 1315–1327 RSC.
- J. Yeow, R. Chapman, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 RSC.
- J. B. Tan, H. Sun, M. G. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- Y. W. Pei, N. C. Dharsana, J. A. Van Hensbergen, R. P. Burford, P. J. Roth and A. B. Lowe, Soft Matter, 2014, 10, 5787–5796 RSC.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- J. B. Tan, Y. H. Bai, X. C. Zhang and L. Zhang, Polym. Chem., 2016, 7, 2372–2380 RSC.
- I. Chaduc, M. Girod, R. Antoine, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2012, 45, 5881–5893 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- I. Chaduc, E. Reynaud, L. Dumas, L. Albertin, F. D'Agosto and M. Lansalot, Polymer, 2016, 106, 218–228 CrossRef CAS.
- J. L. de la Haye, X. W. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef.
- F. L. Hatton, M. Ruda, M. Lansalot, F. D'Agosto, E. Malmström and A. Carlmark, Biomacromolecules, 2016, 17, 1414–1424 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |