
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient synthesis and characterisation of the amyloid beta peptide, Aβ1–42, using a double linker system†
Johanes K.
Kasim
 a,
Iman
Kavianinia
abc,
Jin
Ng
ad,
Paul W. R.
Harris
*abc,
Nigel P.
Birch
ad and
Margaret A.
Brimble
*abc
a,
Iman
Kavianinia
abc,
Jin
Ng
ad,
Paul W. R.
Harris
*abc,
Nigel P.
Birch
ad and
Margaret A.
Brimble
*abc
aSchool of Biological Sciences, The University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand. E-mail: paul.harris@auckland.ac.nz
bMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, 3 Symonds St, Auckland 1010, New Zealand
cSchool of Chemical Sciences, The University of Auckland, 23 Symonds Street, Auckland 1010, New Zealand. E-mail: m.brimble@auckland.ac.nz
dBrain Research New Zealand Rangahau Roro Aotearoa and Centre for Brain Research, Auckland 1010, New Zealand
First published on 26th November 2018
Abstract
The amyloidogenic Aβ42 peptide was efficiently prepared using a double linker system, markedly improving solubility and chromatographic peak resolution, thus enabling full characterisation using standard techniques. The tag was readily cleaved with sodium hydroxide and removed by aqueous extraction, affording Aβ42 in high purity and yield for biophysical characterisation studies.
Alzheimer's disease (AD) is a degenerative disorder of the brain that presents as the leading cause of dementia worldwide.1 One of the disease's neuropathological features is senile plaques,2 which predominantly consist of amyloid beta (Aβ) peptide. Aβ deposition is central to the ‘amyloid cascade hypothesis’ first proposed by Hardy and Higgins, which postulates that aberrant amyloid metabolism precedes other biochemical alterations associated with the disease.3 Endogenous Aβ peptide is derived from enzymatic processing of the transmembrane Amyloid Precursor Protein (APP) by the secretase family of enzymes. APP processing occurs via two main pathways: non-amyloidogenic and amyloidogenic (Fig. 1).
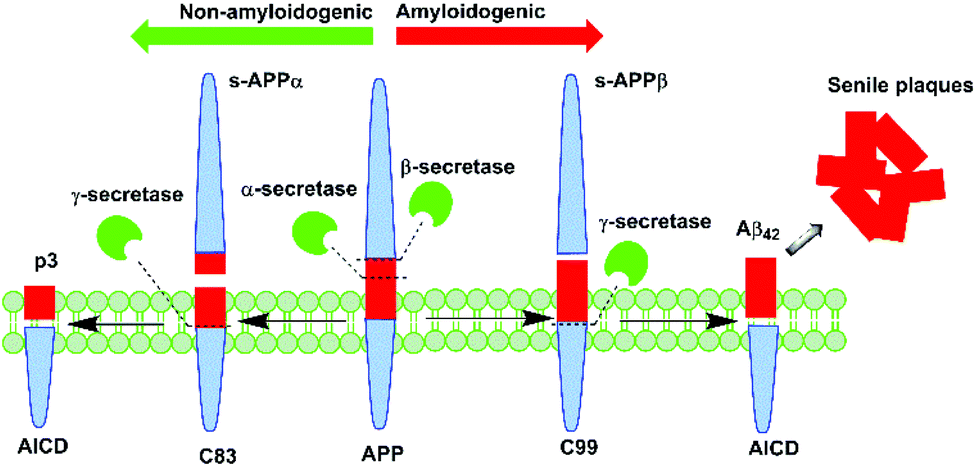 | ||
| Fig. 1 Aβ peptide is generated in vivo from APP processing by β- and γ-secretase via the amyloidogenic pathway. | ||
It is understood that in the disease state, there is a preferential shift towards the latter process, which results in the excessive production of Aβ peptide, leading to its gradual aggregation into plaques. In this pathway, APP is initially cleaved by β-secretase to yield C99, a 99-residue peptide fragment that is further processed by γ-secretase to afford either the 40- or 42-residue variant of Aβ peptide.4 The present knowledge is that the longer variant of the peptide, Aβ42, which is preferentially produced in AD, displays greater neurotoxicity and propensity to aggregate than does the 40-residue variant, Aβ40.5 Thus, it appears that Aβ42 may present as a more valuable target for novel or repurposed pharmaceutical agents in the treatment of AD.
In order to establish a more comprehensive understanding of the extent of contribution of Aβ42 in AD, it is desirable to develop methods that enable synthesis of the peptide in useful quantities and purity for further analysis. However, the peptide's natural inclination to aggregate during solid phase peptide synthesis (SPPS) and in solution has continually hampered its efficient synthesis and characterisation.6 Various methods have been adopted to successfully synthesise Aβ42. These primarily employ Fmoc based SPPS, and include the use of DBU as Fmoc deprotection reagent,7 DMSO as a coupling cosolvent,8 use of the ‘O-acyl isopeptide’ method,9 use of poly(ethyleneglycol)-based, low-loading resins such as ChemMatrix,10 high temperature SPPS,11 high-efficiency solid phase peptide synthesis (HE-SPPS),12 linking of lysine residues to the peptide C-terminus that can be readily removed after purification using immobilised carboxypeptidase B,13 incorporation of an oligoethyleneglycol-containing photocleavable lysine tag14 and, most recently, the development of an Aβ42 oligomer mimic containing an oxime switch, which enables a regulated shift between oligomeric and fibrillar states of the peptide.15
Even though the extensive range of synthetic procedures to access Aβ42 outlined above have advanced the field, there are nevertheless ongoing challenges, including effective recovery of the peptide since crude yields are typically very low. The characterisation of Aβ42 also remains a persisting conundrum to date. Conventional reversed phase-high performance liquid chromatography (RP-HPLC) analysis (room temperature, TFA-containing mobile phases) of crude Aβ42 yields a broad, unresolved, and asymmetrical chromatographic peak, indicative of Aβ42 aggregation. Alternative techniques that have been developed for the analysis of ‘difficult peptides’ include the modification of peptide net charge using alkaline buffers,16 high temperature HPLC,17 and the incorporation of removable tags that effectively address solubility issues,18,19 in some cases permitting HPLC analysis under otherwise unfavourable acidic conditions.
In an exploratory synthesis of Aβ42, we also encountered significant challenges with both the synthesis and purification of the peptide. We therefore sought to mitigate these shortcomings by improving the efficiency of the synthetic protocol employed. Herein, we describe the Fmoc SPPS of Aβ42 using pseudoprolines as an aggregation disruptor, together with a C-terminal lysine solubilising tag, linked via the base-labile HMBA linker, to improve overall tractability. This enabled us to readily prepare multi-milligram amounts of Aβ42 using standard purification techniques.
We began by preparing Aβ42 using a linear Fmoc SPPS protocol on a 0.1 mmol scale, employing 20% piperidine in DMF (v/v) for Fmoc deprotection and O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU)/N-methylmorpholine (NMM) as the coupling reagents, but only managed to recover 33% crude yield (150 mg, based on 0.1 mmol ChemMatrix resin loading, see ESI Fig. S1†). Pseudoprolines introduce ‘kinks’ in the peptide sequence,20 reducing the likelihood of peptide aggregation on resin, and affording a correspondingly higher crude yield. To the best of our knowledge, there are no reports on Aβ42 synthesis using this approach. Repeating the synthesis of Aβ42 with pseudoprolines afforded a crude yield greater than that obtained previously under standard Fmoc/tBu SPPS (259 mg, 57% based on 0.1 mmol ChemMatrix resin loading, see ESI Fig. S2†).
The propensity of Aβ42 to aggregate during HPLC, however, rendered fractionation of the pure peptide from its closely-eluting peptidic impurities extremely difficult. As such, purification by RP-HPLC using acidic mobile phases required execution in relatively small batches (up to 5 mg crude per batch), which represented a major bottleneck to access this biologically important peptide. Subsequently, we devised an enhanced strategy of incorporating a lysine-based cationic tag, which we envisaged would enhance solubility, and reduce internal aggregation of the peptide during the purification process. Previously, this tag has been successfully incorporated in the synthesis of the aggregating cancer protein NY-ESO-121 and the peptide hormone vesiculin.22 Our synthesis of Aβ42 peptide 1 is outlined in Scheme 1, and was undertaken using the PEG-based ChemMatrix resin, utilising the Fmoc/tBu SPPS strategy.
 | ||
| Scheme 1 Preparation of synthetic Aβ42 employing a double linker system, with pseudoprolines replacing underlined residues in the resin-bound peptide sequence. | ||
The synthesis commenced with anchoring the Rink amide linker to ChemMatrix resin, using N,N′-diisopropylcarbodiimide (DIC)/6-chloro-1-hydroxybenzotriazole (6-Cl-HOBt) as coupling reagents, followed by removal of the temporary Fmoc protecting group on resin using 20% piperidine in DMF (v/v). The hexalysine tag was then assembled by sequential attachment of Fmoc-Lys(Boc)-OH using HATU and N,N′-diisopropylethylamine (DIPEA) to form peptidyl resin 2. Following Fmoc deprotection, HMBA linker was coupled to the N-terminus of the preceding lysine residue using DIC/6-Cl-HOBt to complete the double linker system 3. Peptide assembly was then initiated by coupling Fmoc-Ala-OH to the free hydroxyl group using DIC/4-dimethylaminopyridine (DMAP) to generate the base-labile ester bond of 4. Elongation of the peptide sequence, with pseudoproline incorporation as indicated, was then continued using HATU/NMM as coupling reagents on a Tribute Peptide Synthesizer (Protein Technologies Inc., US) to yield the resin-bound peptide 5. Following final Fmoc deprotection, the peptide was cleaved from the resin using a standard cleavage cocktail (TFA/TIPS/EDT/H2O, 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2.5:2.5; v/v/v/v), precipitated in cold diethyl ether, and lyophilised to afford crude 6 (303 mg, 56% crude yield based on 0.1 mmol ChemMatrix resin loading, see ESI Fig. S3†).
:1:2.5:2.5; v/v/v/v), precipitated in cold diethyl ether, and lyophilised to afford crude 6 (303 mg, 56% crude yield based on 0.1 mmol ChemMatrix resin loading, see ESI Fig. S3†).
Purification of 6 (90 mg, 0.017 mmol) was undertaken using RP-HPLC in acidic medium (0.1% TFA), using a semi-preparative C3 column at room temperature. Pure peptide-linker 6 (16.5 mg, 18.3% yield, 92% purity, see ESI Fig. S4†) was then treated with 1 M NaOH for 1 hour to hydrolyse the ester bond and remove the linker from the peptide; the reaction was then quenched with neat TFA, and the peptide recovered by lyophilisation. Suspension in cold diethyl ether and washing with water completely removed the water-soluble HMBA-Lys6-CONH2, and pure Aβ42 was reconstituted in 1:1 v/v acetonitrile and water, then lyophilised to afford 1 (13.8 mg, 92% purity, see ESI Fig. S5†). The pure peptide was immediately stored at −20 °C to prevent oxidation of the single methionine residue in the peptide sequence, a phenomenon understood to hinder the peptide's fibril-forming properties.23 LC profiles of crude Aβ42 with the 3 synthesis strategies attempted is presented in Fig. 2 below, followed by HPLC of pure Aβ42 (Fig. 3).
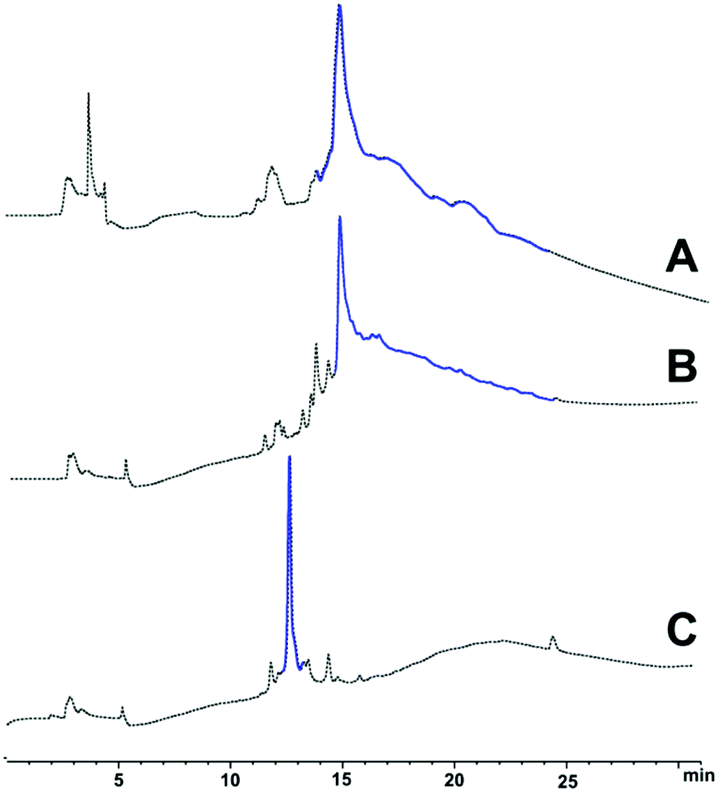 | ||
| Fig. 2 LC profiles of (A) crude Aβ42 (standard Fmoc/tBu SPPS), (B) crude Aβ42 employing pseudoprolines, and (C) crude Aβ42-HMBA-Lys6-CONH2 in acidic medium (0.1% formic acid) at room temperature; regions in blue indicate the time range in which target peptide eluted. | ||
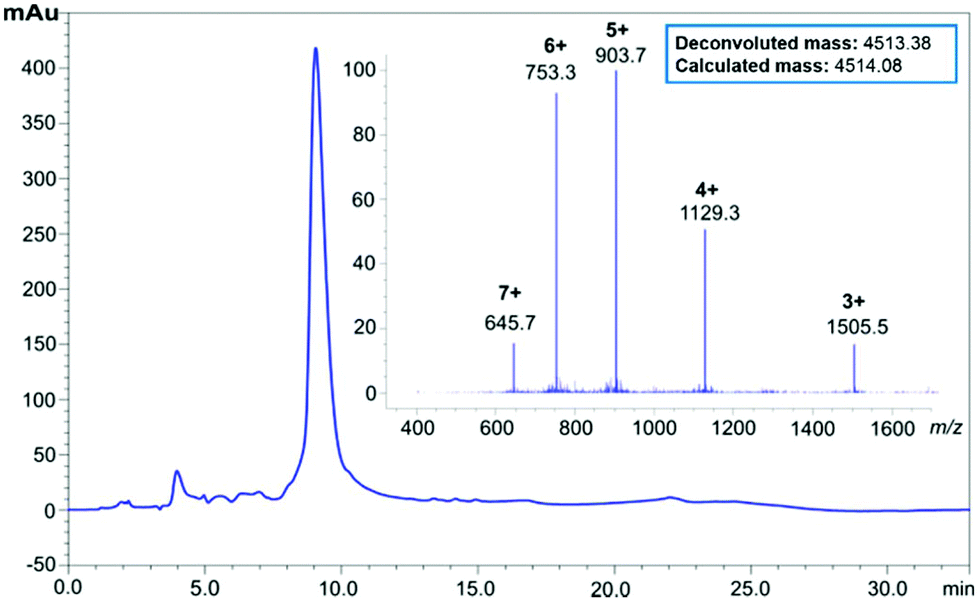 | ||
| Fig. 3 HPLC profile and ESI-MS of pure Aβ42 peptide following removal of the lysine tag. | ||
The presence of positively-charged lysine residues at the C-terminus of the Aβ42 peptide sequence, rendered it more hydrophilic, hence the earlier elution time (ca. 12 minutes, Fig. 2C) compared to the other two (ca. 15 minutes, Fig. 2A and B). Furthermore, the tag also appears to confer increased solubility to the peptide, significantly minimising the likelihood of aggregation, as well as permitting analysis and purification in acidic medium, which is not possible with unmodified Aβ42.
A biophysical assessment of our synthesised Aβ42 was then undertaken. Transmission electron microscopy (TEM) images of Aβ42 in a fibril promoting solution (10 mM HCl at 37 °C)24 were obtained over 3 days (Fig. 4) in order to determine the extent of fibril formation over time. This was followed by conducting a thioflavin T (ThT) assay to determine peptide aggregation kinetics (Fig. 5), as well as secondary structure quantification using circular dichroism (CD) spectroscopy (Fig. 6).
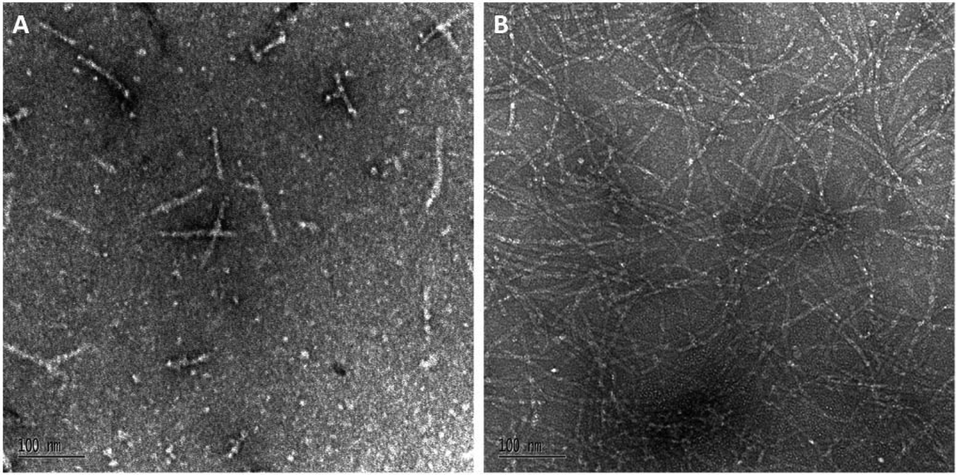 | ||
| Fig. 4 (A) Day 0 and (B) day 3 TEM images of Aβ42 peptide (magnification: ×140000; scale bar: 100 nm). | ||
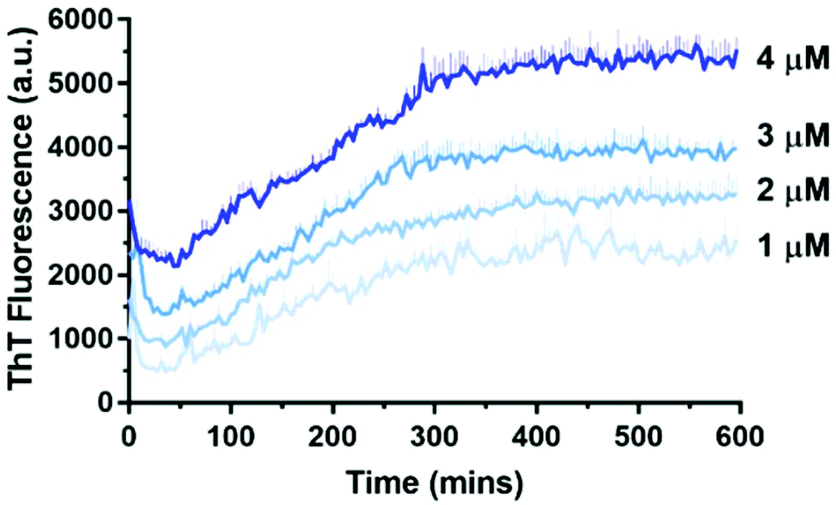 | ||
| Fig. 5 Aβ42 has a concentration-dependent aggregation profile as measured by 10 μM ThT in 10 mM sodium phosphate buffer (pH = 7.4) at 30 °C. Error bars are standard deviation from triplicate wells. | ||
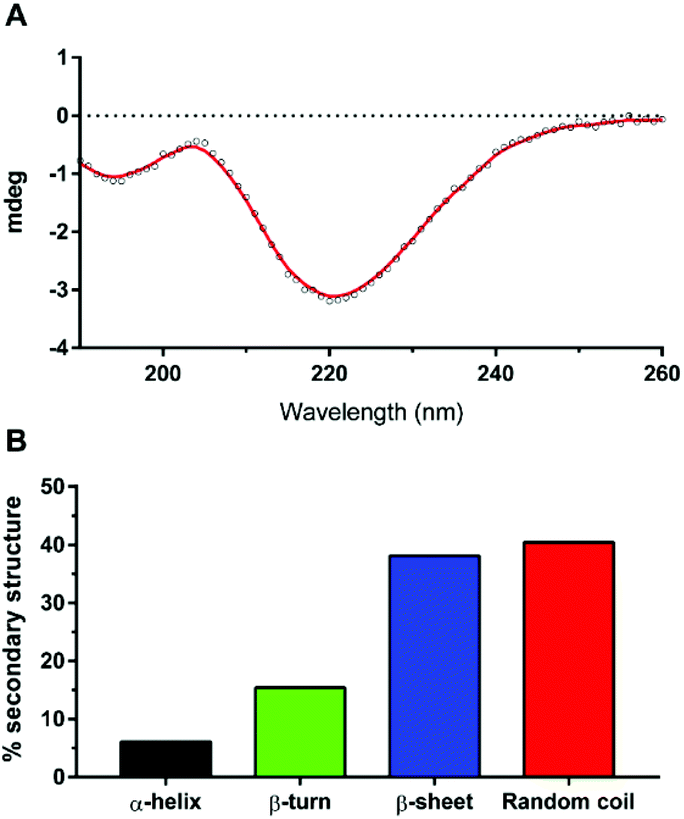 | ||
| Fig. 6 (A) CD spectrum of Aβ42 after 27 minutes in 10 mM sodium phosphate buffer (pH = 7.4) at 30 °C. (B) Secondary structure proportion of Aβ42 with spectral deconvolution from (A) using BeStSel;28 Normalized root-mean square deviation (NRMSD) = 0.0601. | ||
At day 0 (Fig. 4A), our Aβ42 predominantly exists as protofibril (‘worm-like’ morphology, <150 nm) or oligomer (round-shaped morphology), which is expected as fibril formation does not take place immediately following peptide reconstitution. Subsequent incubation of Aβ42 for several days in the fibril promoting solution generated fibrils (ESI Fig. S6†) up to day 3 (Fig. 4B). This fibrillar peptide morphology, which is a signature of Aβ42, is consistent with other TEM images of synthetic Aβ42, such as those published by Smith's group,25 thereby establishing that our Aβ42 is bioequivalent.
ThT is a dye that fluoresces upon binding to β-sheet structures and is commonly used to monitor Aβ42 aggregation, which is characterised by extensive β-sheet formation.26 At higher concentrations of Aβ42, the maximum ThT fluorescence was reached relatively rapidly, whereas at lower concentrations a longer time was required to reach maximum ThT fluorescence. This demonstrated the confirmed concentration-dependent aggregation behaviour of Aβ42, and is consistent with that of Knowles’ group.27
The resulting CD spectrum of our Aβ42 (Fig. 6A) showed a minimum at 220 nm and maximum at 200 nm, which is characteristic of β-sheet formation. There is a relatively high proportion of random coils (Fig. 6B), which may have contributed to the less pronounced maximum. Nevertheless, the peptide predominantly consists of β-sheets and random coils, which are indeed the characteristic secondary structures of amyloid fibrils, as reported recently by Kardos et al.28
We herein reported the successful synthesis of Aβ42 using pseudoproline dipeptides to disrupt on-resin aggregation during SPPS, together with incorporation of a temporary C-terminal hexalysine tag to improve its water solubility and favour the disaggregated state. Use of this new double linker strategy enabled purification and characterisation of Aβ42 using standard HPLC conditions. The procedures employed are straightforward and easily accessible by any laboratory. Furthermore, collated results from biophysical characterisation studies conducted in this work indicate that our Aβ42 peptide behaved similarly to other preparations of synthetic Aβ42. Our methodology ultimately permits the preparation of synthetic Aβ42 in high yield and purity, which is essential to thoroughly examine the underlying mechanism by which unregulated Aβ42 deposition can accelerate AD progression.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Reitz, C. Brayne and R. Mayeux, Nat. Rev. Neurol., 2011, 7, 137–152 CrossRef.
- H. V. Vinters, Annu. Rev. Pathol.: Mech. Dis., 2015, 10, 291–319 CrossRef CAS.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS.
- G. Thinakaran and E. H. Koo, J. Biol. Chem., 2008, 283, 29615–29619 CrossRef CAS.
- Y. Yan and C. Wang, J. Mol. Biol., 2006, 364, 853–862 CrossRef CAS.
- M. Paradis-Bas, J. Tulla-Puche and F. Albericio, Chem. Soc. Rev., 2016, 45, 631–654 RSC.
- A. K. Tickler, C. J. Barrow and J. D. Wade, J. Pept. Sci., 2001, 7, 488–494 CrossRef CAS.
- Y. S. Kim, J. A. Moss and K. D. Janda, J. Org. Chem., 2004, 69, 7776–7778 CrossRef CAS.
- Y. Sohma, Y. Hayashi, M. Kimura, Y. Chiyomori, A. Taniguchi, M. Sasaki, T. Kimura and Y. Kiso, J. Pept. Sci., 2005, 11, 441–451 CrossRef CAS.
- F. García-Martín, M. Quintanar-Audelo, Y. García-Ramos, L. J. Cruz, C. Gravel, R. Furic, S. Côté, J. Tulla-Puche and F. Albericio, J. Comb. Chem., 2006, 8, 213–220 CrossRef.
- B. Bacsa, S. Bosze and C. O. Kappe, J. Org. Chem., 2010, 75, 2103–2106 CrossRef CAS.
- J. M. Collins, K. A. Porter, S. K. Singh and G. S. Vanier, Org. Lett., 2014, 16, 940–943 CrossRef CAS.
- S. Chemuru, R. Kodali and R. Wetzel, Biopolymers, 2014, 102, 206–221 CrossRef CAS.
- J. A. Karas, A. Noor, C. Schieber, T. U. Connell, F. Separovic and P. S. Donnelly, Chem. Commun., 2017, 53, 6903–6905 RSC.
- M. Yamamoto, K. Shinoda, J. Ni, D. Sasaki, M. Kanai and Y. Sohma, Org. Biomol. Chem., 2018, 16, 6537–6542 RSC.
- W. M. Kok, J. M. Cottam, G. D. Ciccotosto, L. A. Miles, J. A. Karas, D. B. Scanlon, B. R. Roberts, M. W. Parker, R. Cappai, K. J. Barnham and C. A. Hutton, Chem. Sci., 2013, 4, 4449–4454 RSC.
- G. Vanhoenacker and P. Sandra, Anal. Bioanal. Chem., 2008, 390, 245–248 CrossRef CAS.
- M. A. Hossain, A. Belgi, F. Lin, S. Zhang, F. Shabanpoor, L. Chan, C. Belyea, H.-T. Truong, A. R. Blair, S. Andrikopoulos, G. W. Tregear and J. D. Wade, Bioconjugate Chem., 2009, 20, 1390–1396 CrossRef CAS.
- C. T. Choma, G. T. Robillard and D. R. Englebretsen, Tetrahedron Lett., 1998, 39, 2417–2420 CrossRef CAS.
- A. Abedini and D. P. Raleigh, Org. Lett., 2005, 7, 693–696 CrossRef CAS.
- P. W. R. Harris and M. A. Brimble, Biopolymers, 2010, 94, 542–550 CrossRef CAS.
- G. M. Williams, G. J. Cooper, K. Lee, L. Whiting and M. A. Brimble, Org. Biomol. Chem., 2013, 11, 3145–3150 RSC.
- L. Hou, I. Kang, R. E. Marchant and M. G. Zagorski, J. Biol. Chem., 2002, 277, 40173–40176 CrossRef CAS.
- W. B. Stine, L. Jungbauer, C. Yu and M. LaDu, Methods Mol. Biol., 2011, 670, 13–32 CrossRef CAS.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS.
- C. Xue, T. Y. Lin, D. Chang and Z. Guo, R. Soc. Open Sci., 2017, 4, 160696 CrossRef PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS.
- A. Micsonai, F. Wien, E. Bulyaki, J. Kun, E. Moussong, Y.-H. Lee, Y. Goto, M. Refregiers and J. Kardos, Nucleic Acids Res., 2018, 46, 315–322 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C8OB02929F |
| This journal is © The Royal Society of Chemistry 2019 |