DOI:
10.1039/C8PY01231H
(Paper)
Polym. Chem., 2019,
10, 94-105
Enzyme triggered disassembly of amphiphilic linear-dendritic block copolymer micelles based on poly[N-(2-hydroxyethyl-L-glutamine)]†
Received
21st August 2018
, Accepted 14th November 2018
First published on 14th November 2018
Abstract
Well-defined enzyme-responsive amphiphilic linear-dendritic diblock copolymers G-b-PHEG and bis-MPA-Gn-b-PHEG (n = 1, 2) based on a hydrophilic linear block of poly(hydroxyethyl L-glutamine) (PHEG) and a hydrophobic dendron derived from cysteamine or an aliphatic polyester of 2,2′-bis(hydroxymethyl)propionic acid (bis-MPA) have been synthesized for the first time via a combination of the “dendron-first” strategy and the ring-opening polymerization of α-amino acid N-carboxyanhydrides (NCA). The obtained copolymers G-b-PHEG and bis-MPA-Gn-b-PHEG (n = 1, 2) were characterized by gel permeation chromatography (GPC), Fourier transfrom-infrared spectroscopy (FT-IR), and 1H NMR spectroscopy. These amphiphilic copolymers are able to form nanospherical micelles or vesicles in aqueous solution as revealed by 1H NMR analyses of the micelles in D2O, fluorescence spectroscopy, transmission electron microscopy (TEM) and particle size analysis. The self-assembled micelles show good enzyme-responsiveness, which can disassemble and release the encapsulated hydrophobic Nile red upon enzymatic activation. In vitro cytotoxicity studies indicate that the copolymers have good compatibility with SMMC-7721 cells. Such smart amphiphilic copolymers could potentially be applied for drug delivery systems and biosensors.
Introduction
Stimuli-responsive polymers that undergo changes in response to applied stimuli such as pH, temperature, light, enzymes, redox potential or electric/magnetic field have developed greatly in recent years as a result of their prospective uses in biotechnology and drug delivery.1–8 Among them, enzyme responsive polymers are especially attractive because of their precise selectivity, high efficiency in enzymatic catalysis and mild reaction conditions (aqueous, pH 5–8, 37 °C).9–11 Amphiphilic enzyme responsive copolymers can self-assemble into micelles with a hydrophobic core and a hydrophilic shell that can disassemble and release the encapsulated hydrophobic molecular cargo (e.g., fluorescent dyes or drugs) upon enzymatic stimuli, allowing for their promising applications in enzyme-triggered localized drug release. So far, there have been a number of reports on amphiphilic enzyme responsive linear–linear block copolymers.12–14 However, enzyme responsive linear-dendritic block copolymers (LDBCs) have seen only limited exploration.15,16 Linear-dendritic block copolymers (LDBCs), originally described by the group of Fréchet in the early 1990s,17 have obtained great interest in the past few decades because they combine chain entangled linear polymers with densely chain-packed dendritic segments, which enables them to possess unique solution, bulk, and especially self-assembling properties.18–20 In particular, amphiphilic LDBCs can assemble into different nanostructures in aqueous solution, which exhibit excellent loading capacity for hydrophobic small molecular medicine and therefore hold great potential for the controlled release of drugs.21,22 To date, the most prominent representatives of amphiphilic LDBCs are conjugates of dendritic segments with poly(ethylene glycol) (PEG) due to the biocompatibility and low toxicity of PEG.23,24 However, in the last few years, studies published in the literature reported on potentially unfavorable effects using PEG and PEG-containing polymers, including possible accumulation in the body due to its non-biodegradability, thermal instability, unexpected changes in pharmacokinetic behavior, and hypersensitivity after intravenous and oral administrations.25 Thus, it is essential to explore possible PEG alternatives that can satisfy the advantages and overcome the disadvantages of PEG.26,27
Synthetic poly(α-amino acid)s are commonly used polymers composed of α-amino acids by the connection of peptide bonds (–CONH–), which can be degraded into α-amino acids and metabolized in the human body by proteolytic enzymes.28,29 Poly(γ-benzyl-L-glutamate) (PBLG) is one of the poly(α-amino acids) which is often used as a drug delivery carrier, tissue engineering material and gene vector.30,31 However, because of the hydrophobic character, uncontrolled degradation and lack of functional groups, the application of PBLG is still limited. The modification of the ester side chains on PBLG, such as aminolysis to get poly[N-(2-hydroxyethyl-L-glutamine)] (PHEG), is an important way to improve the properties of PBLG.32,33 PHEG is a hydrophilic and biodegradable polypeptide, which can markedly improve the cytocompatibility and reduce the cytotoxicity of carriers in gene delivery according to recent studies.34–38
Herein, we report the synthesis of new well-defined enzyme-responsive linear-dendritic block copolymers containing a PHEG chain and a dendron derived from cysteamine or 2,2′-bis(hydroxymethyl)propionic acid (bis-MPA)39–41via a combination of the “dendron-first” strategy and the ring-opening polymerization (ROP) by using a primary amine-containing dendron initiator. Their self-assembly behavior in aqueous solution and the controlled release of encapsulated hydrophobic Nile red upon enzymatic activation were then thoroughly investigated via a combination of 1H NMR spectroscopy, fluorescence spectroscopy, TEM and particle size analysis. In vitro cytotoxicity of the copolymers was also investigated for potential biomedical application.
Experimental
Materials
3,5-Dihydroxybenzoic acid, propargyl bromide, 2-aminoethanethiol hydrochloride, γ-benzyl-L-glutamate, 4-dimethylaminopyridine (DMAP) and di-tert-butyl dicarbonate (Boc2O) were purchased from Aldrich. N-Carboxyanhydride of γ-benzyl-L-glutamate (BLG-NCA),42 methyl 3,5-dihydroxybenzoate,43 2-(Boc-amino) ethanethiol,44 isopropylidene-2,2-bis(oxymethyl)propionic acid45 and isopropylidene-2,2-bis(oxymethyl)propionic anhydride46 were prepared according to a reported method. Tetrahydrofuran (THF) was dried by refluxing over sodium/benzophenone and distilled just prior to use. N,N-Dimethylformamide (DMF) was dried with 4 Å molecular sieves and purified by distillation under reduced pressure. 2,2-Azobisiso-butyronitrile (AIBN, 98%, Sinopharm Chemical Reagent) was recrystallized twice from methanol and other reagents were used as received.
Characterization
1H NMR spectra were obtained using a Bruker DRX-500 spectrometer with CDCl3, DMSO-d6 or D2O as the solvent. Tetramethylsilicone (TMS) was used as the internal standard. ESI-MS spectra were recorded on a Finnigan LCQ-Advantage mass spectrometer. FT-IR spectra were obtained on a Nicolet AVATAR 360 FT-IR spectrometer. The molecular weight and polydispersity of polymers were measured by gel permeation chromatography (GPC). The GPC system consisted of a Waters 2690D separations module and a Waters 2414 refractive index detector (RI). Styragel HR1 and HR4 columns (Waters) were used at 40 °C with poly(methyl methacrylate) as standards and DMF (included 0.05 M LiBr) as a mobile phase at a flow rate of 0.3 mL min−1.
Synthesis of G-b-PHEG
Synthesis of methyl 3,5-bis-(prop-2-ynyloxy) benzoate.
3,5-Bis-(prop-2-ynyloxy) benzoate was synthesized according to a previously published study47 with modifications. Methyl 3,5-dihydroxybenzoate (5 g, 29.75 mmol) and propargyl bromide (5 mL, 63.96 mmol) were dissolved in 100 mL of acetone, followed by the addition of anhydrous K2CO3 (12.30 g, 88.85 mmol). The reaction mixture was stirred for 16 h at room temperature. After filtration, the filtrate was concentrated under reduced pressure and the residue was dissolved in EtOAc (100 mL), and the organic phase was washed with H2O (3 × 50 mL) and dried with anhydrous magnesium sulfate, filtered and evaporated in vacuo. The crude mixture was further purified by silica gel column chromatography using petroleum ether/ethyl acetate (v/v = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent and the product was obtained as off-white crystals in 60.7% yield (4.40 g). 1H NMR in CDCl3, δ(ppm): 7.29 (d, J = 2.5 Hz, 2H, arom H2/H6), 6.81 (t, J = 2.5 Hz, 1H, arom H4), 4.71 (d, J = 2.5 Hz, 4H, 2OCH2), 3.91 (s, 3H, OCH3), 2.54 (t, J = 2.5 Hz, 2H, CH). IR (KBr, cm−1): 3293, 3271 (
:1) as the eluent and the product was obtained as off-white crystals in 60.7% yield (4.40 g). 1H NMR in CDCl3, δ(ppm): 7.29 (d, J = 2.5 Hz, 2H, arom H2/H6), 6.81 (t, J = 2.5 Hz, 1H, arom H4), 4.71 (d, J = 2.5 Hz, 4H, 2OCH2), 3.91 (s, 3H, OCH3), 2.54 (t, J = 2.5 Hz, 2H, CH). IR (KBr, cm−1): 3293, 3271 (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H), 3093 (C–H), 2131 (CC), 1715 (C
C–H), 3093 (C–H), 2131 (CC), 1715 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
Synthesis of G-COOCH3.
Methyl 3,5-bis-(prop-2-ynyloxy) benzoate (0.65 g, 2.66 mmol) was dissolved in dry DMF (30 mL) and 2-(Boc-amino) ethanethiol (2.70 mL, 15.96 mmol) and AIBN (350 mg, 2.13 mmol) were added. The reaction mixture was stirred for 12 h at 80 °C under a nitrogen atmosphere. Then, DMF was removed in a vacuum and the residue was further purified by silica gel column chromatography using petroleum ether/acetone (v/v = 3:1) as the eluent and the product was obtained as a yellowish solidified oil in 83.5% yield (2.12 g). 1H NMR in CDCl3, δ(ppm): 7.20 (d, J = 2 Hz, 2H, arom H2/H6), 6.69 (t, J = 2 Hz, 1H, arom H4), 5.04–5.18 (m, 4H, NHBoc), 4.15–4.25 (m, 4H, 2OCH2), 3.90 (s, 3H, OCH3), 3.26–3.40 (m, 8H, 4NCH2), 3.12–3.20 (m, 2H, 2SCH), 2.87–3.0 (m, 4H, 2SCH2), 2.75–2.81 (m, 4H, 2SCH2), 2.64–2.73 (m, 4H, 2SCH2), 1.44 (s, 36H, Boc). IR (KBr, cm−1): 3421 (N–H), 2977, 2931 (C–H), 1698 (CO), 1599 (CO), 1167 (C–O). ESI-MS(m/z): calculated for C42H72N4O12S4, 952.40; found, [M + H]+ = 953.41, [M + NH4]+ = 970.44, [M + Na]+ = 975.40, [M + K]+ = 991.38.
Synthesis of G-NH2.
A solution of G-COOCH3 (0.82 g, 0.86 mmol) in 10 mL of methanol was added dropwise into a stirred solution of 1,2-diaminoethane (2.6 mL, 39.36 mmol) in 20 mL of methanol at 0 °C. After the addition was completed within 1 h, the mixture was allowed to warm to room temperature and stirred for 96 h. The solvent was removed in a vacuum and the excess 1,2-diaminoethane was removed using an azeotropic mixture of toluene and methanol (v/v = 9:1). After drying in a vacuum for over 48 h, the product was obtained as a yellow solid in 84.1% yield (0.71 g). 1H NMR in CDCl3, δ(ppm): 8.33 (d, J = 7 Hz, 1H, NH), 7.73 (d, J = 2 Hz, 2H, arom H2/H6), 6.66 (t, J = 2.5 Hz, 1H, arom H4), 5.03–5.28 (m, 4H, NH), 4.08–4.28 (m, 4H, 2OCH2), 3.74–3.68 (m, 2H, NCH2), 3.46–3.53 (m, 2H, NCH2), 3.22–3.40 (m, 8H, 4NCH2), 3.10–3.19 (m, 2H, 2SCH), 2.82–2.94 (m, 4H, 2SCH2), 2.73–2.81 (m, 4H, 2SCH2), 2.62–2.72 (m, 4H, 2SCH2), 1.99 (d, J = 2.5 Hz, 2H, NH2), 1.43 (s, 36H, Boc). ESI-MS(m/z): calculated for C43H76N6O11S4, 980.45; found, [M + H]+ = 981.45, [M + NH4]+ = 997.45.
Synthesis of G-b-PBLG.
To a solution of BLG-NCA (2.01 g, 7.64 mmo) in 5 mL of dry DMF was added a solution of G-NH2 (0.50 g, 0.51 mmol) in 3 mL of dry DMF as the initiator. This solution was stirred for 48 h at 30 °C under a nitrogen atmosphere. The crude polymer was subsequently precipitated three times in diethyl ether and dried under vacuum. The product was obtained as a yellow solid in 76.4% yield (1.23 g). 1H NMR in DMSO-d6, δ(ppm): 8.01–8.45 (br, NH), 7.15–7.45 (br, arom H), 6.83–7.15 (br, arom H2/H4/H6), 4.87–5.15 (br, arom-CH2), 3.81–4.40 (br, CH), 3.70–3.77 (br, OCH2), 3.30–3.50 (br, NCH2), 3.18–3.23 (br, SCH), 3.05–3.17 (br, NCH2), 2.94–3.02 (br, NCH2), 2.87–2.93 (br, SCH2), 2.66–2.71 (br, SCH2), 2.56–2.61 (br, SCH2), 1.75–2.45 (br, CH2). IR (KBr, cm−1): 3296 (N–H), 3035, 2933 (C–H), 1735 (CO), 1654 (CO), 1165 (C–O), 746, 697 (Ar).
Synthesis of G-b-PHEG.
1.10 g of G-b-PBLG and 0.50 g of 2-hydroxypyridine were dissolved in 10 mL of dry DMF. Then ca. 8 mL of ethanolamine was added dropwise. After being stirred for 24 h at 40 °C under a nitrogen atmosphere, the solution was precipitated into ca. 200 mL of diethyl ether. The precipitate was redissolved in water, dialyzed (molecular weight cut off 1000), and subsequently freeze-dried and the product was obtained as a yellow solid in 70% yield (0.63 g). 1H NMR in DMSO-d6, δ(ppm): 8.05–8.25 (br, NH), 7.72–7.92 (br, NH), 6.83–7.15 (br, arom H2/H4/H6), 4.55–4.85 (br, OH), 4.12–4.32 (br, CH), 4.05–4.11 (br, OCH2), 3.3–3.59 (br, OCH2 and NCH2), 3.05–3.15 (br, NCH2 and SCH), 2.87–2.94 (br, NCH2), 2.55–2.78 (br, SCH2), 2.0–2.25 (br, CH2CO), 1.65–1.98 (br, CH and CH2). IR (KBr, cm−1): 3301 (O–H), 3080, 2932 (C–H), 1655 (CO), 1167 (C–O).
Synthesis of bis-MPA-Gn-b-PHEG (n = 1, 2)
Synthesis of ketal-G1-NHBoc.
A mixture solution of Boc mono-protected ethylenediamine (4.60 g, 28.72 mmol) and DMAP (0.36 g, 2.95 mmol) in 50 mL of CH2Cl2 was added dropwise to a stirred solution of isopropylidene-2,2-bis(oxymethyl)propionic acid (5 g, 28.72 mmol) and dicyclohexylcarbodiimide (DCC) (5.90 g, 28.72 mmol) in 80 mL of CH2Cl2 at 0 °C under a nitrogen atmosphere. After the addition was completed within 1 h, the mixture was allowed to warm to room temperature and stirred for 24 h. Then, the dicyclohexylurea (DCU) formed was removed by filtration and the solvent was evaporated to give the crude product, which was purified by silica gel column chromatography using petroleum ether/acetone (v/v = 6:1) as the eluent and the product was obtained as a white solid in 77.2% yield (7.01 g). 1H NMR in CDCl3, δ(ppm): 7.33 (t, J = 5.2 Hz, 1H, NH), 4.96 (t, J = 5.2 Hz, 1H, NH), 3.90 (d, J = 12 Hz, 2H, OCH2), 3.77 (d, J = 12 Hz, 2H, OCH2), 3.39 (q, J = 5.2 Hz, 2H, NCH2), 3.30 (q, J = 5.2 Hz, 2H, NCH2), 1.47 (s, 3H, CH3), 1.43 (s, 3H, CH3), 1.42 (s, 9H, 3CH3), 1.02 (s, 3H, CH3).
Synthesis of HO-G1-NHBoc.
To a solution of ketal-G1-NHBoc (10 g, 31.63 mmol) in MeOH was added p-TsOH·H2O (1.20 g, 3.78 mmol). The resulting solution was stirred at room temperature for 15 h. The reaction was quenched with a saturated aqueous NaHCO3 solution, and the aqueous phase was extracted with EtOAc. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in a vacuum. The product was obtained as a white solid in 91.6% yield (8 g). 1H NMR in DMSO-d6, δ(ppm): 7.50 (t, J = 5.2 Hz, 1H, NH), 6.81 (t, J = 5.2 Hz, 1H, NH), 4.66 (s, 2H, 2OH), 3.43 (d, J = 10.8 Hz, 2H, OCH2), 3.41 (d, J = 10.8 Hz, 2H, OCH2), 3.07 (q, J = 6 Hz, 2H, NCH2), 2.98 (q, J = 6 Hz, 2H, NCH2), 1.37 (s, 9H, 3CH3), 0.97 (s, 3H, CH3).
Synthesis of G1-NHBoc.
HO-G1-NHBoc (1 g, 3.62 mmol) was dissolved in 48 mL of a 5:1 CH2Cl2/DMF solution, to which Fmoc-glycine (3.23 g, 10.86 mmol) and DMAP (0.66 g, 5.43 mmol) were added. After dissolution, 2.24 g (10.86 mmol) of DCC in 10 mL of dry dichloromethane were added dropwise into the reaction mixture at 0 °C under a nitrogen atmosphere. After the addition was completed within 1 h, the mixture was allowed to warm to room temperature and stirred for 24 h. Then, the precipitate was filtered off and the solvent was evaporated under reduced pressure to give a crude product, which was purified by silica gel column chromatography using petroleum ether/acetone (v/v = 2:1) as the eluent and the product was obtained as a white solid in 74.2% yield (2.24 g). 1H NMR in DMSO-d6, δ(ppm): 7.90 (d, J = 7.6 Hz, 4H, arom H), 7.77 (m, 3H, NH), 7.70 (d, J = 7.6 Hz, 4H, arom H), 7.41 (t, J = 7.2 Hz, 4H, arom H), 7.32 (t, J = 7.2 Hz, 4H, arom H), 6.77 (t, J = 5.6 Hz, 1H, NH), 4.30 (d, J = 6.8 Hz, 4H, 2OCH2), 4.24 (d, J = 6.4 Hz, 2H, 2 arom-CH), 4.16 (d, J = 3.6 Hz, 4H, 2OCH2), 3.77 (d, J = 6 Hz, 4H, 2NCH2), 3.06 (q, J = 6 Hz, 2H, NCH2), 2.98 (q, J = 6 Hz, 2H, NCH2), 1.34 (s, 9H, 3CH3), 1.10 (s, 3H, CH3). ESI-MS(m/z): calculated for C46H50N4O11, 834.35; found, [M + Na]+ = 857.30.
Synthesis of G1-NH2.
G1-NHBoc (0.48 g, 0.57 mmol) was dissolved in 30 mL of a 1:1 TFA/CH2Cl2 solution at 0 °C. After stirring for 6 h, the solution was neutralized with a saturated aqueous NaHCO3 solution, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous MgSO4 and concentrated in a vacuum. The product was obtained as a pale yellow solid in 78.8% yield (0.33 g). 1H NMR in DMSO-d6, δ(ppm): 7.90 (d, J = 5.2 Hz, 4H, arom H), 7.85 (d, J = 5.6 Hz, 2H, arom H), 7.76 (t, J = 4.8 Hz, 1H, NH), 7.70 (d, J = 5.6 Hz, 2H, arom H), 7.62 (t, J = 5.6 Hz, 1H, NH), 7.53 (t, J = 6.4 Hz, 1H, NH), 7.42 (t, J = 5.6 Hz, 4H, arom H), 7.35 (t, J = 5.6 Hz, 4H, arom H), 4.31 (d, J = 5.2 Hz, 2H, OCH2), 4.24 (t, J = 4.4 Hz, 2H, NCH2), 4.15 (t, J = 8 Hz, 2H, 2Ar-CH), 3.76 (d, J = 5.2 Hz, 2H, OCH2), 3.70 (d, J = 6.8 Hz, 2H, OCH2), 3.63 (d, J = 6.8 Hz, 2H, OCH2), 3.47 (d, J = 8.4 Hz, 2H, OCH2), 3.42 (d, J = 8.4 Hz, 2H, OCH2), 3.06 (s, 2H, NH2), 2.84 (t, J = 4.4 Hz, 2H, NCH2), 1.30 (s, 3H, CH3). ESI-MS(m/z): calculated for C41H42N4O9, 734.30; found, [M + H]+ = 735.30.
Synthesis of bis-MPA-G1-b-PBLG.
To a solution of BLG-NCA (4.30 g, 0.016 mmol) in 10 mL of dry DMF was added a solution of G1-NH2 (0.30 g, 0.41 mmol) in 2 mL of dry DMF as the initiator. This solution was stirred for 48 h at 30 °C under a nitrogen atmosphere. The crude polymer was subsequently precipitated three times in diethyl ether and dried under vacuum. The product was obtained as a white solid in 67.1% yield (3.10 g). 1H NMR in DMSO-d6, δ(ppm): 8.15–8.50 (br, NH), 7.15–7.93 (br, arom H), 4.90–5.20 (br, arom-CH2), 4.07–4.35 (br, OCH2), 3.81–4.0 (br, NCH), 3.74–3.80 (br, NCH2), 3.30–3.40 (br, NCH2), 2.35–2.60 (br, CH2CO), 1.90–2.30 (br, CH and CH2), 1.23 (s, 3H, CH3). IR (KBr, cm−1): 3294 (N–H), 3035, 2952 (C–H), 1735 (CO), 1653 (CO), 746, 697 (Ar).
Synthesis of bis-MPA-G1-b-PHEG.
2.60 g of bis-MPA-G1-b-PBLG and 1.10 g of 2-hydroxypyridine were dissolved in 20 mL of dry DMF. Then ca. 20 mL of ethanolamine was added dropwise. After being stirred for 24 h at 40 °C under a nitrogen atmosphere, the solution was precipitated into ca. 400 mL of diethyl ether. The precipitate was redissolved in water, dialyzed (molecular weight cut off 3500), and subsequently freeze-dried and the product was obtained as a white solid in 78.4% yield (1.63 g). 1H NMR in DMSO-d6, δ(ppm): 8.05–8.23 (br, NH), 7.35–7.98 (br, NH and arom H), 4.64–4.74 (br, OH), 4.12–4.28 (br, OCH2 and NCH2), 3.36–3.42 (br, OCH2 and NCH2), 3.06–3.15 (br, NCH2), 2.05–2.19 (br, CH2CO), 1.65–1.95 (br, CH and CH2), 0.97 (s, CH3). IR (KBr, cm−1): 3424 (O–H), 3092, 2927 (C–H), 1653 (CO).
Synthesis of ketal-G2-NHBoc.
HO-G1-NHBoc (6 g, 21.72 mmol), isopropylidene-2,2-bis(oxymethyl)propionic anhydride (21.60 g, 65.16 mmol) and DMAP (5.31 g, 43.44 mmol) were dissolved in 80 mL of a 3:2 pyridine/CH2Cl2 solution. After stirring for 18 h at room temperature, the excess anhydride was quenched by stirring the reaction mixture with 10 mL of a 1:1 pyridine/water solution overnight. The organic phase was diluted with 100 mL of CH2Cl2 and extracted with 1 M NaHSO4 (3 × 50 mL), 10% Na2CO3 (3 × 50 mL), and saturated brine (50 mL). The organic phase was dried with MgSO4 and filtered, and the filtrate was evaporated to give the crude product, which was purified by silica gel column chromatography using petroleum ether/acetone (v/v = 6:1) as the eluent and the product was obtained as a white solid in 72.5% yield (9.27 g). 1H NMR in CDCl3, δ(ppm): 6.86 (t, J = 5.6 Hz, 1H, NH), 5.34 (t, J = 5.6 Hz, 1H, NH), 4.35 (d, J = 11.2 Hz, 2H, OCH2), 4.31 (d, J = 11.2 Hz, 2H, OCH2), 4.17 (d, J = 12 Hz, 4H, 2OCH2), 3.67 (d, J = 12 Hz, 4H, 2OCH2), 3.34 (q, J = 4.8 Hz, 2H, NCH2), 3.25 (q, J = 4.8 Hz, 2H, NCH2), 1.43 (s, 9H, 3CH3), 1.37 (s, 6H, 2CH3), 1.28 (s, 6H, 2CH3), 1.25 (s, 3H, CH3), 1.10 (s, 6H, 2CH3).
Synthesis of HO-G2-NHBoc.
The ketal-G2-NHBoc (7.54 g, 12.82 mmol) was dissolved in 50 mL of methanol and Dowex-H+ resin (15 g) was added. The reaction was allowed to stir at room temperature for 8 h. Then, the residue of Dowex-H+ was filtered off and the solvent was evaporated under reduced pressure and the product was obtained as a yellow solid in 95.7% yield (6.24 g). 1H NMR in DMSO-d6, δ(ppm): 7.79 (t, 1H, J = 5.6 Hz, NH), 6.80 (t, 1H, J = 5.6 Hz, NH), 4.65 (s, 4H, OH), 4.10 (d, J = 10.8 Hz, 4H, 2OCH2), 3.42 (d, J = 10 Hz, 8H, 4OCH2), 3.08 (q, J = 6 Hz, 2H, NCH2), 2.97 (q, J = 6 Hz, 2H, NCH2), 1.37 (s, 9H, 3CH3), 1.13 (s, 3H, CH3), 1.01 (s, 6H, 2CH3).
Synthesis of G2-NHBoc.
HO-G2-NHBoc (0.20 g, 0.40 mmol) and Fmoc-glycine (0.71 g, 2.40 mmol) were dissolved in 48 mL of a 5:1 CH2Cl2/DMF solution. After dissolution, 0.69 g (3.60 mmol) of EDC and DMAP (0.15 g, 1.20 mmol) were added into the reaction mixture. The mixture was stirred for 12 h at room temperature under a nitrogen atmosphere. Then, the solvent was evaporated under reduced pressure and the residue was dissolved in EtOAc, and the organic phase was washed with 1 M NaHSO4 (3 × 50 mL), 10% Na2CO3 (3 × 50 mL), and saturated brine (50 mL). The organic phase was dried with MgSO4 and filtered, and the filtrate was evaporated to give a yellow-green solid in 92.3% yield (0.60 g). 1H NMR in DMSO-d6, δ(ppm): 9.38 (t, 1H, J = 5.6 Hz, NH), 7.99 (t, 1H, J = 5.6 Hz, NH), 7.89 (d, J = 7.6 Hz, 8H, arom H), 7.76 (t, J = 5.6 Hz, 4H, 4NH), 7.70 (d, J = 7.6 Hz, 8H, arom H), 7.41 (t, J = 7.6 Hz, 8H, arom H), 7.32 (t, J = 7.6 Hz, 8H, arom H), 4.29 (d, J = 6.4 Hz, 8H, 4NCH2), 4.21 (t, J = 6.4 Hz, 4H, 4arom-CH), 4.17 (s, 12H, 6OCH2), 3.76 (d, J = 6 Hz, 8H, 4NCH2), 3.20 (s, 4H, 2NCH2), 1.23 (s, 9H, 3CH3), 1.13 (s, 3H, CH3), 1.11 (s, 6H, 2CH3). ESI-MS(m/z): calculated for C90H92N6O23 = 1624.62; found, [M + Na]+ = 1647.61.
Synthesis of G2-NH2.
G2-NHBoc (2.70 g, 1.66 mmol) was dissolved in 30 mL of a 1:1 TFA/CH2Cl2 solution at 0 °C, after stirring for 6 h, and the solution was then neutralized with a saturated aqueous NaHCO3 solution, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous MgSO4 and concentrated in a vacuum, and the product was obtained as a pale yellow solid in 71.1% yield (1.80 g). 1H NMR in CDCl3, δ(ppm): 7.73 (m, 8H, arom H), 7.60 (m, 8H, arom H), 7.35 (m, 16H, arom H), 5.75 (m, 3H, NH), 5.33 (s, 2H, 2arom-CH), 3.85–4.46 (br, 32H, 6NCH2 and 10OCH2), 3.80 (s, 2H, 2arom-CH), 3.69 (m, 2H, NH2), 3.40 (m, 2H, NH), 1.36 (s, 3H, CH3), 1.28 (s, 6H, 2CH3). ESI-MS(m/z): calculated for C85H84N6O21, 1524.57; found, [M + H]+ = 1525.57.
Synthesis of bis-MPA-G2-b-PBLG.
To a solution of BLG-NCA (3.16 g, 12 mmol) in 8 mL of dry DMF was added a solution of G2-NH2 (0.46 g, 0.30 mmol) in 4 mL of dry DMF as the initiator. This solution was stirred for 48 h at 30 °C under a nitrogen atmosphere. The crude polymer was subsequently precipitated three times in diethyl ether and dried under vacuum. The product was obtained as a white solid in 64.4% yield (2.33 g). 1H NMR in DMSO-d6, δ(ppm): 8.05–8.55 (br, NH), 7.05–7.93 (br, arom H), 4.85–5.20 (br, arom-CH2), 4.10–4.35 (br, OCH2), 3.81–4.10 (br, NCH), 3.71–3.80 (br, NCH2), 3.25–3.45 (br, NCH2), 2.35–2.65 (br, COCH2), 1.75–2.33 (br, CH and CH2), 1.24 (s, CH3), 1.12 (s, CH3). IR (KBr, cm−1): 3293 (N–H), 3034, 2954 (C–H), 1734 (CO), 1653 (CO), 743, 698 (Ar).
Synthesis of bis-MPA-G2-b-PHEG.
2.10 g of bis-MPA-G1-b-PBLG and 0.90 g of 2-hydroxypyridine were dissolved in 18 mL of dry DMF. Then ca. 18 mL of ethanolamine was added dropwise. After being stirred for 24 h at 40 °C under a nitrogen atmosphere, the solution was precipitated into ca. 400 mL of diethyl ether. The precipitate was redissolved in water, dialyzed (molecular weight cut off 3500), and subsequently freeze-dried and the product was obtained as a white solid in 65% yield (1.16 g). 1H NMR in DMSO-d6, δ(ppm): 7.35–8.30 (br, NH and arom H), 4.64–4.80 (br, OH), 4.10–4.35 (br, OCH2 and NCH2), 3.36–3.48 (br, CH2O and NCH2), 3.03–3.20 (br, NCH2), 2.06–2.25 (br, CH2CO), 1.60–1.98 (br, CH and CH2), 1.24 (s, CH3), 0.98 (s, CH3). IR (KBr, cm−1): 3418 (O–H), 3093, 2935 (C–H), 1653 (CO).
Critical micelle concentration (CMC)
Fluorescence measurements were conducted with a Hitachi F-4500 luminescence spectrophotometer using pyrene as a fluorescent probe. Firstly, the micelles were prepared from G-b-PHEG and bis-MPA-Gn-b-PHEG (n = 1, 2) according to the literature.3 Then, aliquots of pyrene solution (2 × 10−4 M in acetone, 5 μL) were added to volumetric flasks. After acetone was evaporated, aqueous solutions of the copolymer at different concentrations were added to the flasks to make the final pyrene concentration of 2 × 10−7 M. The solutions containing pyrene were kept at room temperature for 24 h before measurements.
Dye Nile red loading and release
G-b-PHEG (10 mg) was dissolved in PBS buffer (50 mM, pH 6.5) to give a concentration of 250 mM. The solution was sonicated for 30 min and then filtered through a 0.45 μm nylon syringe filter. 120 μL of 5 mM Nile red in acetone was poured to 3 mL of PBS and then 250 mM G-b-PHEG stock solution was added. After stirring for 6 h at 250 rpm and standing overnight, the mixture was centrifuged for 10 min at 10000 rpm, and the supernatant liquid was gathered for further experiment. 1.0 mL (or 1.5 mL) of this solution was accurately transferred to a quartz cuvette and a fluorescence emission scan was performed (t = 0), and then 1 mL (or 0.5 mL) of papain stock solution (80 μM, prepared as described by Romberg et al.48) was added to give a final concentration of 40 μM and 20 μM of papain. Fluorescence scans were performed every 30 min. The emission spectra were recorded from 575 to 800 nm with an excitation wavelength of 550 nm. Excitation and emission slit widths were kept at 10 and 20 nm, respectively. bis-MPA-Gn-b-PHEG (n = 1, 2) was treated as mentioned above.
Particle size and transmission electron microscopy (TEM) measurements
Particle size measurements were performed with a ZataPALS particle size analyzer (Brookhaven Instruments Corp) operating at room temperature. A drop of micellar solution (1 mg mL−1, or Nile red loaded micelles, or dealt with papain stock solution) was placed on a FORMVAR-coated copper grid and dried before TEM measurement using a JEM-2100 transmission electron microscope at an acceleration voltage of 200 kV.
In vitro cytotoxicity testing
The in vitro cytotoxicity of G-b-PHEG and bis-MPA-Gn-b-PHEG was estimated by MTS assay against the human liver cancer cell line SMMC-7721 according to the literature.49
Results and discussion
Synthesis of the dendritic initiators
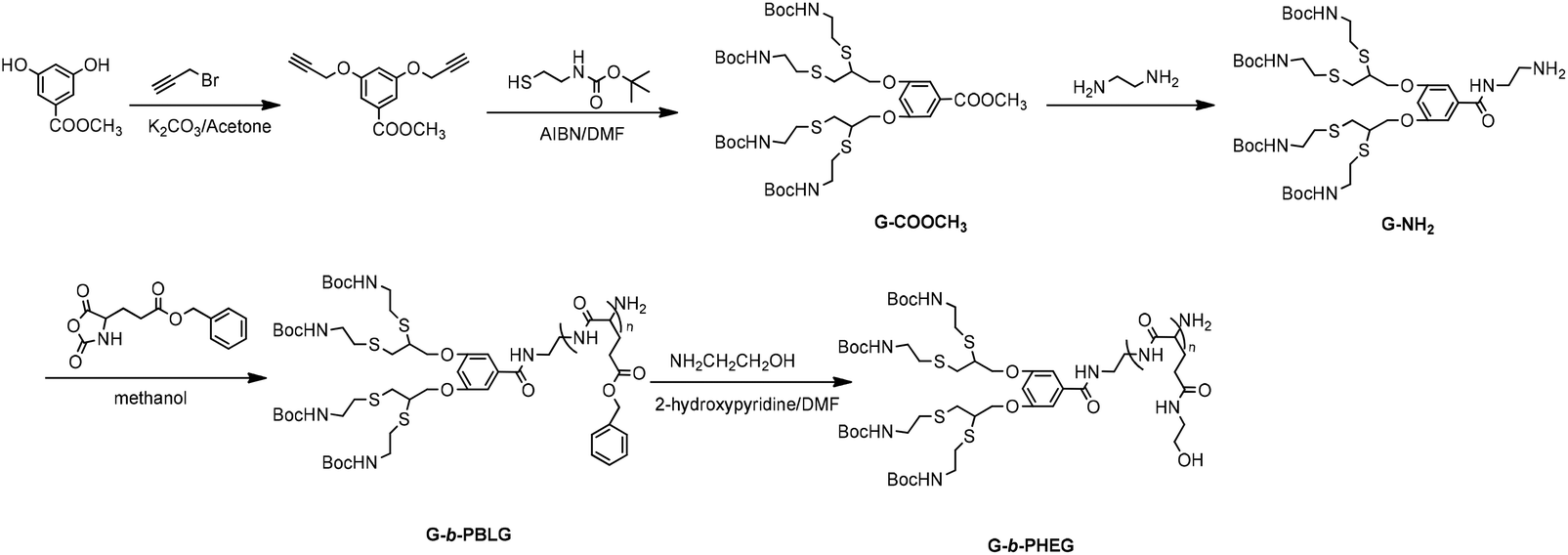
The synthesis procedures for the dendritic initiators are shown in Schemes 1 and 2. In Scheme 1, a dendron derived from cysteamine having an amino focal point G-NH2 was first synthesized by a thiol–yne reaction between 3,5-bis-(prop-2-ynyloxy) benzoate and 2-(Boc-amino) ethanethiol. In Scheme 2, isopropylidene-2,2-bis(oxymethyl)propionic acid was reacted with Boc mono-protected ethylenediamine in the presence of DCC/DMAP by aminolysis to afford the first-generation ketal-protected 2,2′-dimethylolpropionamide derivatives (ketal-G1-NHBoc). Then, the ketal was deprotected using TsOH·H2O to give the first-generation 2,2′-dimethylolpropionamide derivatives with terminal hydroxyl groups HO-G1-NHBoc. The second-generation 2,2′-dimethylolpropionamide derivatives with terminal hydroxyl groups HO-G2-NHBoc were synthesized via a two-step method consisting of (1) the condensation of HO-G1-NHBoc with isopropylidene-2,2-bis(oxymethyl)propionic anhydride and (2) the deprotection of ketal by DOWEX-H+. HO-Gn-NHBoc were converted into two-end protection, which can allow the selective deprotection by using a TFA–CH2Cl2 system and then dendritic initiators having an amino focal point Gn-NH2 were synthesized. We used 1H NMR, FTIR and ESI-MS to confirm the structure of all new compounds.
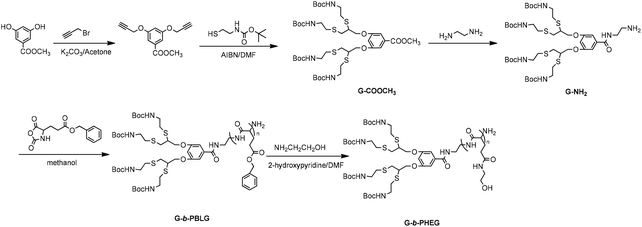 |
| | Scheme 1 Synthesis of amphiphilic linear-dendritic copolymer G-b-PHEG. | |
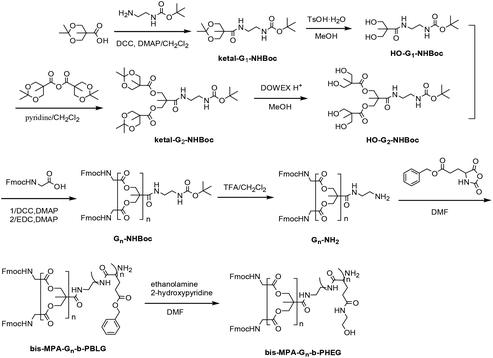 |
| | Scheme 2 Synthesis of amphiphilic linear-dendritic copolymer bis-MPA-Gn-b-PHEG (n = 1, 2). | |
Synthesis of linear-dendritic copolymers
It has been well-established that primary amine residues can be employed for the ROP of α-amino acid NCA, resulting in the formation of polypeptide polymers. The linear-dendritic diblock copolymers G-b-PBLG and bis-MPA-Gn-b-PBLG (n = 1, 2) based on a linear block of poly(benzyl-L-glutamate) (PBLG) and a dendron derived from cysteamine or an aliphatic polyester of 2,2′-dimethylolpropionic acid (bis-MPA) have been synthesized via a combination of the “dendron-first” strategy and the ring-opening polymerization of α-amino acid N-carboxyanhydrides (NCA). Then, G-b-PHEG and bis-MPA-Gn-b-PHEG (n = 1, 2) were synthesized via the aminolysis of PBLG with 2-aminoethanol in the presence of 2-hydroxypyridine as the catalyst. The products were characterized by FTIR and 1H NMR spectroscopy, and GPC.
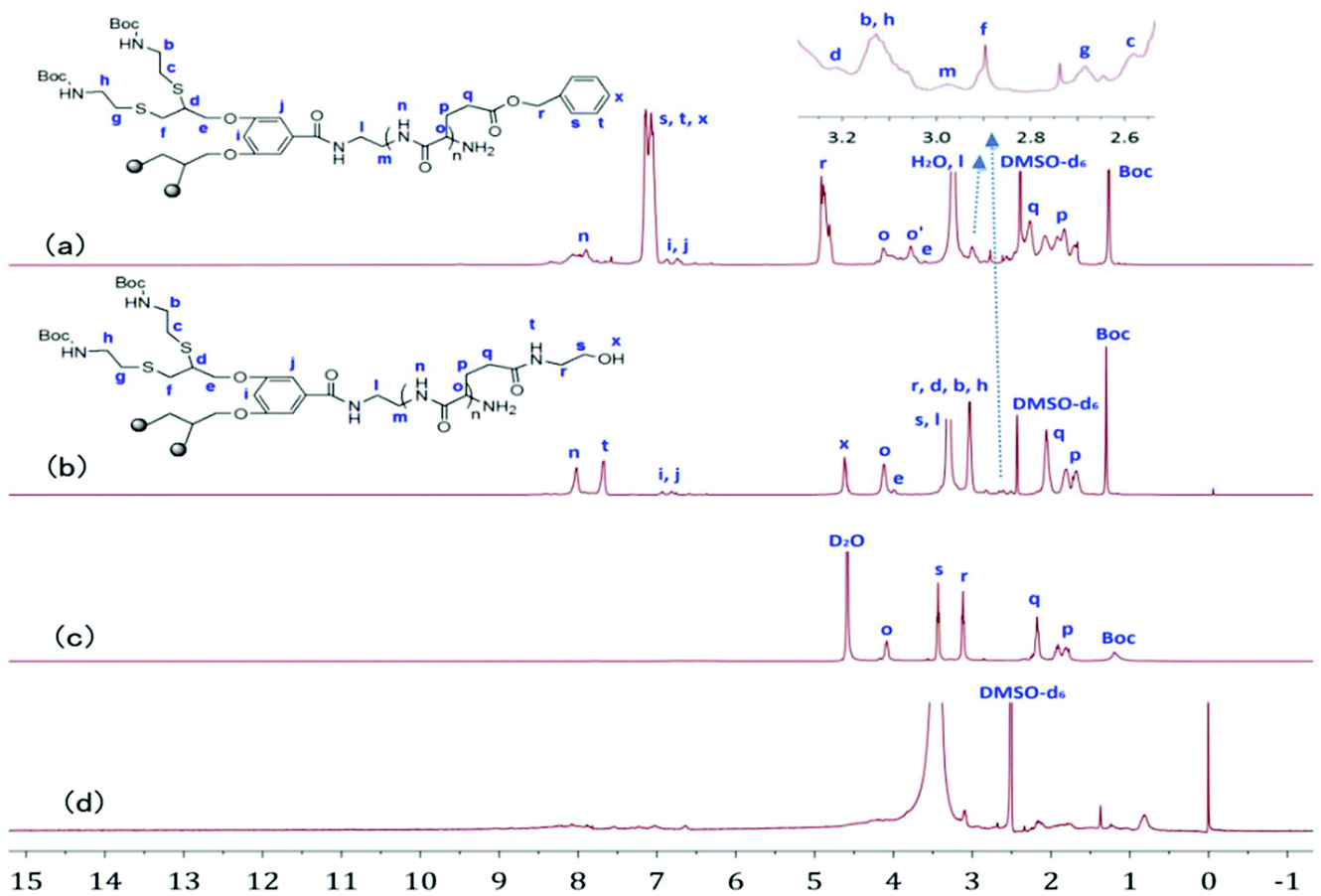
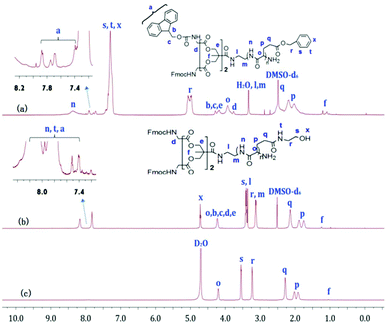
Fig. 1a and b show the 1H NMR spectra of G-b-PBLG and G-b-PHEG in DMSO-d6. All characteristic resonances in cysteamine dendron blocks and linear poly(α-amino acid)s blocks can be found. For G-b-PBLG (Fig. 1a), the peaks marked with letters from n to x could be assigned to the characteristic signals of the protons in PBLG repeat-units, such as n at 8.01–8.45 ppm (br, NH), s, t and x at 7.15–7.45 ppm (br, arom H), r at 4.87–5.15 ppm (br, arom-CH2), o at 3.81–4.40 ppm (br, CH), and p and q at 1.75–2.45 ppm (br, CH2). The resonance signals of dendron protons of the tert-butoxycarbonyl group (Boc), methylene groups (b, c, e, f, g, h, l, m), the methine group (d) and the phenyl group (i, j) occurred at 1.36, 2.58, 2.68, 2.89, 2.98, 3.13, 3.21, 3.73, 6.93, and 7.04 ppm, respectively. Compared to those of G-b-PBLG, we can clearly see that signals characteristic of the benzyloxy group at 7.15–7.45 and 4.87–5.15 ppm in the 1H NMR spectrum of G-b-PHEG disappeared, indicating the complete removal of the benzyloxy group. The IR spectra of G-b-PBLG and G-b-PHEG are shown in Fig. S1.† For G-b-PBLG, the absorption peak at 3296 cm−1 was assigned to the νN–H stretch vibration, the peak at 1735 cm−1 was assigned to the νCO stretch vibration, the peaks at 1654 cm−1 (νCO) were attributed to the amide group, and the absorptions at 746 and 697 cm−1 from the phenyl group were characteristic of the PBLG block carrying protecting groups. After the reaction with 2-aminoethanol, the peak at 1735 cm−1 of the νCO stretch vibration and the peaks at 746 and 697 cm−1 of the phenyl group in the IR spectrum (Fig. S1†) of G-b-PHEG totally disappeared, demonstrating the elimination of the phenyl group.
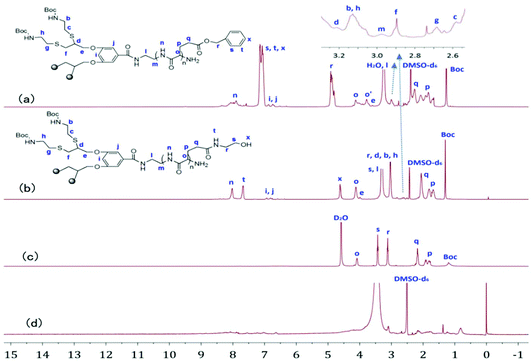 |
| | Fig. 1
1H NMR spectra of G-b-PBLG (a), and G-b-PHEG before (b, c) and after (d) the enzyme treatment in DMSO-d6 and D2O. | |
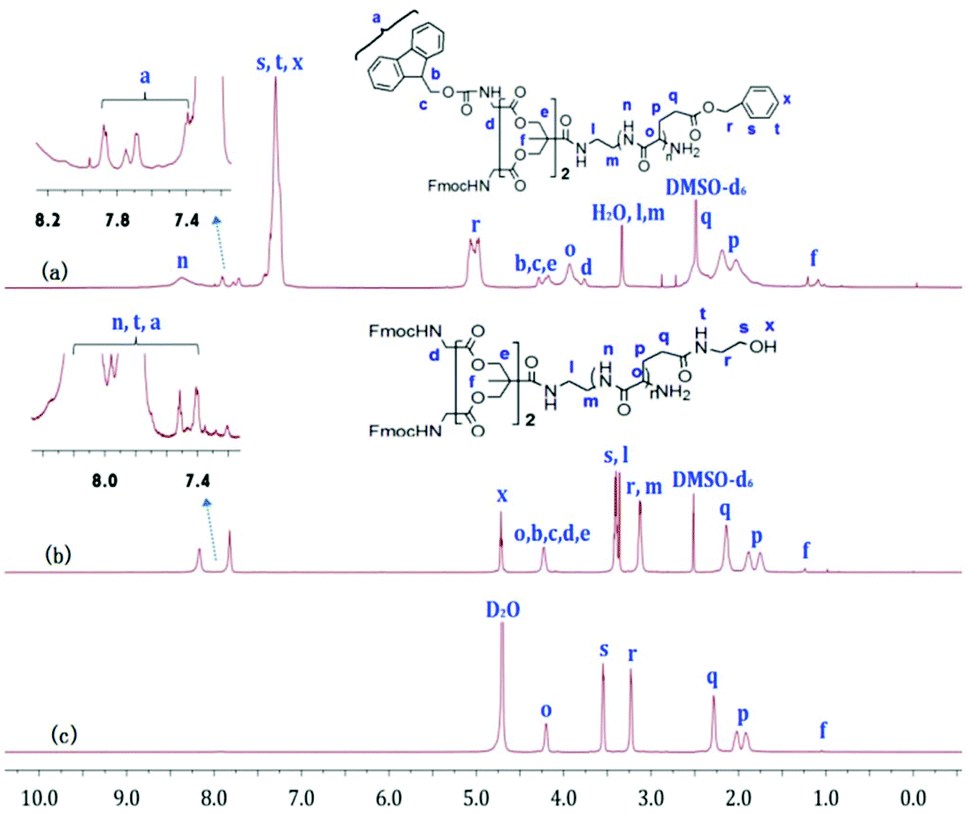
Fig. 2a, b and 3a, b show the 1H NMR spectra of bis-MPA-Gn-b-PBLG and bis-MPA-Gn-b-PHEG, respectively. In the case of bis-MPA-Gn-b-PBLG, the signals at 8.15–8.50, 7.15–7.40, 4.90–5.20, 3.80–4.0, 2.35–2.60 and 1.90–2.30 ppm were ascribed to the protons of PBLG repeat-units of the amide group (n), the phenyl group (s, t, x), the methylene group of benzyl (r), the α-methine group (o), the β-methylene group (p) and the γ-methylene group (q). Signals at 7.35–7.93, 4.07–4.35, 3.73–3.81, 3.10–3.50, and 1.23 ppm are all characteristic signals of the dendron. When two generations of block copolymers of bis-MPA-Gn-b-PBLG (n = 1, 2) were reacted with 2-aminoethanol, we found that signals characteristic of the benzyloxy group of the PBLG building block at about 7.05–7.50 and 4.85–5.20 ppm (Fig. 2b and 3b) in the 1H NMR spectra of bis-MPA-Gn-b-PHEG disappeared as well. In the FTIR spectra, we see that the δC–H vibration of the benzyl at about 743 and 698 cm−1 disappeared and appeared at the vibration of O–H from bis-MPA-Gn-b-PHEG (n = 1, 2) at about 3418 cm−1. It shows that PBLG has been reacted with 2-aminoethanol to get the linear-dendritic diblock copolymers bis-MPA-Gn-b-PHEG (n = 1, 2).
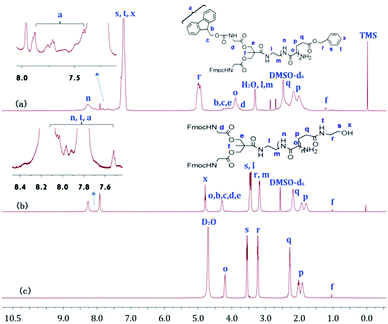 |
| | Fig. 2
1H NMR spectra of bis-MPA-G1-b-PBLG (a), and bis-MPA-G1-b-PHEG in DMSO-d6 (b) and D2O (c). | |
 |
| | Fig. 3
1H NMR spectra of bis-MPA-G2-b-PBLG (a), and bis-MPA-G2-b-PHEG in DMSO-d6 (b) and D2O (c). | |
GPC analysis revealed monomodal peaks for the copolymers (Fig. 4) with narrow molecular weight distributions (Mw/Mn ≤ 1.20; Table 1). Based on the integral ratio of the peaks of the methyl protons of Boc in the cysteamine dendron at 1.36 ppm and the methylene protons r of benzyloxy in PBLG at 4.87–5.15 ppm in the 1H NMR spectrum of G-b-PHEG (Fig. 1a), the degree of polymerization (DP) of G-b-PBLG was calculated to be 8, which was in fairly good agreement with the theoretical DP. The theoretical degree of polymerization was calculated by the method in which the molecular weight of the initiator is subtracted from the theoretical molecular weight of the copolymers and then divided by the molecular weight of BLG. It can be calculated that bis-MPA-G1-b-PBLG and bis-MPA-Gn-b-PBLG have DPs of 32 and 33 by the integral ratio of the peaks of f with o (Fig. 2a) and f with r (Fig. 3a), respectively. Furthermore, on the basis of DPs, we can conclude that the molecular weights (Mn,NMR) of G-b-PHEG and bis-MPA-Gn-b-PHEG (n = 1, 2) are lower than their precursor polymers, which are in accordance with their Mn,th theoretical values. It should be noted that the Mn,GPC values of G-b-PHEG and bis-MPA-Gn-b-PHEG were quite different from their Mn,th and Mn,NMR values (Table 1). The reason may be attributed to the difference in the hydrodynamic volume of the amphiphilic linear-dendritic block copolymers and the linear standards used for calibration.50–52
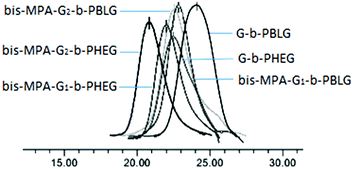 |
| | Fig. 4 GPC traces of copolymers. | |
Table 1 Characterization of copolymers
| Sample |
M
n,tha (g mol−1) |
M
n,GPCb (g mol−1) |
M
n,NMR (g mol−1) |
PDI = Mw/Mn |
Conversion (%) |
DPc |
|
The theoretical molecular weight was calculated by the formula: Mn,th = Mmonomer × ([monomer]/[initiation]) × Conversion% + Minitiation.
M
n and PDI were determined by GPC.
The degree of polymerization of the copolymers was calculated using Mn,NMR.
|
| G-b-PBLG |
2915 |
2840 |
2733 |
1.20 |
49 |
8 |
| G-b-PHEG |
2539 |
8518 |
2357 |
1.05 |
— |
— |
| Bis-MPA-G1-b-PBLG |
7832 |
7670 |
7747 |
1.08 |
67 |
32 |
| Bis-MPA-G1-b-PHEG |
6328 |
10950 |
6245 |
1.07 |
— |
— |
| Bis-MPA-G2-b-PBLG |
8307 |
9295 |
8756 |
1.17 |
64 |
33 |
| Bis-MPA-G2-b-PHEG |
6756 |
16696 |
7205 |
1.09 |
— |
— |
Formation of micelles from amphiphilic linear-dendritic block copolymers
G-b-PHEG and bis-MPA-Gn-b-PHEG are composed of a hydrophobic dendron derived from cysteamine with Boc end groups or an aliphatic polyester of 2,2′-dimethylolpropionic acid (bis-MPA) and hydrophilic poly(hydroxyethyl L-glutamine) (PHEG). Therefore, they are expected to be able to self-assemble into micelles in an aqueous solution. To examine the aqueous assembly of the amphiphilic block copolymers, 1H NMR spectroscopy, transmission electron microscopy (TEM), particle size analysis and fluorescence spectroscopy were employed. The 1H NMR spectra of G-b-PHEG in D2O (Fig. 1c) showed that the intensity of the proton peak at 1.36 ppm originating from tert-butoxy-carbonyl (Boc) was dramatically reduced compared to the one in DMSO-d6 (Fig. 1b). The 1H NMR spectra of bis-MPA-Gn-b-PHEG in D2O (Fig. 2c and 3c) showed that the benzene proton peaks of the Fmoc protecting group at about 7.01–8.20 ppm disappeared, indicating that the mobility of the dendron segments has been restricted. Similar phenomena were also reported in other micelle systems.53 These results suggested that amphiphilic G-b-PHEG and bis-MPA-Gn-b-PHEG diblock copolymers may associate to form the core–shell micellar structure in water in which the dendron derived from cysteamine with Boc end groups or an aliphatic polyester of 2,2′-dimethylolpropionic acid (bis-MPA) constituted the micellar inner core and the PHEG block constituted the micellar outer shell. The characteristics of the block copolymer micelles in the aqueous phase were further investigated by fluorescence techniques using pyrene as a probe. Fig. S2 and Fig. 5 show the emission spectra of pyrene in the G-b-PHEG and bis-MPA-Gn-b-PHEG solutions at various concentrations and the intensity ratio of I384/I373versus the logarithm of the concentrations of G-b-PHEG and bis-MPA-Gn-b-PHEG. Below a certain concentration, I384/I373 is constant, and above this concentration, an abrupt increase was observed with increasing copolymer concentrations, indicating the formation of micelles and the transfer of pyrene into the hydrophobic core of micelles. The concentration at the abrupt increase in the ratio of I384/I373 was defined as the critical micelle concentration (CMC). The CMC value is 0.045, 0.195, and 0.053 mg mL−1 for G-b-PHEG, bis-MPA-G1-b-PHEG and bis-MPA-G2-b-PHEG, respectively. The CMC values of G-b-PHEG and bis-MPA-G2-b-PHEG are low, providing evidence for an apparent stability of micelles and allowing their use in very dilute aqueous milieu such as body fluids.54
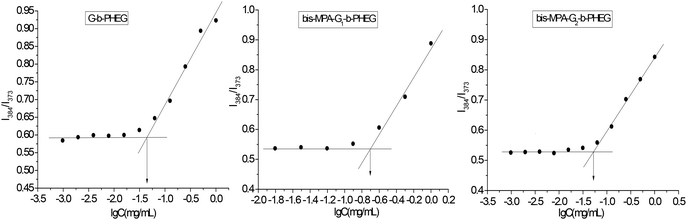 |
| | Fig. 5 Plots of I384/I373versus logarithm of the concentration of G-b-PHEG, bis-MPA-G1-b-PHEG and bis-MPA-G2-b-PHEG. | |
The micellar morphology and size were studied using a transmission electron microscope (TEM). As shown in Fig. 6a, G-b-PHEG self-assembles into spherical micelles in aqueous solution and the total size of the observed micelles is not uniform with diameters ranging from 60 to 140 nm. Bis-MPA-G2-b-PHEG aggregates to form vesicles with a mean diameter of 100–250 nm and a wall thickness of 10 nm (Fig. 6c).
 |
| | Fig. 6 TEM images of the formed micelles by G-b-PHEG before (a) and after (b) the enzyme treatment, bis-MPA-G2-b-PHEG (c, without Nile red), loaded with Nile red (d) and loaded with Nile red after the enzyme treatment (e). | |
Analysis of enzymatically degraded copolymers
To investigate the enzyme-responsive disassembly of the copolymer micelles, different concentrations of papain stock solution were introduced into the micellar aqueous solutions at 37 °C in pH 6.5 PBS. The enzyme-responsive feature can be clearly evidenced with the naked eye. For G-b-PHEG, after 54 h of incubation, the colorless and transparent copolymer micelle solution slowly turned turbid (Fig. 7a). The turbidity of the solution increased with the concentration of papain solution. The turbid solution was freeze-dried and subsequently analyzed by 1H NMR spectroscopy. As shown in Fig. 1d, the resonance signals of the protons of the amino acid segment (PHEG) almost disappeared, and the absorption peaks of dendron blocks at 6.83–7.15, 4.05–4.11, 2.55–3.25 and 1.36 ppm were observed. TEM analysis showed that the spherical nanostructures could no longer be observed for G-b-PHEG after the enzyme treatment of the micellar solution (Fig. 6b). These results suggested that hydrophilic PHEG can be totally degraded by papain. This change in amphiphilicity should result in the destabilization of the micellar aggregates, leading to their disassembly and release of the insoluble dendron.
 |
| | Fig. 7 Variation of the color of the blank micelle solution (a), and Nile red loaded micelle solution (b) of G-b-PHEG since the addition of different concentrations of papain after 54 h of incubation. | |
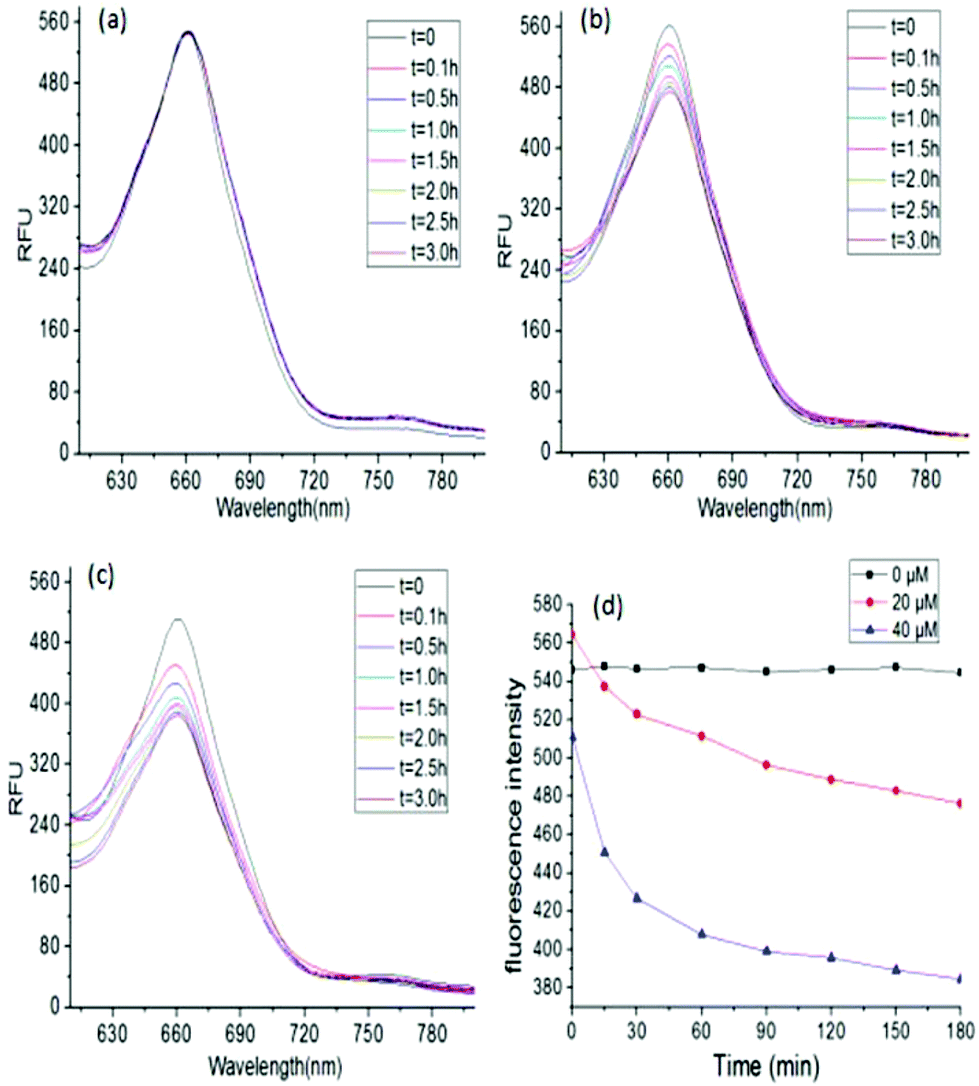
The encapsulation of a hydrophobic dye, Nile red, by G-b-PHEG and bis-MPA-Gn-b-PHEG micelles and the enzyme-induced release were also investigated. Fig. 7b shows that the encapsulation of Nile red by micelles made the micellar solution appear pink. Upon the addition of an 80 μM solution of papain, after 54 h of incubation, the color of the solution gradually became lighter from 0 to 40 μM and generated a precipitate (Fig. 7b) due to the released Nile red which is insoluble in water. Fig. 8 and 9 show the fluorescence emission spectra of the micellar solution with the encapsulated Nile red after the addition of an 80 μM solution of papain. For comparison, the emission spectrum of the blank micelle solution prepared under the same conditions is also shown. Time-dependent decreases in fluorescence were observed for G-b-PHEG and bis-MPA-Gn-b-PHEG in the presence of papain, indicating that the Nile red molecules were released from the hydrophobic core of the micelles. For G-b-PHEG, during the first 3 h of incubation, the fluorescence intensity decreases rapidly and the concentration of 40 μM of papain was much faster than 20 μM (Fig. 9d) of papain, it should be noted that there were no decreases in the absence of papain. TEM measurements further support the enzyme triggered disassembly of the micelles. Fig. 6d shows that the size and the wall thickness of the vesicles made from bis-MPA-Gn-b-PHEG with entrapped Nile red dye become bigger than those from the copolymer without Nile red. The reason may be the encapsulation of the hydrophobic dye. But the vesicles could not be observed from the aqueous solution after treatment with papain (Fig. 6e). Moreover, we found that the disassembly of the micelles was dependent on the generation of dendrons in bis-MPA-Gn-b-PHEG (Fig. 10): the higher the generation, the slower the disassembly rate. These experiments demonstrated that the micelles can disassemble and release the encapsulated molecular cargo upon enzymatic activation. In addition, the disassembly and release rates depended on the concentration of papain and the generation of dendrons in LDBCs.
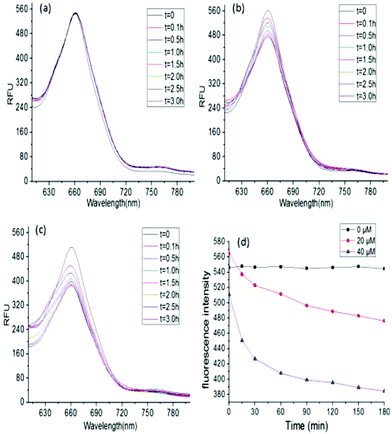 |
| | Fig. 8 Fluorescence spectra of Nile red in G-b-PHEG micelles at different concentrations of papain, 0 μM (a), 20 μM (b) and 40 μM (c). The effect of the concentrations of papain in G-b-PHEG on the disassembly rates (d). | |
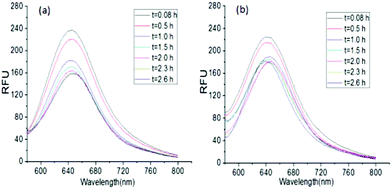 |
| | Fig. 9 Fluorescence spectra of Nile red in bis-MPA-G1-b-PHEG (a) and bis-MPA-G2-b-PHEG (b) micelles after the incubation with 80 μM papain. | |
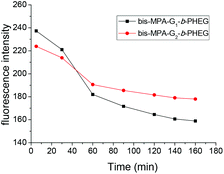 |
| | Fig. 10 Generation of dendrons in bis-MPA-Gn-b-PHEG has an effect on the disassembly rates. | |
The enzyme-induced disassembly was also confirmed using particle size measurements. The particle size of G-b-PHEG micelles showed that the micellar diameters were much larger than those examined by TEM (Fig. 6 and Table 2). This may be because of the particle size of micelles in an aqueous solution and TEM reflects the conformations in the dry state. In addition, we found that the particle size of copolymer micelles became bigger when hydrophobic Nile red was successfully loaded into the spherical aggregates (Table 2). After the enzyme treatment of the micellar solution with Nile red or without Nile red, the particle size of G-b-PHEG and bis-MPA-Gn-b-PHEG micelles became much more larger.11,12 It means that after the enzyme-responsive disassembly, the residual hydrophobic dendron may undergo aggregation in aqueous solutions.55
Table 2 The size of G-b-PHEG and bis-MPA-Gn-b-PHEG micellesa
| Sample |
Concentration of papain |
Effective diameter (nm) |
|
The size of the micelles was measured using a ZataPALS particle size analyzer.
|
| G-b-PHEG |
0 μM (blank micelles) |
380.3 |
| 20 μM (blank micelles) |
1286 |
| 40 μM (blank micelles) |
1443.6 |
| 0 μM (Nile red loaded micelles) |
1388.6 |
| 20 μM (Nile red loaded micelles) |
1899.5 |
| 40 μM ( Nile red loaded micelles) |
2095.2 |
| Bis-MPA-G1-b-PHEG |
0 μM (blank micelles) |
204.1 |
| Bis-MPA-G2-b-PHEG |
0 μM (blank micelles) |
245.8 |
| Bis-MPA-G1-b-PHEG |
40 μM (Nile red loaded micelles) |
807.8 |
| Bis-MPA-G2-b-PHEG |
40 μM (Nile red loaded micelles) |
3035.2 |
In vitro cytotoxicity of linear-dendritic block copolymers
The potential of the obtained linear-dendritic block copolymers (LDBCs) as a drug carrier was investigated from cytotoxicity studies. The in vitro cytotoxicity of G-b-PHEG and bis-MPA-G2-b-PHEG to the human liver cancer cell line SMMC-7721 was determined by MTS assay. As shown in Table 3, the cell viabilities were >95% for G-b-PHEG and >92% for bis-MPA-G2-b-PHEG at concentrations ranging from 50 to 800 μg mL−1 after 48 h incubation at 37 °C when compared with control values (without any polymer additive) (p < 0.05). The results suggest that the LDBCs have good biocompatibility with SMMC-7721 cells.
Table 3 Cell viability values after 48 h of incubation with G-b-PHEG and bis-MPA-G2-b-PHEG at 37 °C
| Concentration (μg mL−1) |
50 |
100 |
200 |
400 |
800 |
| Viability (%, G-b-PHEG) |
98.28 |
95.76 |
96.11 |
95.46 |
96.51 |
| Viability (%, bis-MPA-G2-b-PHEG) |
97.54 |
94.32 |
98.04 |
92.70 |
96.19 |
Conclusions
In summary, well-defined enzyme-responsive amphiphilic linear-dendritic diblock copolymers G-b-PHEG and bis-MPA-Gn-b-PHEG based on a hydrophilic linear block of poly(hydroxyethyl L-glutamine) (PHEG) and a hydrophobic dendron derived from cysteamine or an aliphatic polyester of 2,2′-dimethylolpropionic acid (bis-MPA) were successfully designed and synthesized for the first time via a combination of the “dendron-first” strategy and the ring-opening polymerization of α-amino acid N-carboxyanhydrides (NCA). These block copolymers assembled into nanospherical micelles or vesicles in the aqueous phase owing to their amphiphilic characteristics. The self-assembled micelles show good responsiveness in the presence of papain. After papain solution was added to the micellar aqueous solution, the dye molecules were released into the aqueous environment upon the disassembly of the micelles and their fluorescence intensity decreased, leading to the solutions becoming turbid and precipitate. The disassembly of the micelles was dependent on the concentration of papain and the generation of dendrons in LDBCs. The higher the enzyme concentration is, the faster the disassembly rate is, and the higher the generation, the slower the disassembly rate. In vitro cytotoxicity studies indicate that these LDBCs are well tolerated at the concentration tested in SMMC-7721 cell cultures. Due to the enzyme responsiveness and biocompatibility, these copolymers may have great potential in the field of drug delivery.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the support for this study from the National Natural Science Foundation of China (21264017 and 21564017).
Notes and references
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- V. Yesilyurt, R. Ramireddy and P. S. Thayumanavan, Angew. Chem., Int. Ed., 2011, 50, 3038–3042 CrossRef CAS PubMed.
- Y. M. Bi, C. X. Yan, L. D. Shao, Y. F. Wang, Y. C. Ma and G. Tang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3240–3250 CrossRef CAS.
- Y. Li, W. Xiao, K. Xiao, L. Berti, J. Luo, H. P. Tseng, G. Fung and K. S. Lam, Angew. Chem., Int. Ed., 2012, 51, 2864–2869 CrossRef CAS PubMed.
- L. L. Wu, Y. Zou, C. Deng, R. Cheng, F. H. Meng and Z. Y. Zhong, Biomaterials, 2013, 34, 5262–5272 CrossRef CAS.
- K. R. Raghupathi, M. A. Azagarsamy and S. Thayumanavan, Chem. – Eur. J., 2011, 17, 11752–11760 CrossRef CAS.
- M. Zelzer, S. J. Todd, A. R. Hirst, T. O. McDonald and R. V. Ulijn, Biomater. Sci., 2012, 1, 11–39 RSC.
- J. Liu, C. Detrembleur, M. De Pauw-Gillet, S. Mornet, E. Duguet and C. Jérôme, Polym. Chem., 2014, 5, 799–813 RSC.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- P. D. Thornton, R. J. Mart and R. V. Ulijn, Adv. Mater., 2010, 19, 1252–1256 CrossRef.
- T. H. Ku, M. P. Chien, M. P. Thompson, R. S. Sinkovits, N. H. Olson, T. S. Baker and N. C. Gianneschi, J. Am. Chem. Soc., 2011, 133, 8392–8395 CrossRef CAS.
- J. Y. Rao and A. Khan, J. Am. Chem. Soc., 2013, 135, 14056–14059 CrossRef CAS.
- J. M. Hu, G. Q. Zhang and S. Y. Liu, Chem. Soc. Rev., 2012, 43, 5933–5949 RSC.
- G. J. Habraken, M. Peeters, P. D. Thornton, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 3761–3769 CrossRef CAS.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS.
- I. Rosenbaum, A. J. Harnoy, E. Tirosh, M. Buzhor, M. Segal, L. Frid, R. Shaharabani, R. Avinery, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2015, 137, 2276–2284 CrossRef CAS.
- I. Gitsov, K. L. Wooley and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2010, 31, 1200–1202 CrossRef.
- F. Wurm and H. Frey, Prog. Polym. Sci., 2011, 36, 1–52 CrossRef CAS.
- M. G. Jeong, J. C. M. van Hest and K. T. Kim, Chem. Commun., 2012, 48, 3590–3592 RSC.
- N. Kalva, N. Parekh and A. V. Ambade, Polym. Chem., 2015, 6, 6826–6835 RSC.
- G. Whitton and E. R. Gillies, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 148–172 CrossRef CAS.
- H. Cai, G. Jiang, C. Chen, Z. Li, Z. Shen and X. Fan, Macromolecules, 2014, 47, 146–151 CrossRef CAS.
- A. S. Herves, R. Riguera and E. F. Megia, New J. Chem., 2012, 43, 205–210 RSC.
- W. Zhang, W. Jiang, D. Zhang, G. Bai, P. Lou and Z. Hu, Polym. Chem., 2015, 6, 2274–2282 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2011, 42, 6288–6308 Search PubMed.
- B. Guillerm, V. Darcos, V. Lapinte, S. Monge, J. Coudaneb and J. Robina, Chem. Commun., 2012, 48, 2879–2881 RSC.
- L. Yin, M. C. Dalsin, A. Sizovs, T. M. Reineke and M. A. Hillmyer, Macromolecules, 2012, 45, 4322–4332 CrossRef CAS.
- C. C. Lee and J. M. J. Fréchet, Macromolecules, 2005, 39, 476–481 CrossRef.
- C. Deng, G. Z. Rong, H. Y. Tian, Z. H. Tang, X. S. Chen and X. B. Jing, Polymer, 2005, 46, 653–659 CrossRef CAS.
- J. S. Guo, Y. B. Huang, X. B. Jing and X. S. Chen, Polymer, 2009, 50, 2847–2855 CrossRef CAS.
- J. D. Han, J. X. Ding, Z. C. Wang, S. F. Yan, X. L. Zhuang, X. S. Chen and J. B. Yin, Sci. China: Chem., 2013, 56, 729–738 CrossRef CAS.
- J. M. Metselaar, P. Bruin, L. W. T. de Boer, T. de Vringer, C. Snel, C. Oussoren, M. H. M. Wauben, D. J. A. Crommelin, G. Storm and W. E. Hennink, Bioconjugate Chem., 2003, 14, 1156–1164 CrossRef CAS.
- J. Y. Rao, Y. F. Zhang, J. Y. Zhang and S. Y. Liu, Biomacromolecules, 2008, 9, 2586–2593 CrossRef CAS.
- L. Chen, H. Y. Tian, J. Chen, X. S. Chen, Y. B. Huang and X. B. Jing, J. Gene Med., 2010, 12, 64–76 CrossRef CAS.
- K. D. Winne, E. Roseeuw, J. Pagnaer and E. Schacht, J. Bioact. Compat. Polym., 2004, 19, 439–451 CrossRef.
- A. Marre, K. Hoste, D. Bruneel and E. Schacht, J. Bioact. Compat. Polym., 1996, 11, 85–99 CrossRef.
- B. Romberg, J. M. Metselaar, L. Baranyi, C. J. Snel, R. Bünger, W. E. Hennink, J. Szebeni and G. Storm, Int. J. Pharm., 2007, 331, 186–189 CrossRef CAS.
- J. Chen, H. Y. Tian, Z. P. Guo, L. Lin, X. Dong, X. J. Zhu and X. S. Chen, J. Appl. Polym. Sci., 2011, 123, 2257–2265 CrossRef.
- W. W. Zhang, W. W. Jiang, D. L. Zhang, G. Y. Bai, P. X. Lou and Z. G. Hu, Polym. Chem., 2015, 6, 2274–2282 RSC.
- A. Carlmark, E. Malmstrom and M. Malkoch, Chem. Soc. Rev., 2013, 44, 5858–5879 RSC.
- C. C. Lee and J. M. J. Fréchet, Macromolecules, 2005, 39, 476–481 CrossRef.
- W. H. Daly and D. Poché, Tetrahedron Lett., 1988, 29, 5859–5862 CrossRef CAS.
- A. J. Brouwer, S. J. E. Mulders and R. M. J. Liskamp, Eur. J. Org. Chem., 2010, 1903–1915 Search PubMed.
- Y. Z. Kim and J. P. Kim, Synth. Commun., 2002, 32, 1601–1605 CrossRef CAS.
- J. Movellan, P. Urban, E. Moles, J. M. de la Fuente, T. Sierra, J. L. Serrano and X. F. Busquets, Biomaterials, 2014, 35, 7940–7950 CrossRef CAS.
- E. R. Gillies and J. M. J. Frechet, J. Am. Chem. Soc., 2002, 124, 14137–14146 CrossRef CAS.
- I. Dijkgraaf, A. Y. Rijiders, A. Soede, A. C. Dechesne, G. W. van Esse, A. J. Brouwer, F. H. Corstens, O. C. Boerman, D. T. Rijkers and R. M. Liskamp, Org. Biomol. Chem., 2007, 5, 935–944 RSC.
- B. Romberg, J. M. Metselaar, T. de Vringer, K. Motonaga, J. J. K. van den Bosch, C. Oussoren, G. Storm and W. E. Hennink, Bioconjugate Chem., 2005, 16, 767–774 CrossRef CAS.
- X. Peng, Z. Xiang, F. Du, J. Tan, L. Zhong and R. Sun, Cellulose, 2018, 25, 245–257 CrossRef CAS.
- H. Lee, J. A. Lee, Z. Poon and P. T. Hammond, Chem. Commun., 2008, 32, 3726–3728 RSC.
- B. S. Sumerlin, D. Neugebauer and K. Matyjaszewski, Macromolecules, 2005, 38, 702–708 CrossRef CAS.
- F. Samperi, S. Battiato, C. Puglisi, A. Scamporrino, V. Ambrogi, L. Ascione and C. Carfagna, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3615–3630 CrossRef CAS.
- J. Luo, K. Xiao, Y. Li, J. S. Lee, L. Shi, Y. Tan, L. Xing, R. H. Cheng, G. Liu and K. S. Lam, Bioconjugate Chem., 2010, 21, 1216–1224 CrossRef CAS.
- H. Wei, X. Z. Zhang, Y. Zhou, S. X. Cheng and R. X. Zhuo, Biomaterials, 2006, 27, 2028–2034 CrossRef CAS.
- M. Y. Qi, G. Z. Wu, M. L. Sha and Y. S. Liu, Radiat. Phys. Chem., 2008, 77, 1248–1252 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01231h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*