
Recent advances in stimuli-responsive polymeric micelles via click chemistry
Yu
Dai
a,
Xin
Chen
 *b and
Xiaojin
Zhang
*a
*b and
Xiaojin
Zhang
*a
aEngineering Research Center of Nano-Geomaterials of Ministry of Education, Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, China. E-mail: zhangxj@cug.edu.cn
bSchool of Chemical Engineering and Technology, Shanxi Key Laboratory of Energy Chemical Process Intensification, Xi'an Jiao Tong University, Xi'an 710049, China. E-mail: chenx2015@xjtu.edu.cn
First published on 21st September 2018
Abstract
Stimuli-responsive polymeric micelles have served as an emerging and interesting platform for poorly soluble drug delivery towards cancer therapy in response to exogenous stimuli (variations in temperature, light, electric field, magnetic field, or ultrasound intensity) and endogenous stimuli (changes in pH, enzyme concentration, or redox gradients). Click chemistry, an attractive synthetic methodology for conjugation, has emerged in nearly all fields of current chemistry including the construction of stimuli-responsive polymeric micelles. Here we summarize a number of pioneering studies in the area of stimuli-responsive polymeric micelles via click chemistry and divide them into six major sections in view of stimulus types (temperature, light, ultrasound, pH, enzymes, and redox). The design, synthesis, and biomedical applications of stimuli-responsive polymeric micelles via click chemistry are highlighted. Recent achievements and further perspectives in this area are briefly discussed as well.
 Yu Dai | Dr Yu Dai is currently an associate professor at China University of Geosciences (Wuhan). She received her BSc degree (2007) and PhD degree (2012) from Wuhan University (Shengli Chen's group). She then joined China University of Geosciences (Wuhan) in 2012. She worked as a postdoctoral fellow in Prof. Kai Sun's group at the University of Michigan, Ann Arbor from June 2013 to August 2014. Her scientific interests are focused on chemical and biosensor fields. |
 Xin Chen | Dr Xin Chen is currently a professor at Xi'an Jiao Tong University. He received his BSc degree (2007) from Anhui University, Master's degree (2010) from the University of Science and Technology of China, and PhD degree (2012) from the University of New South Wales in Australia (Justin Gooding's group). He then worked as a postdoctoral fellow in Prof. Peter X. Ma's group at the University of Michigan, Ann Arbor. He joined Xi'an Jiao Tong University in 2015. His scientific interests are focused on nanomaterials for theranostics. |
 Xiaojin Zhang | Dr Xiaojin Zhang is currently a professor at China University of Geosciences (Wuhan). He received his BSc degree (2007) and PhD degree (2012) from Wuhan University (Renxi Zhuo's group). He then worked as a postdoctoral fellow in Prof. Peter X. Ma's group at the University of Michigan, Ann Arbor. He joined China University of Geosciences (Wuhan) in 2016. His scientific interests are focused on biomedical polymers. |
1. Introduction
Polymeric micelles formed by the self-assembly of amphiphilic polymers in selective solvents have become one of the most attractive vehicles for poorly soluble drug delivery towards cancer therapy in recent years.1 Compared with traditional drug delivery systems, polymeric micelles exhibit some significant advantages such as small particle size, good stability, enhanced permeability and retention (EPR) effects, improved solubility of poorly soluble drugs, and reduced toxicity of drugs.2 More importantly, polymeric micelles or amphiphilic polymers are easily functionalized by the incorporation of biological macromolecules (e.g. protein, antibody, and gene) and small molecules (e.g. folic acid and peptide) to afford targeted drug delivery.3–6 Biodegradable polymers such as peptide7 and polycarbonate8 could be introduced into amphiphilic polymers to reduce the toxicity of polymeric micelles. What's more, stimuli-responsive polymeric micelles in response to various exogenous and endogenous stimuli have emerged as a promising platform for on-demand or triggered drug delivery to be able to control drug bio-distribution.9Exogenous stimuli mainly include temperature, light, electric field, magnetic field, and ultrasound.10 Temperature either can be an external stimulus from heating from the outside or acts as an internal stimulus from pathological lesions inducing naturally elevated temperature.11 Light can be controlled from the outside of the system with great ease and convenience without the use of any additional reagents.12 Electric and magnetic fields have attracted rapidly increasing scientific and technological interest due to their easy operation and industrial needs.13 Ultrasound, particularly high-intensity focused ultrasound (HIFU, frequency range 0.8–3.5 MHz), is an emerging and interesting external trigger for the controlled release of anticancer drugs from polymeric micelles towards cancer therapy.14 Endogenous stimuli typically include pH, enzymes and redox.15 pH gradients in tumor microenvironments (e.g. ∼6.5 for tumor tissues, ∼5.5 for endosomes, and ∼5.0 for lysosomes) can serve as an endogenous stimulus to achieve maximal drug potency.16 Enzymes such as proteases, oxidoreductases, and phospholipases are promising biological triggers because of the mild conditions and exceptional selectivity of enzyme-catalyzed reactions.17 Redox potentials that have distinct differences between tumor and normal tissues as well as between the extracellular and intracellular compartments have been utilized in the construction of redox-responsive polymers for drug delivery.18 The rapid development of stimuli-responsive polymeric micelles has benefited from the significant progress of nanotechnologies and synthetic methodologies, in particular click chemistry.19
Click chemistry, first fully described by Sharpless et al. in 2001,20 describes a series of specific and controllable reactions that are required to meet certain criteria such as simple reaction conditions, high chemical yields, available starting materials, only inoffensive byproducts, and high atom economy. Several reactions, e.g. copper-catalyzed azide–alkyne cycloaddition (CuAAC), strain-promoted azide–alkyne cycloaddition (SPAAC), thiol–ene reaction, thiol–yne reaction, Diels–Alder reaction, nucleophilic substitution, carbonyl-chemistry-like formation, and Michael addition reaction, have been identified in click chemistry.21 Click chemistry has a growing number of applications in nearly all fields of current chemistry from drug development to materials science,22e.g. polymer and materials science,23,24 the labeling of biological molecules,25 drug discovery and diverse chemical-biology applications,26 nanosensors for biochemical assays,27 functional aliphatic polycarbonates,28 and polymeric scaffolds.29
To the best of our knowledge, however, a lot of pioneering studies in the area of stimuli-responsive polymeric micelles via click chemistry have not been excellently reviewed to date. In view of the stimulus type as shown in Fig. 1, these studies can be divided into six major sections: (1) temperature-responsive polymeric micelles, (2) light-responsive polymeric micelles, (3) ultrasound-responsive polymeric micelles, (4) pH-responsive polymeric micelles, (5) enzyme-responsive polymeric micelles, and (6) redox-responsive polymeric micelles. This review highlights recent advances in stimuli-responsive polymeric micelles via click chemistry including their design, synthesis, and biomedical applications. Each section may be subdivided according to the click site in the polymer such as the main chain and side chain. Finally, the possible directions and further perspectives in this emerging field are briefly discussed as well.
 | ||
| Fig. 1 Types of stimuli-responsive polymeric micelles via click chemistry highlighted in this review. | ||
2. Exogenous stimuli
2.1. Temperature-responsive polymeric micelles
Temperature-responsive polymeric micelles exhibit tunable drug release rate and cellular uptake to accumulate at tumor tissues through exogenous administration of heat in combination with a hyperthermia system.30 Click chemistry has been utilized to synthesize oligo(ethylene glycol)-grafted polypeptides31 or aliphatic polycarbonates32 through click coupling of pendant groups for the fabrication of temperature-responsive polymeric micelles. For example, Zhuang et al. reported temperature-responsive polypeptide micelles via CuAAC for antitumor drug delivery as shown in Fig. 2.31 Temperature-responsive polypeptides were synthesized by grafting azide-terminated oligo(ethylene glycol) onto poly(γ-propargyl-L-glutamate). The polypeptides self-assembled into polymeric micelles with a temperature-responsive shell. Doxorubicin (DOX, an antitumor drug) could be loaded into the core of polymeric micelles. Temperature-responsive polypeptide micelles were stable under physiological conditions and drug release from polymeric micelles could be accelerated with a decrease of temperature. Poly(N-isopropylacrylamide) (PNIPAM), a typical temperature-responsive polymer, has been frequently used for the synthesis of amphiphilic polymers via click chemistry.33–37 These polymers are able to form temperature-responsive polymeric micelles for drug delivery.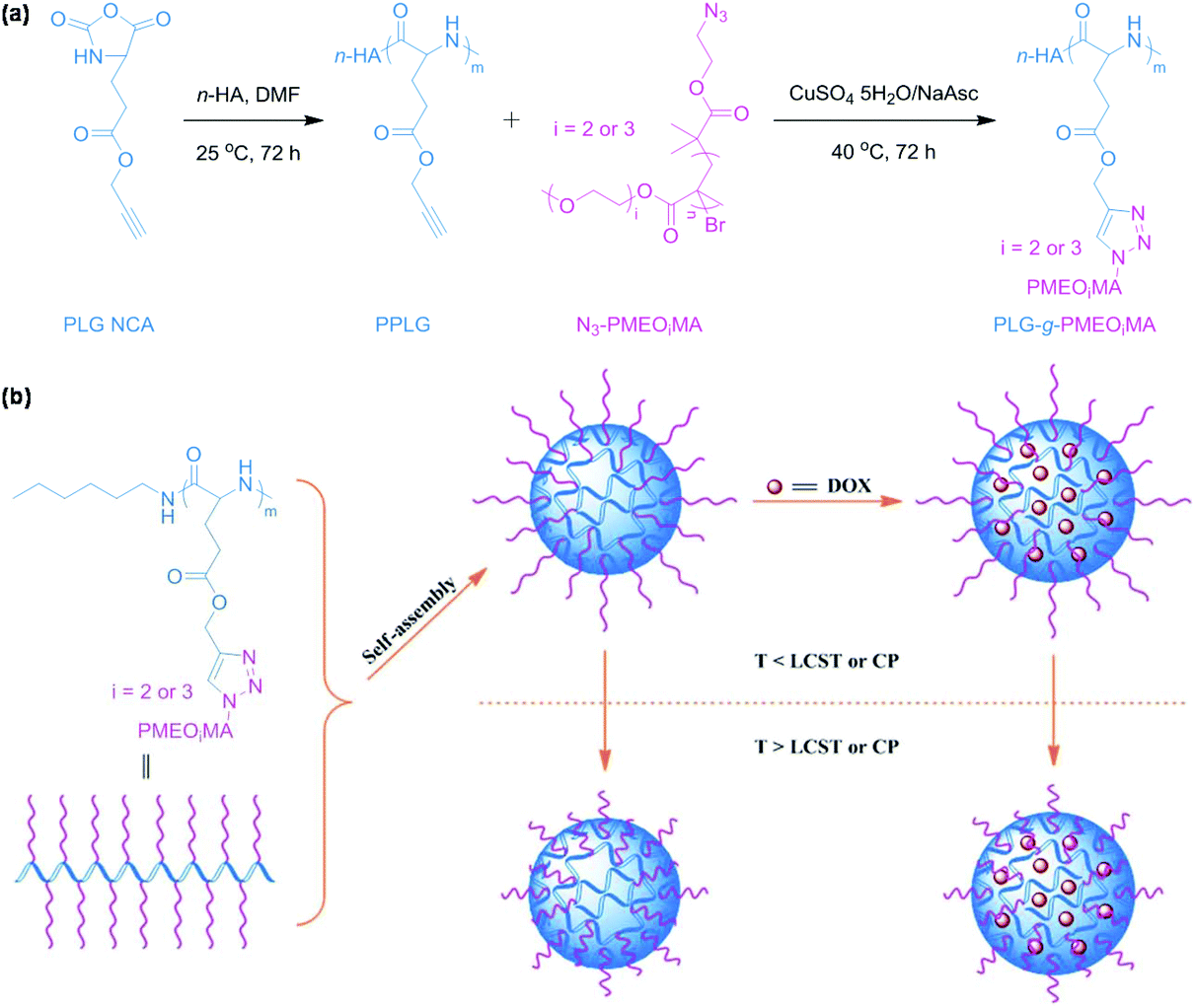 | ||
| Fig. 2 (a) Synthesis of temperature-responsive polypeptides PLG-g-PMEOiMA. (b) Schematic illustration of the self-assembly of polypeptides, DOX loading and temperature-responsiveness of polymeric micelles. Reprinted with permission.31 Copyright (2013) The Royal Society of Chemistry. | ||
2.2. Light-responsive polymeric micelles
Light-responsive polymeric micelles have attracted increasing attention in both academia and industry, particularly in biomedical fields, because the photo-reaction processes can be controlled from the outside of the system with great ease and convenience.38 Photoisomerization and photocleavage are the two most common strategies in light-responsive polymeric micelles for on-demand drug delivery.39 For instance, Lu et al. presented light-responsive poly(carbonate)s micelles through introducing a spiropyran chromophore onto the side chain of poly(carbonate)s via CuAAC as shown in Fig. 3.40 Light-responsive poly(carbonate)s PEG113-b-PMPCn-SP were synthesized via CuAAC of the azide-bearing spiropyran and amphiphilic block copolymer PEG113-b-PMPCn with pendant alkyne groups. PEG113-b-PMPCn-SP self-assembled to form polymeric micelles that disrupted within 10 min under UV light (365 nm) irradiation owing to the transformation of hydrophobic spiropyran into hydrophilic merocyanine. Visible light (620 nm) irradiation would allow polymeric micelles to aggregate again. Light-responsive poly(α-hydroxy acids) micelles were also prepared through introducing a spiropyran chromophore onto the side chain of poly(α-hydroxy acids) via CuAAC.41 Light-responsive poly(methacrylate) micelles were prepared through introducing a spiropyran chromophore onto the side chain of poly(methacrylate)s via CuAAC.42Trans–cis photoisomerization of azobenzene in response to UV and visible light was used in the design of light-responsive polymeric micelles based on azobenzene-bearing poly(carbonate)s43 and azobenzene-modified poly(α-hydroxy acids)44 synthesized via CuAAC. Irreversible photocleavage of o-nitrobenzyl esters in response to UV light was also utilized in the construction of light-responsive polymeric micelles based on amphiphilic poly(α-hydroxy acids) with pendent o-nitrobenzyl ester45 and amphiphilic dextran-g-poly(o-nitrobenzyl acrylate) glycopolymers46 prepared via CuAAC.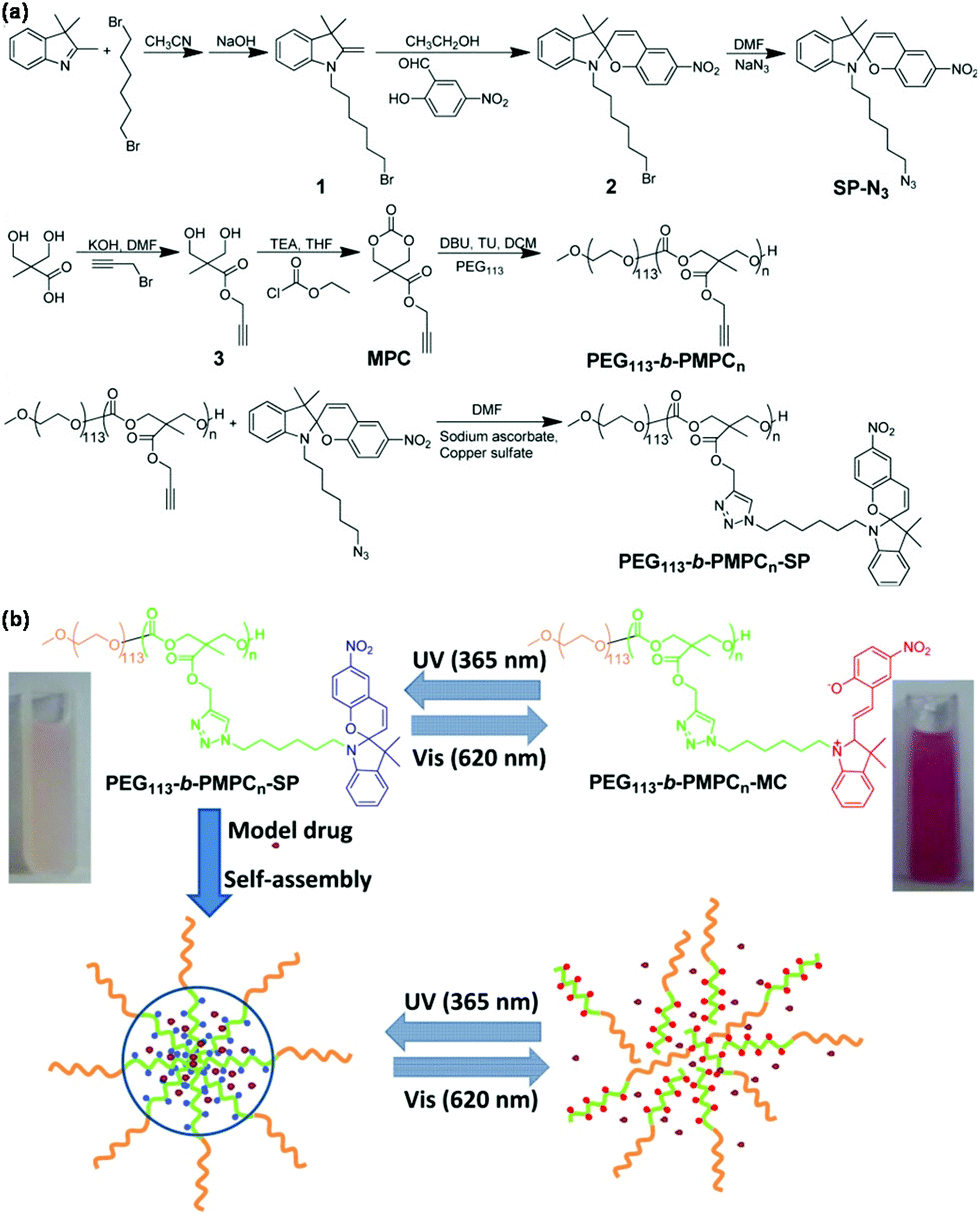 | ||
| Fig. 3 (a) Synthesis of light-responsive poly(carbonate)s PEG113-b-PMPCn-SP. (b) Schematic illustration of the self-assembly of poly(carbonate)s, coumarin-102 loading and light-responsiveness of polymeric micelles. Reprinted with permission.40 Copyright (2014) Wiley Periodicals, Inc. | ||
Hydrophobic naphthopyran was coupled with hydrophilic PEG via CuAAC to synthesize an amphiphilic linear block copolymer for the fabrication of reversibly light-responsive polymeric micelles based on the photochromism of naphthopyran with on-demand control of light irradiation.47 Hydrophobic azobenzene was conjugated to a polyhedral oligomeric silsesquioxane (POSS) unit with a PEG chain via a thiol–ene reaction to prepare an amphiphilic tadpole-shaped polymer for the construction of reversibly light-responsive polymeric micelles based on trans–cis photoisomerization of azobenzene.48 Ambade et al. synthesized a visible light-cleavable BODIPY-linked polymer to develop irreversibly light-responsive polymeric micelles as shown in Fig. 4.49 Boron dipyrromethene (BODIPY, a fluorescent dye molecule) acted as the junction in an amphiphilic linear block copolymer to conjugate PEG via CuAAC and initiate atom transfer radical polymerization (ATRP) of styrene. Polymeric micelles, by self-assembly of the polymer, disassembled under visible light irradiation after photocleavage of the B–O bond in BODIPY to release the encapsulated Nile red.
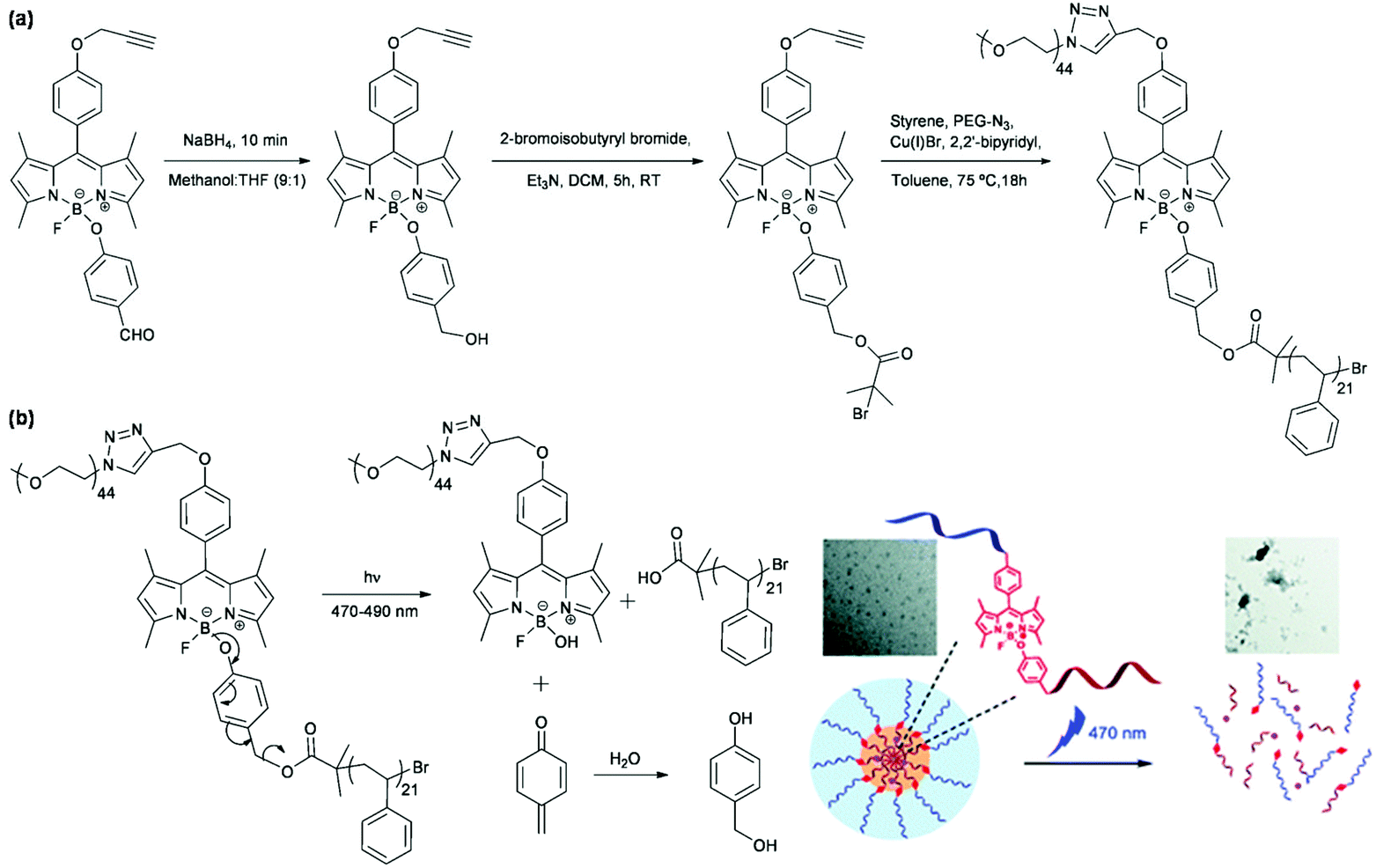 | ||
| Fig. 4 (a) Synthesis of a visible light-cleavable BODIPY-linked polymer. (b) Schematic illustration of the self-assembly of the polymer, Nile red loading and light-responsiveness of polymeric micelles. Reprinted with permission.49 Copyright (2015) The Royal Society of Chemistry. | ||
2.3. Ultrasound-responsive polymeric micelles
Ultrasound-responsive nanocarriers such as bubbles, droplets, liposomes, emulsions, and micelles have become an interesting platform for triggering drug release by adjusting ultrasound time, intensity, and location, especially using high-intensity focused ultrasound (HIFU).50 Click chemistry is a convenient method to construct ultrasound-responsive polymeric micelles with weak bonds. For example, Xia et al. developed ultrasound-responsive polymeric micelles of poly(ethylene glycol)-b-poly(propylene glycol) synthesized via CuAAC as shown in Fig. 5.51 The amphiphilic block copolymer poly(ethylene glycol)-b-poly(propylene glycol) with multiple ester bonds and 1,2,3-triazole moiety in the main chain (PEG-click-PPG) were synthesized. PEG-click-PPG could form polymeric micelles in aqueous solution by self-assembly. Polymeric micelles were disrupted by HIFU to trigger the release of Nile red in the micelles because multiple ester bonds were broken under HIFU.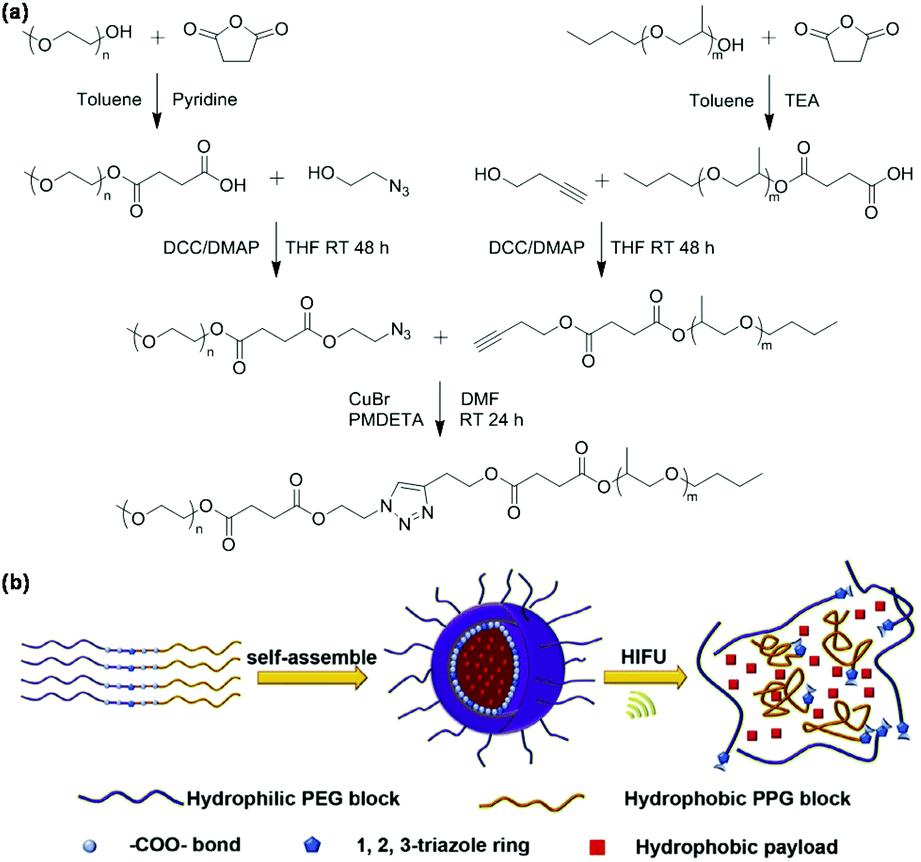 | ||
| Fig. 5 (a) Synthesis of the PEG-click-PPG copolymer. (b) Schematic illustration of the self-assembly of PEG-click-PPG, Nile red loading and ultrasound-responsiveness of polymeric micelles. Reprinted with permission.51 Copyright (2015) Elsevier B.V. | ||
3. Endogenous stimuli
3.1. pH-Responsive polymeric micelles
pH-Responsive nanocarriers have been widely developed to trigger drug release in response to highly acidic tumor microenvironments owing to the active metabolism of tumor cells.52 Oxime linkage is a robust tool for the design of pH-responsive nanocarriers.53 For instance, Shunmugam et al. developed pH-responsive polymeric micelles with doxorubicin-conjugated oxime linkage for targeted drug delivery as shown in Fig. 6.54 A biotin-terminated and doxorubicin-bearing copolymer was synthesized using a combination of ring-opening polymerization (ROP) and CuAAC. The biotin and doxorubicin-conjugated copolymer formed polymeric micelles and released the drugs under weakly acidic conditions owing to the unique oxime linkage. Schiff-base linkage can serve as a pH-responsive linker in polymer chemistry.55 Benzaldehyde-containing amphiphilic block polynorbornenes were synthesized for further conjugation with drugs containing amino groups via Schiff-base linkage with the result of pH-triggered drug release from polymeric micelles under acidic conditions (e.g. pH 4.0).56 Other pH-sensitive linkers such as benzoic–imine linkage,57 hydrazone linkage,58 acetal linkage,59 and β-thiopropionate linkage60 could be employed in the design of pH-responsive polymeric micelles. Attaching carboxylic acid groups to side chains of the polymer by thiol–ene reaction or thiol–yne reaction is a facile and convenient procedure to prepare pH-responsive polymeric micelles for oral delivery.61–63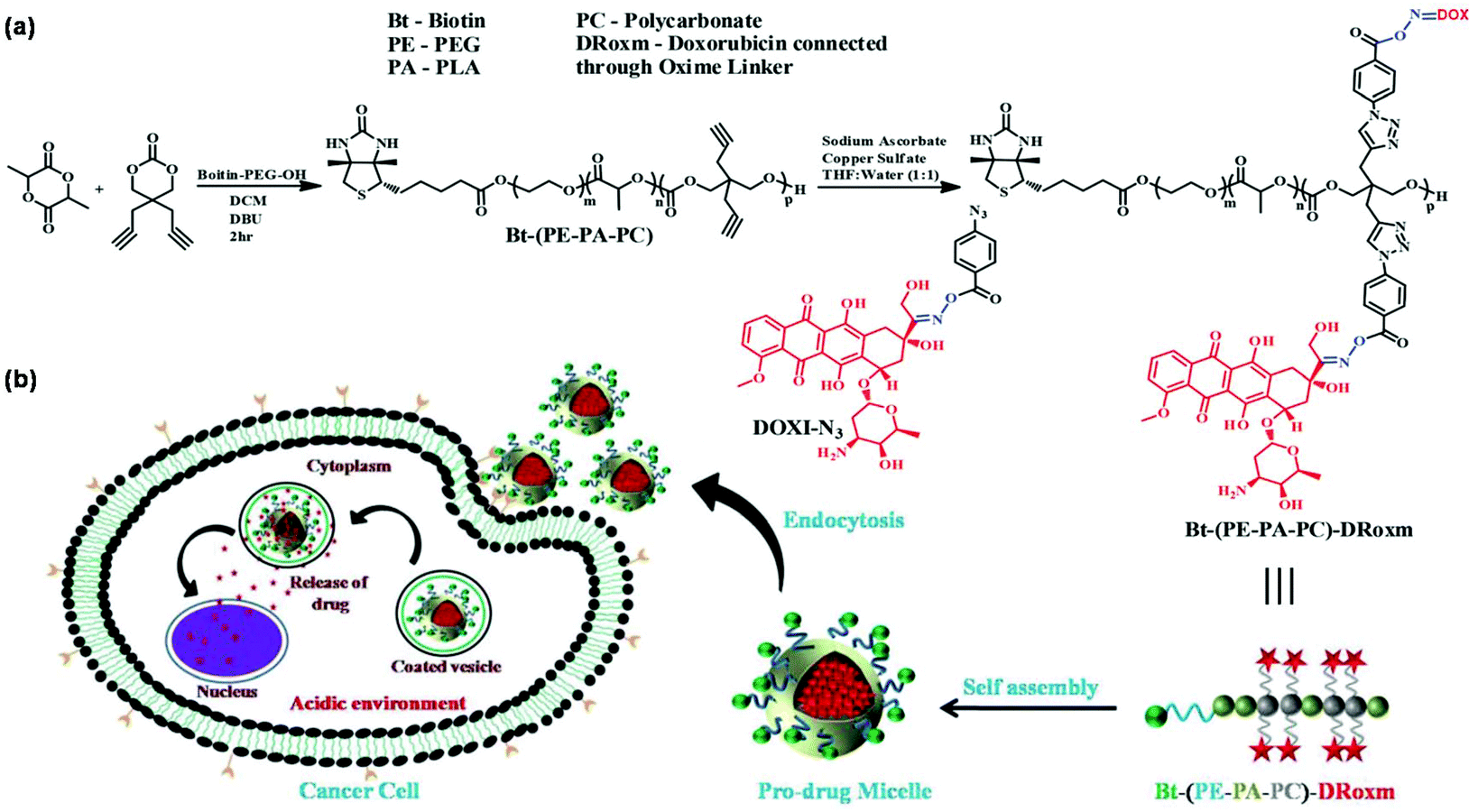 | ||
| Fig. 6 (a) Synthesis of the biotin and doxorubicin-conjugated copolymer. (b) Schematic illustration of the self-assembly of the polymer and pH-responsiveness of polymeric micelles. Reprinted with permission.54 Copyright (2016) The Royal Society of Chemistry. | ||
Zhong et al. reported acid degradable poly(acetal urethane) (PAU) for the synthesis of pH-responsive triblock copolymers via thiol–ene reaction for pH-triggered intracellular drug delivery as shown in Fig. 7.64 PEG-PAU-PEG triblock copolymers were synthesized by polycondensation of a diacetal-containing diol and lysine diisocyanate, followed by thiol–ene reaction for the conjugation of PEG. PEG-PAU-PEG triblock copolymers formed polymeric micelles and endo/lysosomal pH triggered micelle swelling and intracellular drug release. POSS-based acid-labile polyacetal dendrimers were reported to be functionalized by PEG for pH-responsive polymeric micelles.65 Amphiphilic block copolymers with conjugation of a hydrophilic dextran block and a hydrophobic acetalated dextran block via CuAAC self-assembled into polymeric micelles that were stable at physiological pH but disrupted under weakly acidic conditions.66
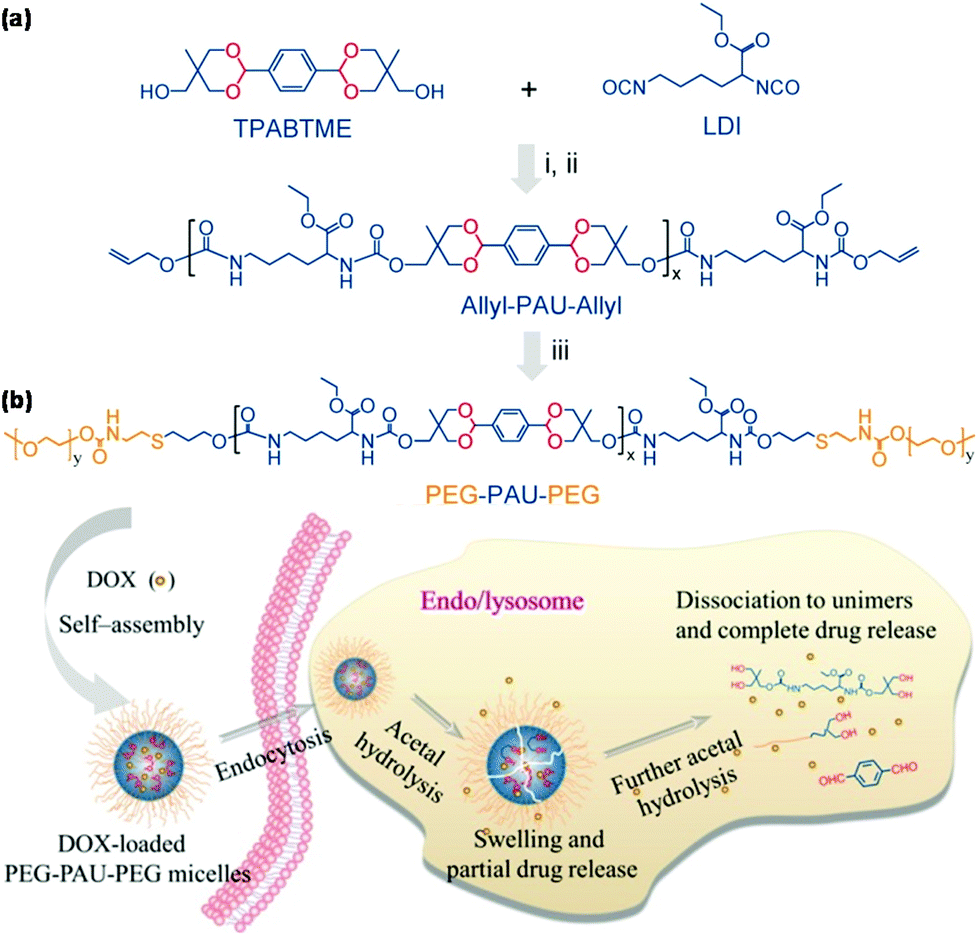 | ||
| Fig. 7 (a) Synthesis of PEG-PAU-PEG triblock copolymers. (b) Schematic illustration of the self-assembly of PEG-PAU-PEG triblock copolymers, DOX loading and pH-responsiveness of polymeric micelles. Reprinted with permission.64 Copyright (2015) American Chemical Society. | ||
3.2. Enzyme-responsive polymeric micelles
Enzymes as a trigger for stimuli-responsive degradation and disassembly of polymeric micelles have several significant advantages such as their high selectivity and natural existence in all living organisms.67 Matrix metalloproteases (MMPs, a large family of endopeptidases) that are ubiquitously overexpressed at tumor sites are capable of degrading all kinds of extracellular matrix proteins.68 MMPs commonly act as an endogenous stimulus.69 The hydrophilic PVGLIG peptide that could be cleavable in the presence of MMP-2 and MMP-9 was coupled to hydrophobic poly(trimethylene carbonate) via thiol–ene reaction for the fabrication of MMP-responsive polymeric micelles.70 Ge et al. constructed enzyme-responsive polymeric micelles with a MMP-responsive peptide as a linker in the main chain of block copolymers as shown in Fig. 8.71 The MMP-responsive peptide-linked block copolymer PEG-GPLGVRGDG-P(BLA-co-Asp) was synthesized via CuAAC followed by ROP of N-carboxyanhydride initiated by terminal amino groups and partial hydrolysis of PBLA. Polymeric micelles of PEG-GPLGVRGDG-P(BLA-co-Asp) encapsulated DOX efficiently and rapidly released the drugs inside tumor cells because of MMP-triggered dePEGylation. Similarly, the peptide-linked amphiphilic block copolymer PEG-GPLGVRGDG-PDLLA was used to prepare MMP-responsive polymeric micelles for intelligent systemic anticancer drug delivery.72 Cathepsin B (a lysosomal cysteine protease) that is overexpressed in tumor cells has also been used in the construction of enzyme-responsive polymeric micelles.73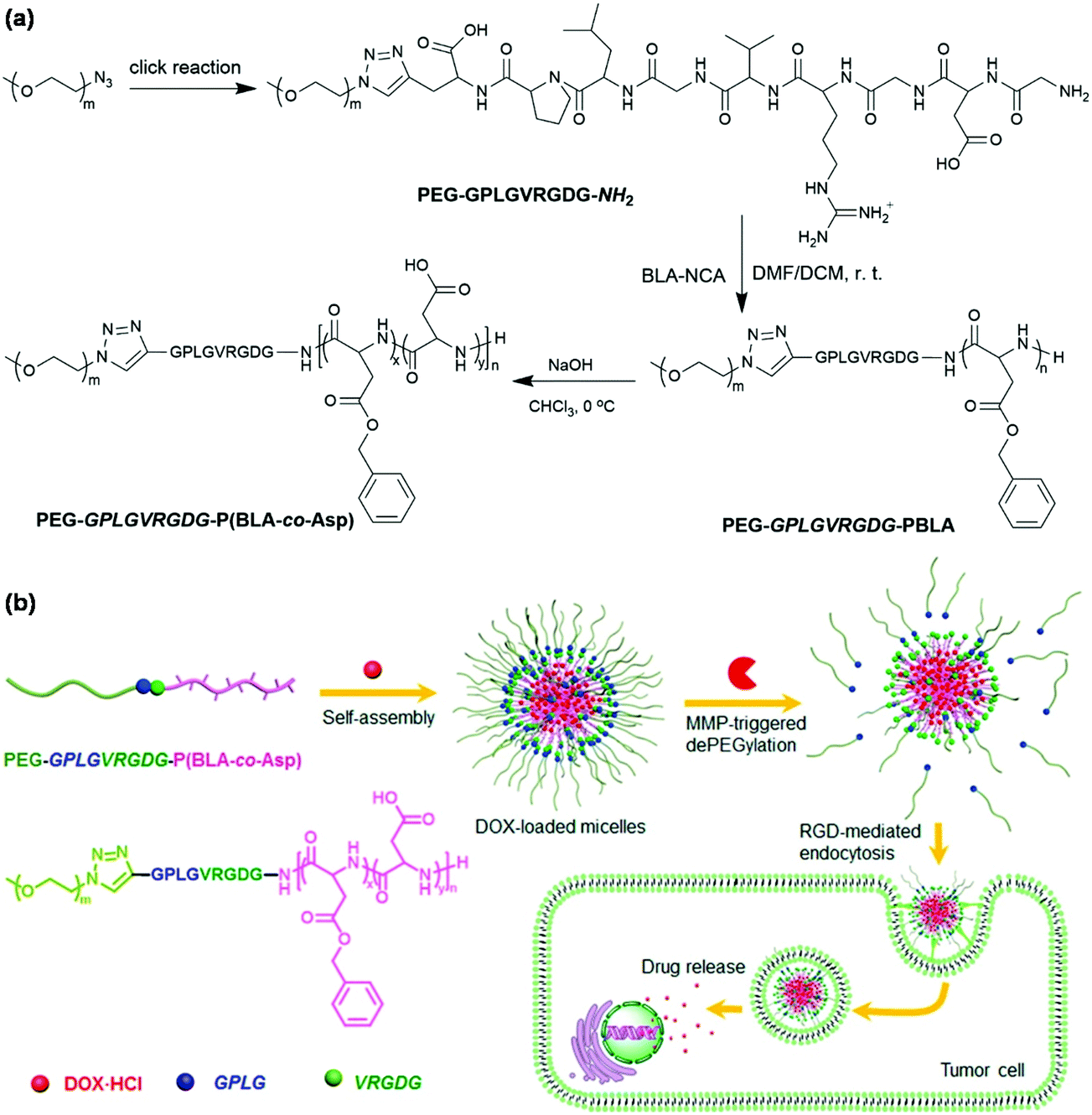 | ||
| Fig. 8 (a) Synthesis of MMP-responsive block copolymer, PEG-GPLGVRGDG-P(BLA-co-Asp). (b) Schematic illustration of self-assembly of PEG-GPLGVRGDG-P(BLA-co-Asp), DOX loading and MMP-responsiveness of polymeric micelles. Reprinted with permission.71 Copyright (2016) American Chemical Society. | ||
3.3. Redox-responsive polymeric micelles
Redox-responsive polymeric micelles have been explored for triggered intracellular drug release owing to high redox potential in tumor tissues, particularly inside tumor cells.74 Disulfide bonds can be cleaved by glutathione (GSH) that exists in the cytosol, mitochondria, and nucleus of tumor cells at a high concentration.75 For example, Ni et al. developed a reduction-cleavable polymer-camptothecin (CPT) prodrug for GSH-triggered drug delivery as shown in Fig. 9.76 The redox-responsive polymeric prodrug (PBYP-g-ss-CPT)-b-PEEP was synthesized via CuAAC of the pendant alkynes of polyphosphoester and azide group modified CPT with a disulfide bond. The prodrug could self-assemble into polymeric micelles that were disrupted under a tumor reducing environment to cause a rapid release of CPT owing to the cleavage of disulfide bonds. Other similar cases of reduction-responsive polymeric prodrugs were also reported.77,78 Reduction-responsive polymeric prodrugs could be further crosslinked by bis(azidoethyl) disulfide, a reduction-sensitive crosslinker, to enhance the stability and triggered drug release.79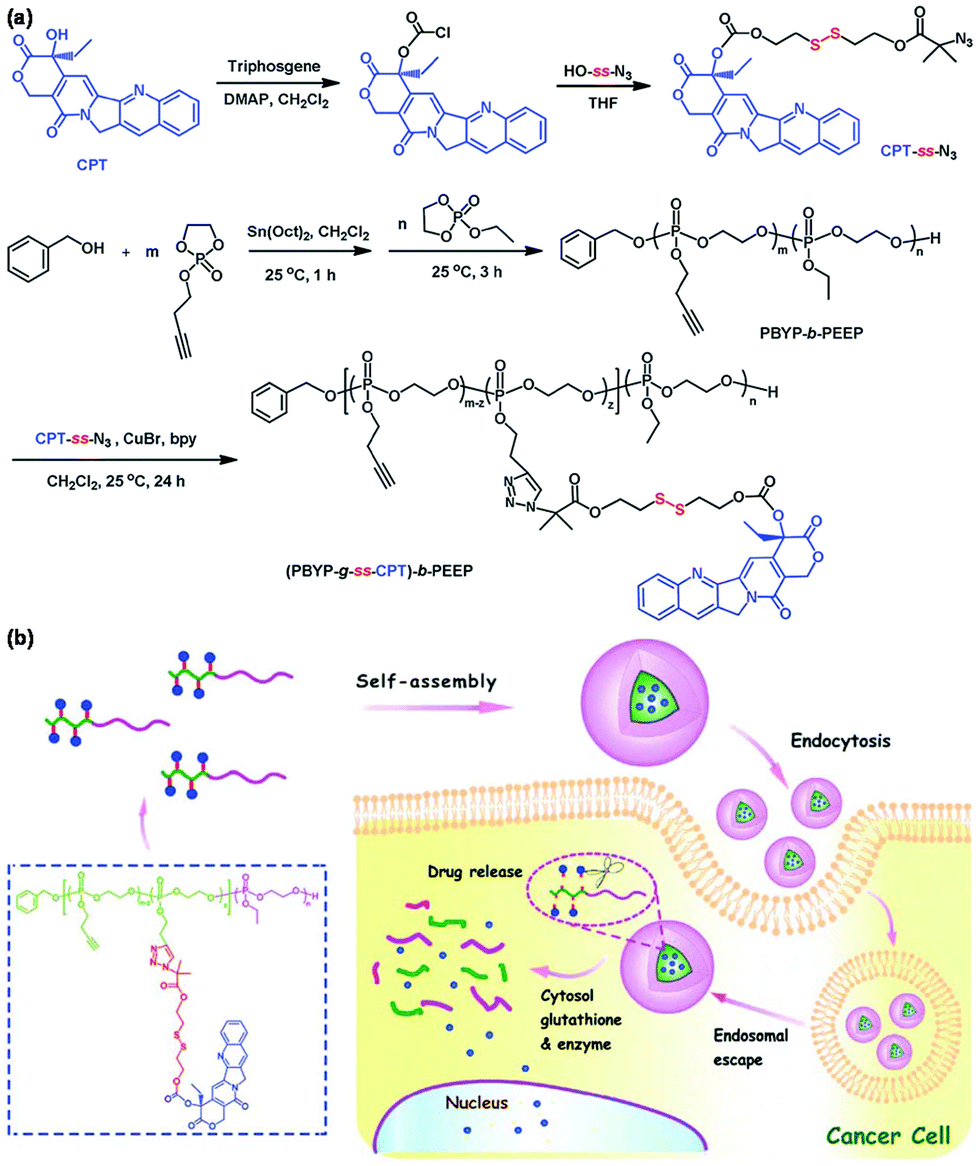 | ||
| Fig. 9 (a) Synthesis of the reduction-responsive polymeric prodrug (PBYP-g-ss-CPT)-b-PEEP. (b) Schematic illustration of the self-assembly of (PBYP-g-ss-CPT)-b-PEEP and GSH-responsiveness of polymeric micelles. Reprinted with permission.76 Copyright (2015) The Royal Society of Chemistry. | ||
The disulfide bond can also be designed in the polymer backbone via click polymerization80–83 or click coupling.84–87 Recently, Kim et al. reported an amphiphilic miktoarm block copolymer with a singlet oxygen (1O2)-labile stereospecific β-aminoacrylate junction for triggered drug release as shown in Fig. 10.88 The amphiphilic miktoarm block copolymer PEG-b-PCL2 was synthesized through a combination of amino-yne click chemistry and ROP using 1O2-labile stereospecific β-aminoacrylate as a linkage. Chlorin e6 (Ce6, a hydrophobic photosensitizer) and DOX were encapsulated into PEG-b-PCL2 micelles. Ce6 induced the 1O2-generation upon red laser irradiation to cause dissociation of polymeric micelles owing to photocleavage of β-aminoacrylate for on-demand drug release.
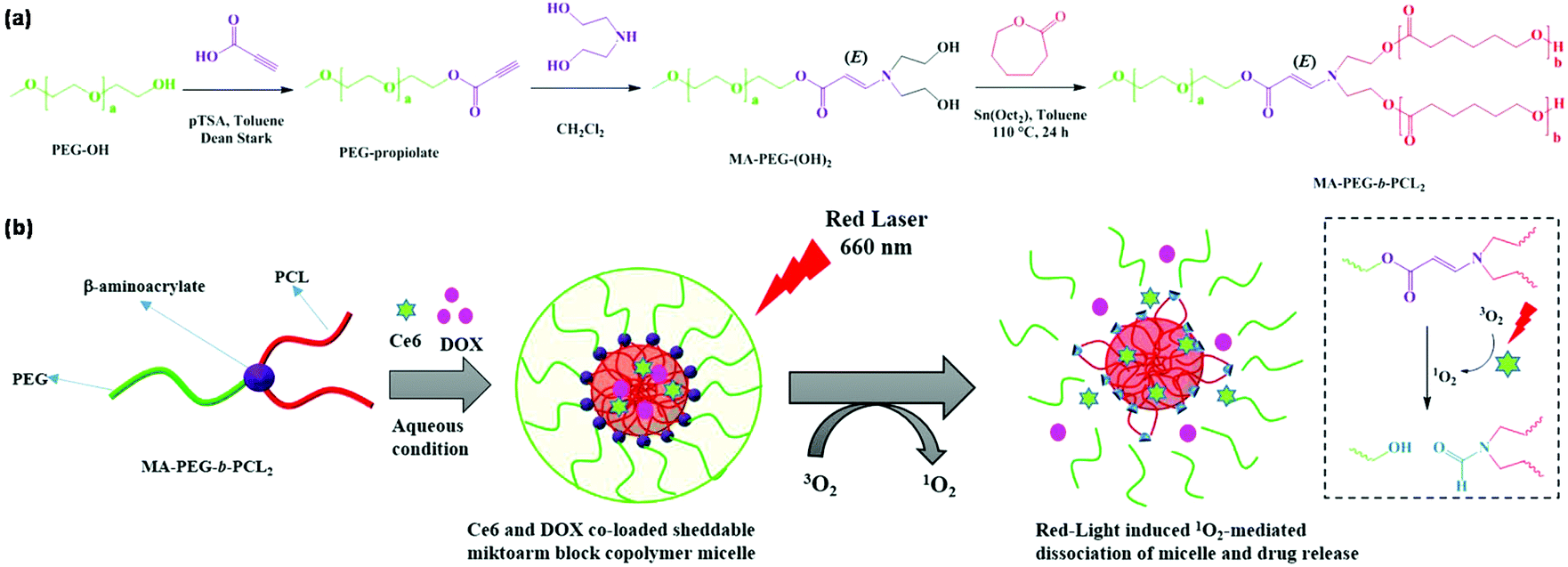 | ||
| Fig. 10 (a) Synthesis of the amphiphilic miktoarm block copolymer PEG-b-PCL2. (b) Schematic illustration of the self-assembly of PEG-b-PCL2, DOX loading and 1O2-responsiveness of polymeric micelles. Reprinted with permission.88 Copyright (2018) American Chemical Society. | ||
Diazide or dialkyne compounds with a disulfide bond have been widely used to further crosslink polymeric micelles to prevent dissociation of the micelles at high dilution and to redox-trigger their disruption for on-demand drug release in a more reductive environment.89–100 As an example, we developed redox-responsive micelles with cores crosslinked via CuAAC as shown in Fig. 11.98 The amphiphilic block copolymer mPEG-b-P(DTC-ADTC) was synthesized by ROP of 2,2-dimethyltrimethylene carbonate (DTC) and 2,2-bis(azidomethyl)trimethylene carbonate (ADTC) with monomethoxy PEG as an initiator. mPEG-b-P(DTC-ADTC) could self-assemble to afford polymeric micelles for further core-crosslinking through click coupling of pendant azide groups of the polycarbonate block and dialkyne compounds with a disulfide bond. Dithiol bis(propanoic dihydrazide),101 2,2′-dithiodiethanol diacrylate,102 and dithiobismaleimidoethane103 could act as a crosslinker to fabricate reduction-responsive core-crosslinked micelles via hydrazine formation, thiol–ene reactions, and Diels–Alder reactions, respectively.
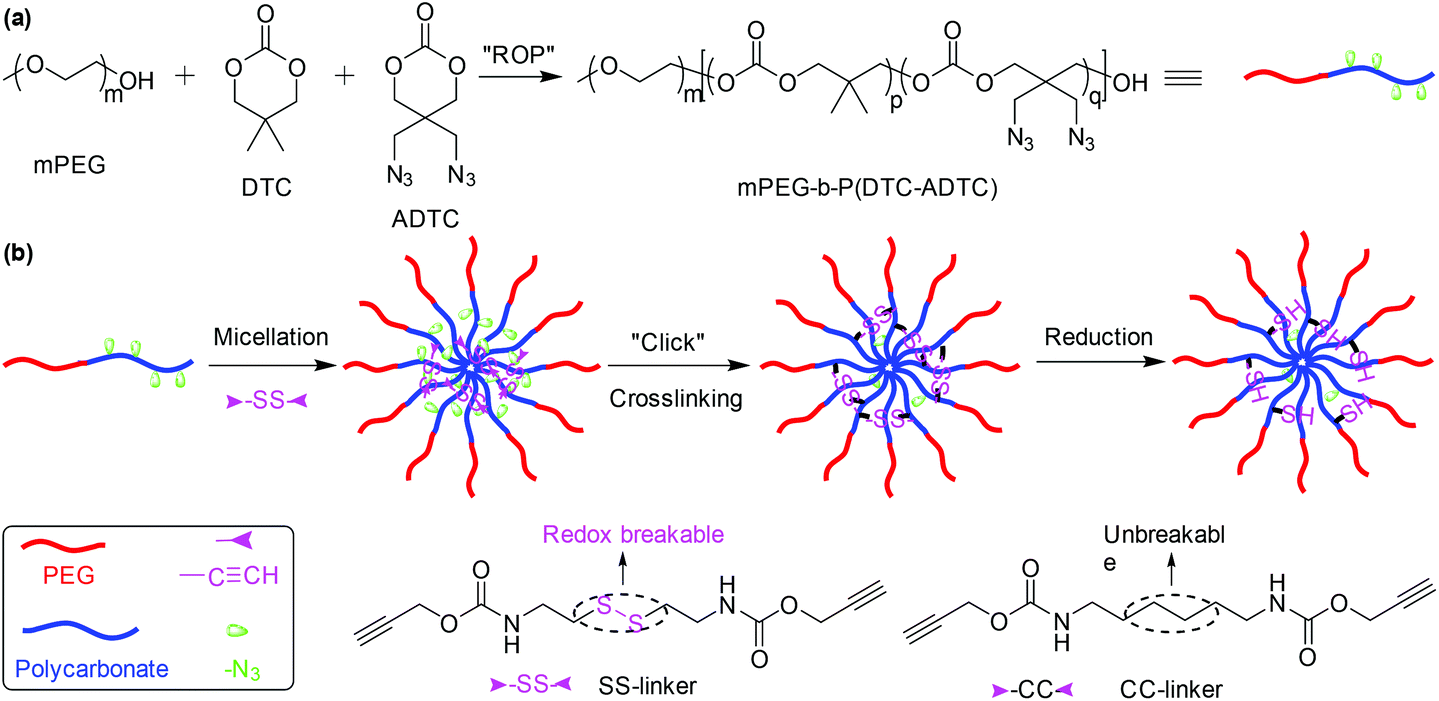 | ||
| Fig. 11 (a) Synthesis of the amphiphilic block copolymer mPEG-b-P(DTC-ADTC). (b) Schematic illustration of the self-assembly of mPEG-b-P(DTC-ADTC, methotrexate loading and reduction-responsiveness of polymeric micelles. Reprinted with permission.98 Copyright (2016) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
4. Summary and outlook
A lot of preclinical studies on stimuli-responsive polymeric micelles via click chemistry have been published, which shows that they can serve as an efficient platform for drug delivery towards cancer therapy. The translation from basic research to clinical practice of stimuli-responsive polymeric micelles via click chemistry is already being considered, since they show good biocompatibility, low toxicity, triggered drug release, and enhanced systemic circulation.104 Although remarkable results in this area have been achieved in the past decade, several challenges still remain to be addressed.105 Firstly, sensitivity and specificity in response to either exogenous or endogenous stimuli need to be further enhanced, particularly for intracellular rapid drug release to overcome multi-drug resistance. Secondly, stimuli-responsive polymeric micelles with shapes other than spherical micelles deserve more extensive investigation, which probably enhance cellular uptake efficiency and improve tumor penetration depth. Thirdly, stimuli-responsive polymeric micelles modified with target ligands might exhibit enhanced drug delivery to specific cells and tissues. Finally, the simplified design of stimuli-responsive polymeric micelles is likely to promote their clinical translation, since a number of cases have been reported in the paper whereas only small quantities of them enter the clinical stage. New developments are expected to emerge along with combined interdisciplinary efforts to afford stimuli-responsive polymeric micelles with improved therapeutic outcomes. We believe that stimuli-responsive polymeric micelles could become an efficient method for clinical cancer therapy in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51703209 and 21603196), the Natural Science Foundation of Hubei Province (No. 2017CFB217), and the Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (No. CUG170601 and CUGL170406).Notes and references
- A. Gothwal, I. Khan and U. Gupta, Pharm. Res., 2016, 33, 18–39 CrossRef CAS.
- S. Biswas, P. Kumari, P. M. Lakhani and B. Ghosh, Eur. J. Pharm. Sci., 2016, 83, 184–202 CrossRef CAS PubMed.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomedicine, 2010, 6, 714–729 CrossRef CAS PubMed.
- Y. F. Zhang, Y. X. Huang and S. Li, AAPS PharmSciTech, 2014, 15, 862–871 CrossRef CAS PubMed.
- C. Deng, Y. J. Jiang, R. Cheng, F. H. Meng and Z. Y. Zhong, Nano Today, 2012, 7, 467–480 CrossRef CAS.
- Y. Dai, H. Q. Wang and X. J. Zhang, J. Nanopart. Res., 2017, 19, 298 CrossRef.
- N. Dube, A. D. Presley, J. Y. Shu and T. Xu, Macromol. Rapid Commun., 2011, 32, 344–353 CrossRef CAS.
- Y. Dai and X. J. Zhang, Polym. Chem., 2017, 8, 7429–7437 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS.
- H. Hatakeyama, Chem. Pharm. Bull., 2017, 65, 612–617 CrossRef CAS PubMed.
- M. Karimi, P. S. Zangabad, A. Ghasemi, M. Amiri, M. Bahrami, H. Malekzad, H. G. Asl, Z. Mandieh, M. Bozorgomid, A. Ghasemi, M. Boyuk and M. R. Hamblin, ACS Appl. Mater. Interfaces, 2016, 8, 21107–21133 CrossRef CAS.
- X. J. Zhang, X. Ma, K. Wang, S. J. Lin, S. T. Zhu, Y. Dai and F. Xia, Macromol. Rapid Commun., 2018, 39, 1800142 CrossRef.
- W. L. Zhang and H. J. Choi, Polymers, 2014, 6, 2803–2818 CrossRef CAS.
- T. Manouras and M. Vamvakaki, Polym. Chem., 2017, 8, 74–96 RSC.
- H. C. Chen, D. Y. Liu and Z. J. Guo, Chem. Lett., 2016, 45, 242–249 CrossRef CAS.
- W. Wu, L. Luo, Y. Wang, Q. Wu, H. B. Dai, J. S. Li, C. Durkan, N. Wang and G. X. Wang, Theranostics, 2018, 8, 3038–3058 CrossRef CAS.
- Q. Y. Hu, P. S. Katti and Z. Gu, Nanoscale, 2014, 6, 12273–12286 RSC.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- A. Kakkar, G. Traverso, O. C. Farokhzad, R. Weissleder and R. Langer, Nat. Rev. Chem., 2017, 1, 0063 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- M. D. Best, Biochemistry, 2009, 48, 6571–6584 CrossRef CAS.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- Y. P. Chen, Y. L. Xianyu, J. Wu, B. F. Yin and X. Y. Jiang, Theranostics, 2016, 6, 969–985 CrossRef CAS.
- Y. Dai, X. J. Zhang and F. Xia, Macromol. Rapid Commun., 2017, 38, 1700357 CrossRef.
- Y. Zou, L. Zhang, L. Yang, F. Zhu, M. M. Ding, F. Lin, Z. Wang and Y. W. Li, J. Controlled Release, 2018, 273, 160–179 CrossRef CAS PubMed.
- J. Akimoto, M. Nakayama and T. Okano, J. Controlled Release, 2014, 193, 2–8 CrossRef CAS.
- J. X. Ding, L. Zhao, D. Li, C. S. Xiao, X. L. Zhuang and X. S. Chen, Polym. Chem., 2013, 4, 3345–3356 RSC.
- A. W. Thomas, P. K. Kuroishi, M. M. Perez-Madrigal, A. K. Whittaker and A. P. Dove, Polym. Chem., 2017, 8, 5082–5090 RSC.
- X. J. Wan, T. Liu and S. Y. Liu, Biomacromolecules, 2011, 12, 1146–1154 CrossRef CAS.
- A. J. de Graaf, E. Mastrobattista, T. Vermonden, C. F. van Nostrum, D. T. S. Rijkers, R. M. J. Liskamp and W. E. Hennink, Macromolecules, 2012, 45, 842–851 CrossRef CAS.
- H. H. Liu, W. D. Pan, M. Tong and Y. L. Zhao, Polym. Chem., 2016, 7, 1603–1611 RSC.
- H. H. Huo, X. Y. Ma, Y. Q. Dong and F. J. Qu, Eur. Polym. J., 2017, 87, 331–343 CrossRef CAS.
- S. Vandewalle, M. Van De Walle, S. Chattopadhyay and F. E. Du Prez, Eur. Polym. J., 2018, 98, 468–474 CrossRef CAS.
- Y. Huang, R. J. Dong, X. Y. Zhu and D. Y. Yan, Soft Matter, 2014, 10, 6121–6138 RSC.
- C. S. Linsley and B. M. Wu, Ther. Delivery, 2017, 8, 89–107 CrossRef CAS.
- D. Hu, H. Peng, Y. L. Niu, Y. F. Li, Y. C. Xia, L. Li, J. W. He, X. Y. Liu, X. N. Xia, Y. B. Lu and W. J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 750–760 CrossRef CAS.
- Y. L. Niu, Y. F. Li, Y. B. Lu and W. J. Xu, RSC Adv., 2014, 4, 58432–58439 RSC.
- C. Ventura, P. Thornton, S. Giordani and A. Heise, Polym. Chem., 2014, 5, 6318–6324 RSC.
- D. Hu, Y. F. Li, Y. L. Niu, L. Li, J. W. He, X. Y. Liu, X. N. Xia, Y. B. Lu, Y. Q. Xiong and W. J. Xu, RSC Adv., 2014, 4, 47929–47936 RSC.
- Y. F. Li, Y. L. Niu, D. Hu, Y. W. Song, J. W. He, X. Y. Liu, X. N. Xia, Y. B. Lu and W. J. Xu, Macromol. Chem. Phys., 2015, 216, 77–84 CrossRef CAS.
- X. Y. Liu, J. W. He, Y. L. Niu, Y. F. Li, D. Hu, X. N. Xia, Y. B. Lu and W. J. Xu, Polym. Adv. Technol., 2015, 26, 449–456 CrossRef CAS.
- S. M. A. Soliman, L. Colombeau, C. Nouvel, J. Babin and J. L. Six, Carbohydr. Polym., 2016, 136, 598–608 CrossRef CAS.
- M. D. Zhang, D. H. Li, G. Wang, J. X. Li and H. Z. Sun, Dyes Pigm., 2014, 105, 232–237 CrossRef CAS.
- X. Wang, Y. Y. Yang, P. Y. Gao, F. Yang, H. Shen, H. X. Guo and D. C. Wu, ACS Macro Lett., 2015, 4, 1321–1326 CrossRef CAS.
- N. G. Patil, N. B. Basutkar and A. V. Ambade, Chem. Commun., 2015, 51, 17708–17711 RSC.
- Y. Z. Zhao, L. N. Du, C. T. Lu, Y. G. Jin and S. P. Ge, Int. J. Nanomed., 2013, 8, 1621–1633 Search PubMed.
- F. Y. Li, C. Xie, Z. G. Cheng and H. S. Xia, Ultrason. Sonochem., 2016, 30, 9–17 CrossRef CAS.
- J. Liu, Y. R. Huang, A. Kumar, A. Tan, S. B. Jin, A. Mozhi and X. J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed.
- Y. Jin, L. Song, Y. Su, L. J. Zhu, Y. Pang, F. Qiu, G. S. Tong, D. Y. Yan, B. S. Zhu and X. Y. Zhu, Biomacromolecules, 2011, 12, 3460–3468 CrossRef CAS.
- M. N. Ganivada, P. Kumar, P. Kanjilal, H. Dinda, J. Das Sarma and R. Shunmugam, Polym. Chem., 2016, 7, 4237–4245 RSC.
- Y. Xin and J. Y. Yuan, Polym. Chem., 2012, 3, 3045–3055 RSC.
- G. R. Qiu, L. Zhao, X. Liu, Q. X. Zhao, F. F. Liu, Y. Liu, Y. W. Liu and H. B. Gu, React. Funct. Polym., 2018, 128, 1–15 CrossRef CAS.
- J. Wei, X. Y. Shuai, R. Wang, X. L. He, Y. W. Li, M. M. Ding, J. H. Li, H. Tan and Q. Fu, Biomaterials, 2017, 145, 138–153 CrossRef CAS.
- X. T. Cao, C. M. Q. Le, H. H. P. Thi, G. D. Kim, Y. S. Gal and K. T. Lim, eXPRESS Polym. Lett., 2017, 11, 832–845 CrossRef CAS.
- S. S. Patil and P. P. Wadgaonkar, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1383–1396 CrossRef CAS.
- K. Dan and S. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 7300–7305 CrossRef CAS.
- M. R. Nabid and I. Omrani, Mater. Sci. Eng., C, 2016, 69, 532–537 CrossRef CAS.
- K. Jong, B. Z. Ju and S. F. Zhang, J. Biomater. Sci., Polym. Ed., 2017, 28, 1525–1537 CrossRef CAS.
- Y. Sun, X. Q. Du, J. L. He, J. Hu, M. Z. Zhang and P. H. Ni, J. Mater. Chem. B, 2017, 5, 3771–3782 RSC.
- F. S. Huang, R. Cheng, F. H. Meng, C. Deng and Z. Y. Zhong, Biomacromolecules, 2015, 16, 2228–2236 CrossRef CAS.
- D. Huang, F. Yang, X. Wang, H. Shen, Y. You and D. C. Wu, Polym. Chem., 2016, 7, 6154–6158 RSC.
- B. B. Breitenbach, I. Schmid and P. R. Wich, Biomacromolecules, 2017, 18, 2839–2848 CrossRef CAS.
- A. J. Harnoy, M. Buzhor, E. Tirosh, R. Shaharabani, R. Beck and R. J. Amir, Biomacromolecules, 2017, 18, 1218–1228 CrossRef CAS.
- J. Y. Xiong and H. L. Gao, J. Microencapsulation, 2017, 34, 440–453 CrossRef CAS.
- H. X. Wang, X. Z. Yang, C. Y. Sun, C. Q. Mao, Y. H. Zhu and J. Wang, Biomaterials, 2014, 35, 7622–7634 CrossRef CAS.
- D. Bacinello, E. Garanger, D. Taton, K. C. Tam and S. Lecommandoux, Biomacromolecules, 2014, 15, 1882–1888 CrossRef CAS.
- W. D. Ke, J. J. Li, K. J. Zhao, Z. S. Zha, Y. Han, Y. H. Wang, W. Yin, P. Zhang and Z. S. Ge, Biomacromolecules, 2016, 17, 3268–3276 CrossRef CAS.
- W. D. Ke, Z. S. Zha, J. F. Mukerabigwi, W. J. Chen, Y. H. Wang, C. X. He and Z. S. Ge, Bioconjugate Chem., 2017, 28, 2190–2198 CrossRef CAS.
- N. Li, H. Cai, L. Jiang, J. N. Hu, A. Bains, J. Hu, Q. Y. Gong, K. Luo and Z. W. Gu, ACS Appl. Mater. Interfaces, 2017, 9, 6865–6877 CrossRef CAS.
- H. L. Sun, F. H. Meng, R. Cheng, C. Deng and Z. Y. Zhong, Antioxid. Redox Signaling, 2014, 21, 755–767 CrossRef CAS.
- R. Cheng, F. Feng, F. H. Meng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS.
- Q. Q. Zhang, J. L. He, M. Z. Zhang and P. H. Ni, J. Mater. Chem. B, 2015, 3, 4922–4932 RSC.
- D. L. Cao, J. L. He, J. Y. Xu, M. Z. Zhang, L. Zhao, G. X. Duan, Y. W. Cao, R. H. Zhou and P. H. Ni, Polym. Chem., 2016, 7, 4198–4212 RSC.
- X. Q. Du, Y. Sun, M. Z. Zhang, J. L. He and P. H. Ni, ACS Appl. Mater. Interfaces, 2017, 9, 13939–13949 CrossRef CAS PubMed.
- X. Q. Yi, D. Zhao, Q. Zhang, J. Q. Xu, G. D. Yuan, R. X. Zhuo and F. Li, Polym. Chem., 2016, 7, 5966–5977 RSC.
- D. H. Han, X. Tong and Y. Zhao, Langmuir, 2012, 28, 2327–2331 CrossRef CAS PubMed.
- Y. J. Wu, H. H. Kuang, Z. G. Xie, X. S. Chen, X. B. Jing and Y. B. Huang, Polym. Chem., 2014, 5, 4488–4498 RSC.
- X. J. Zhang and Y. Dai, J. Nanopart. Res., 2018, 20, 147 CrossRef.
- X. J. Zhang, H. Q. Wang and Y. Dai, J. Nanopart. Res., 2018, 20, 126 CrossRef.
- J. Xuan, D. H. Han, H. S. Xia and Y. Zhao, Langmuir, 2014, 30, 410–417 CrossRef CAS.
- H. Y. Fan, Y. X. Li, J. X. Yang and X. D. Ye, J. Phys. Chem. B, 2017, 121, 9708–9717 CrossRef CAS.
- M. Q. Wang, Y. F. Wang, S. J. Zhao, X. L. Zhang and H. Wei, Macromol. Chem. Phys., 2018, 219, 1800061 CrossRef.
- X. S. Fan, X. Y. Wang, M. Y. Cao, C. G. Wang, Z. G. Hu, Y. L. Wu, Z. B. Li and X. J. Loh, Polym. Chem., 2017, 8, 5611–5620 RSC.
- G. Saravanakumar, H. Park, J. Kim, D. Park, S. Pramanick, D. H. Kim and W. J. Kim, Biomacromolecules, 2018, 19, 2202–2213 CrossRef CAS PubMed.
- S. Cajot, N. Lautram, C. Passirani and C. Jerome, J. Controlled Release, 2011, 152, 30–36 CrossRef CAS.
- H. Wang, L. Tang, C. L. Tu, Z. Y. Song, Q. Yin, L. C. Yin, Z. H. Zhang and J. J. Cheng, Biomacromolecules, 2013, 14, 3706–3712 CrossRef CAS.
- Z. H. Zhang, L. C. Yin, C. L. Tu, Z. Y. Song, Y. F. Zhang, Y. X. Xu, R. Tong, Q. Zhou, J. Ren and J. J. Cheng, ACS Macro Lett., 2013, 2, 40–44 CrossRef CAS.
- Y. L. Cheng, C. L. He, C. S. Xiao, J. X. Ding, K. X. Ren, S. J. Yu, X. L. Zhuang and X. S. Chen, Polym. Chem., 2013, 4, 3851–3858 RSC.
- Y. Han, J. J. Li, M. H. Zan, S. Z. Luo, Z. S. Ge and S. Y. Liu, Polym. Chem., 2014, 5, 3707–3718 RSC.
- A. Guerry, S. Cottaz, E. Fleury, J. Bernard and S. Halila, Carbohydr. Polym., 2014, 112, 746–752 CrossRef CAS.
- W. P. Zhu, Y. Wang, X. Cai, G. Y. Zha, Q. J. Luo, R. Sun, X. D. Li and Z. Q. Shen, J. Mater. Chem. B, 2015, 3, 3024–3031 RSC.
- Y. H. Kim, X. T. Cao, S. S. Hong, Y. S. Gal and K. T. Lim, Mol. Cryst. Liq. Cryst., 2016, 635, 107–113 CrossRef CAS.
- Y. C. Xia, H. He, X. Y. Liu, D. Hu, L. C. Yin, Y. B. Lu and W. J. Xua, Polym. Chem., 2016, 7, 6330–6339 RSC.
- X. J. Zhang, H. Dong, S. L. Fu, Z. L. Zhong and R. X. Zhuo, Macromol. Rapid Commun., 2016, 37, 993–997 CrossRef CAS.
- X. T. Cao, Y. H. Kim, J. M. Park and K. T. Lim, Eur. Polym. J., 2016, 78, 264–273 CrossRef CAS.
- Y. Dai, H. Q. Wang and X. J. Zhang, J. Nanopart. Res., 2017, 19, 383 CrossRef.
- X. L. Hu, J. Tian, T. Liu, G. Y. Zhang and S. Y. Liu, Macromolecules, 2013, 46, 6243–6256 CrossRef CAS.
- Y. Huang, R. Sun, Q. J. Luo, Y. Wang, K. Zhang, X. L. Deng, W. P. Zhu, X. D. Li and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 99–107 CrossRef CAS.
- C. M. Q. Le, H. H. P. Thi, X. T. Cao, G. D. Kim, C. W. Oh and K. T. Lim, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3741–3750 CrossRef CAS.
- A. S. Deshmukh, P. N. Chauhan, M. N. Noolvi, K. Chaturvedi, K. Ganguly, S. S. Shukla, M. N. Nadagouda and T. M. Aminabhavi, Int. J. Pharm., 2017, 532, 249–268 CrossRef CAS.
- Q. Zhou, L. Zhang, T. H. Yang and H. Wu, Int. J. Nanomed., 2018, 13, 2921–2942 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |