
A Viedma ripening route to an enantiopure building block for Levetiracetam and Brivaracetam†
Iaroslav
Baglai
a,
Michel
Leeman
b,
Richard M.
Kellogg
b and
Willem L.
Noorduin
 *a
*a
aAMOLF, Science Park 104, 1098 XG Amsterdam, The Netherlands. E-mail: noorduin@amolf.nl
bSyncom BV, Kadijk 3, 9747 AT Groningen, The Netherlands
First published on 5th November 2018
Abstract
A simple route to enantiomerically pure (S)-2-aminobutyramide – the chiral component of the anti-epileptic drugs Levetiracetam and Brivaracetam has been developed. This approach is based on the rational design and application of a Viedma ripening process. The practical potential of the process is demonstrated on a large scale.
Simple synthetic routes to enantiomerically pure building blocks are essential for the cost-effective production of pharmaceutical compounds. Levetiracetam 1a and Brivaracetam 1b are commonly used for the treatment of epilepsy.1,2 The essential building block for both of these drugs is enantiomerically pure (S)-2-aminobutyramide 2. A key step in the preparation is the introduction of the desired absolute (S) configuration at the chiral center.3–7 In the initial route developed by the originator UCB, chromatographic separation of the enantiomers of racemic 1a by simulated moving bed technology was used to produce enantiomerically pure 1a.4 Currently, however, most generic producers of 1a,b start with the single enantiomer of the unnatural amino acid (S)-2-aminobutyric acid amide 2 (Scheme 1), prepared by classical resolution of the racemate using diastereomeric salt formation with enantiomerically pure D-(−)-mandelic acid or L-(+)-tartaric acid.5–7 For all of these resolution techniques, the theoretical yield of the desired (S) enantiomer is limited to 50%. To overcome this yield restriction, the undesired (R) enantiomer may be “externally” racemized and resolved again, although a drawback is that this often requires a multi-step process. To our knowledge, attempts to develop a classical resolution combined with in situ racemization for the obtainment of enantiomerically pure 2 have not yet been successful, let alone been developed into an industrially viable process. A simple and robust process to produce only the (S)-enantiomer in high yield and enantiomeric excess (ee) from the racemate is thus very desirable.
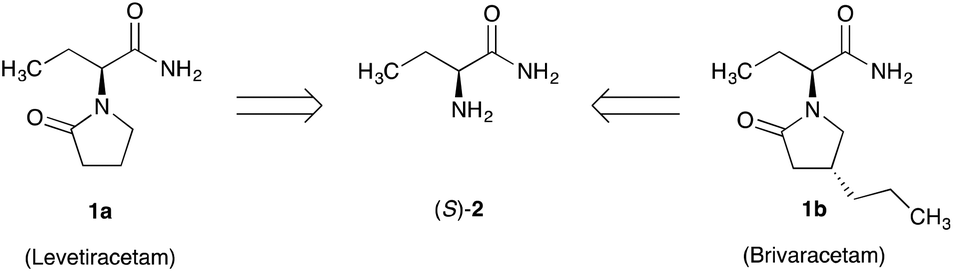 | ||
| Scheme 1 (S)-2-Aminobutyramide (S)-2, a chiral building block for Levetiracetam 1a and Brivacetam 1b. | ||
We here present a simple and efficient route to the building block (S)-2-aminobutyramide 2 with ee > 99% in virtually quantitative yield. Our approach relies on a recently discovered deracemization technique that combines simple grinding of crystals with racemization in solution. This attrition-enhanced deracemization (Viedma ripening) allows total conversion of a racemate to a single enantiomer of the desired chirality.8–14 Under near-equilibrium conditions a solid phase of the racemate is entirely converted into a solid phase of the enantiomer of choice by grinding of a slurry. The fundamental basis for this remarkable process is understood although the details remain the subject of continuing discussion. However, there is consensus that the process has two prerequisites: (1) the enantiomers of the racemate must crystallize as a conglomerate, i.e. a mechanical mixture of enantiopure crystals, and (2) racemization in the solution in contact with the crystals must occur, i.e. the enantiomers must interconvert in the liquid phase. These requirements limit the choice of suitable molecules. Satisfactory racemization conditions are often not obvious, and despite the recent progress made in calculating crystal structures,15 conglomerate formation remains still unpredictable and statistically occurs only for 5–10% of chiral organic compounds.16 As a result, it remains challenging to design new suitable compounds for attrition-enhanced deracemization.
To the best of our knowledge, in the currently employed synthetic routes to compounds 1a,b none of the reported intermediates or final products crystallizes as a conglomerate. Since conglomerate behavior is essential, but unpredictable, we need a library approach to design a new conglomerate intermediate that racemizes in solution.
Recently we developed an α-aminonitrile-based methodology that allows the absolute asymmetric synthesis of highly sterically hindered α-amino acids via formation of intermediate racemizable conglomerates.17 Moreover, we demonstrated the crucial role of balanced physicochemical properties (crystallinity/acidity) of conglomerates suitable for Viedma ripening. However, based on these results we envisaged that low crystallinity and/or high solubility of analogous α-aminobutyronitrile imines would likely hinder their application in a Viedma ripening process.
Considering this analysis, and relying on previous experience,18,19 we designed a library of amino acid amide 2 derivatives to identify a racemizable conglomerate. In short, we allowed racemate 2 to react with a selection of aromatic aldehydes to yield a library of imines (Schiff bases) 3a–f (Scheme 2). All products were nicely crystalline probably owing to the hydrogen bonding network.
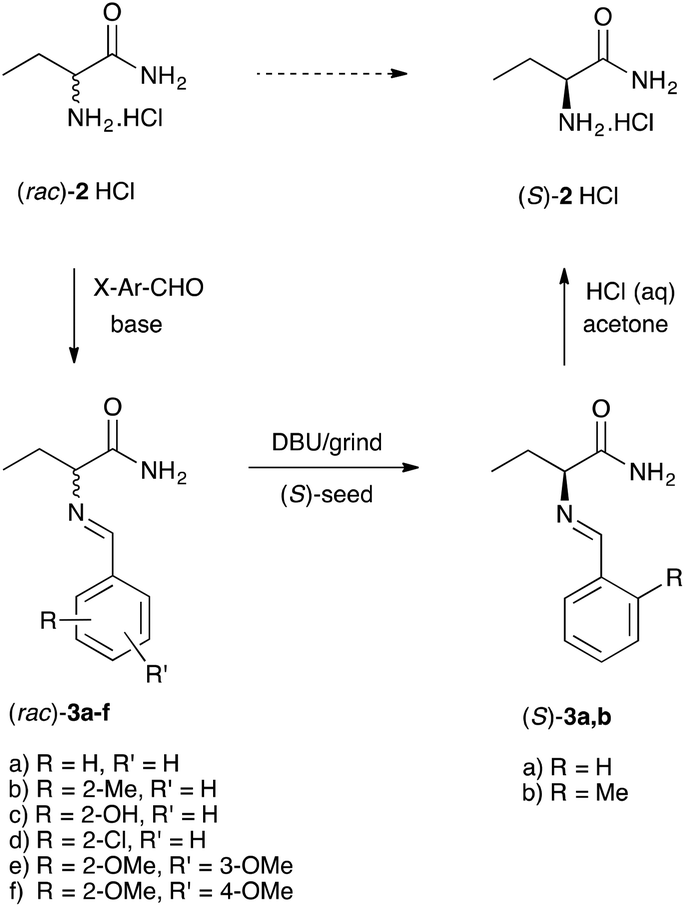 | ||
| Scheme 2 Schematic illustration of the imines library preparation and deracemization of the identified conglomerates 3a,b. | ||
To identify the library entries that crystallize as conglomerates, the imines were screened by second harmonic generation (SHG) measurements,20,21 followed by comparison of the racemate and enantiomerically pure form using X-Ray powder diffraction (XRPD) and differential scanning calorimetry (DSC). From this screen, the imines 3a,b formed with benzaldehyde and with 2-methylbenzaldehyde, respectively, were identified as conglomerates as can be judged from the identical XRPD diffractograms (Fig. 1) of the enantiomerically pure and racemic crystals. For economic reasons, compound 3a was chosen for detailed investigation although from preliminary experiments it was established that 3b also undergoes complete deracemization on attrition (ESI†).
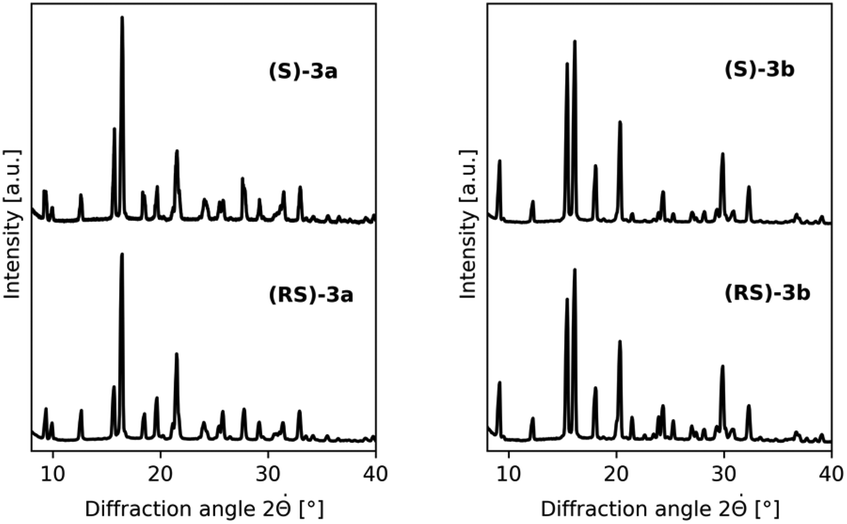 | ||
| Fig. 1 Conglomerate identification: as expected for conglomerate formation, XRPD diffractograms of the racemates and enantiomerically pure forms of 3a (left) and 3b (right) are identical. | ||
As anticipated, despite the lower acidity of imines 3 as compared to the previously reported phenylglycinamides derivatives,22–24 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was found to racemize (S)-3a at ambient temperature at rates suitable for deracemization. To optimize the racemization conditions, we examined the racemization of (S)-3a with DBU in multiple solvents. As illustrated in Fig. 2, the racemization rate is dependent on solvent and is clearly fastest in acetonitrile and toluene.
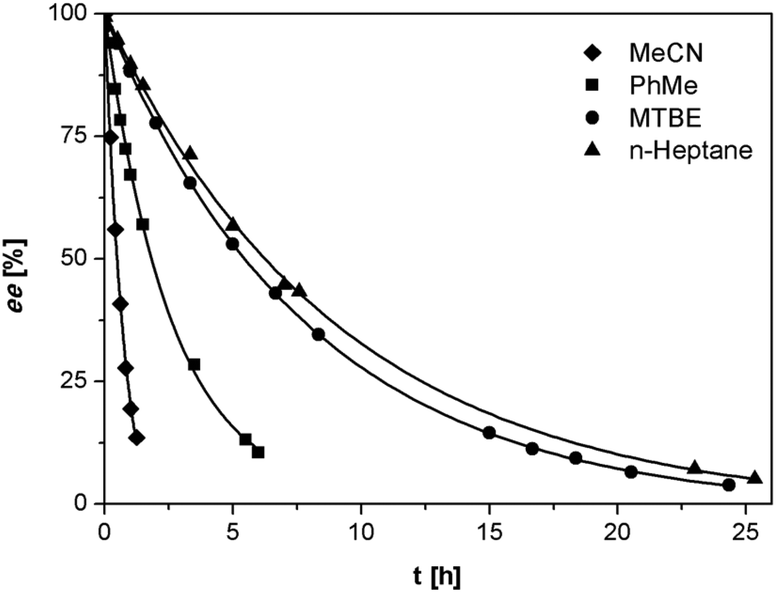 | ||
| Fig. 2 Racemization of (S)-3a (0.5 mg mL−1) with DBU (3.3 μL mL−1) in various solvents at ambient temperature. | ||
We investigated the deracemization of 3a by attrition (Viedma ripening) in all four solvents. In the initial small-scale experiments, a reaction vessel was placed in a temperature controlled ultrasonic bath and sonicated at 20 °C. To direct the outcome of the deracemization the solid phase was seeded with the desired (S) enantiomer to obtain approximately 10–15% starting ee in (S) in the solid phase. The enrichment in the solid phase during the process was monitored by first isolating the solid phase using filtration, which promptly stopped the racemization. Subsequently, the enantiomeric excess of the solid phase was analyzed using HPLC on a chiral column. Within 6 hours, complete deracemization was observed in acetonitrile. Despite lower rates, experiments in toluene and methyl tert-butyl ether also led to complete deracemization. In n-heptane only a slight enrichment of the solid phase was detected before the slurry turned into a “glue-like” sticky substance after 3 days of attrition (Fig. 3). This solvent effect might be due to the fact that heptane is a poorer emulgator than acetonitrile and, to a lesser extent, toluene. The latter solvents have both a polar and less polar side, which assists in stabilizing the crystal surface, thus hindering destabilization of the dispersion, especially under high attrition rates. These observations suggest that an ideal solvent for Viedma ripening not only supports swift racemization and crystallization/dissolution, but also acts as a dispersion agent.
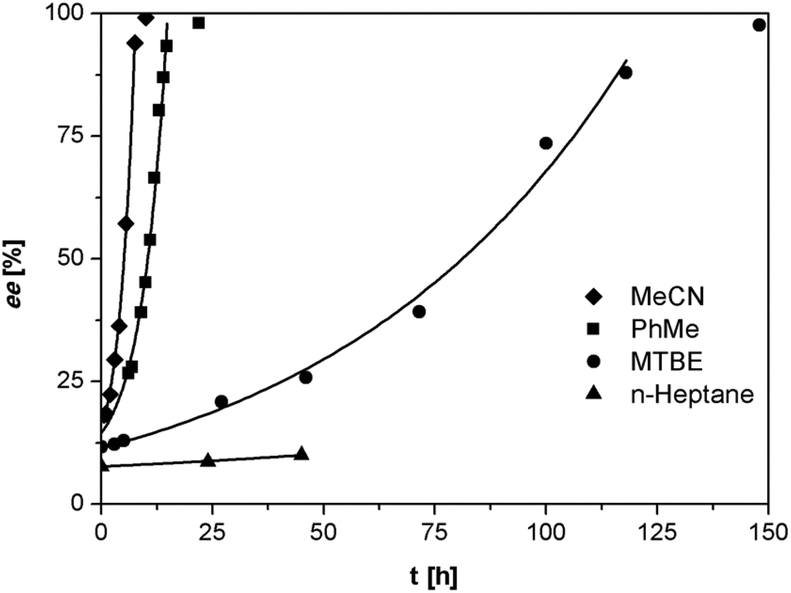 | ||
| Fig. 3 Enantiomeric enrichment in the solid phase during attrition-enhanced deracemization of compound 3a in various solvents. Lines are a fit of the exponential deracemization rate (ee of the isolated solids excluded from the fit). Conditions: 100 mg of the solid phase per 1.0 mL of solvent, [DBU] = 40 μL mL−1, 20 °C. | ||
The deracemization reaction rate k can be estimated from the relation ee(t) = ee(0)exp(kt), in which ee(0) and ee(t) are the enantiomeric excess in the solid phase at the beginning of the experiment t = 0 and time t respectively.25 As expected, the time evolution of the ee in the solid phase follows the exponential behavior that is typical for this process (Fig. 3).
Although racemization and deracemization are somewhat more rapid in acetonitrile compared to toluene, the formation of small amounts of an unidentified and undesired side-product as well as the relatively high solubility of 3a made the former solvent less attractive for a continuous large-scale process. Further optimization of the reaction conditions was performed using toluene as a solvent, in which both racemization and deracemization occurred with suitable rates for practical application.
To demonstrate the potential of practical application of this method we performed our process on a relatively large scale – starting with 15 g of 3a. To obtain the desired (S)-enantiomer, we created an initial ee of 14% in the solid phase by seeding with the (S)-enantiomer. Despite the relatively high solid content compared to the initial experiments, complete deracemization of the solid phase was achieved within 60 hours at 20 °C (Fig. S1, ESI†), providing the desired (S)-3a in 77% isolated yield and 99% ee. The racemic material remaining in the liquid phase can be easily recovered. Furthermore, we confirmed that such filtered liquid phase can be recycled to perform another deracemization experiment (ESI†). The process is thus virtually quantitative in an economic operation procedure.
Finally, to obtain the desired enantiomerically pure building block (S)-2 for Levetiracetam 1a and Brivaracetam 1b deracemized (S)-3a was hydrolyzed under acidic conditions to provide the HCl salt of (S)-2 with complete preservation of stereochemistry at the chiral center with >99% ee in nearly quantitative yield (ESI†).
In summary, we introduce a practical absolute asymmetric synthesis route to the chiral key precursor for Levetiracetam and Brivacetam. Owing to its simplicity, robustness and virtually quantitative yield, this Viedma ripening approach provides an attractive cost-effective alternative to the current routes. Only mechanical attrition at ambient temperature of the corresponding benzaldehyde Schiff base at near equilibrium is required combined with simple, high yielding condensation chemistry and equally high yielding hydrolyses with low cost mineral acids. Finally, these results show that the invention of a practical Viedma ripening process does not merely rely on the design and identification of a racemizable conglomerate, but rather requires a holistic approach in which a multitude of factors is taken into account (e.g. reagents, reaction conditions, attrition, solvent, etc.).
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by AMOLF funds for Topsector-related research.Notes and references
- B. Abou-khalil, Neuropsychiatr. Dis. Treat., 2008, 4, 507–523 CrossRef CAS.
- L. S. Deshpande and R. J. Delorenzo, Front. Neurol., 2014, 5, 1–5 CrossRef.
- (a) G. Springuel and T. Leyssens, Cryst. Growth Des., 2012, 12, 3374–3378 CrossRef CAS; (b) O. Shemchuk, L. Song, K. Robeyns, D. Braga, F. Grepioni and T. Leyssens, Chem. Commun., 2018, 54, 10890–10892 RSC.
- E. Cavoy, M. Hamende, M. Deleers, J.-P. Canvat and V. Zimmermann, US Pat, 6124473, 2000 Search PubMed.
- W. H. J. Boesten, J. J. C. J. Boonen and P. L. Woestenborghs, Eur. Pat. Appl, 2524910, 2012 Search PubMed.
- A. K. Mandal, S. W. Mahajan, P. Ganguly, A. Chetia, N. D. Chauhan, D. N. Bhatt and R. R. Baria, PCT Int. Pat. Appl, WO 2006103696, 2006 Search PubMed.
- I. Katsumi and K. Nishi, Jpn. Kokai Tokkyo Koho, JP 2007191470, 2007 Search PubMed.
- C. Viedma, Phys. Rev. Lett., 2005, 94, 65504 CrossRef.
- P. S. M. Cheung, J. Gagnon, J. Surprenant, Y. Tao, H. Xu and L. A. Cuccia, Chem. Commun., 2008, 987–989 RSC.
- W. L. Noorduin, T. Izumi, A. Millemaggi, M. Leeman, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158–1159 CrossRef CAS.
- C. Viedma, J. E. Ortiz, T. de Torres, T. Izumi and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274–15275 CrossRef CAS.
- W. L. Noorduin, E. Vlieg, R. M. Kellogg and B. Kaptein, Angew. Chem., Int. Ed., 2009, 48, 9600–9606 CrossRef CAS.
- L.-C. Sögütoglu, R. E. Steendam, H. Meekes, E. Vlieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2015, 44, 6723–6732 RSC.
- W. L. Noorduin, W. J. P. Van-enckevort, H. Meekes, B. Kaptein, R. M. Kellogg, J. C. Tully, J. M. McBride and E. Vlieg, Angew. Chem., Int. Ed., 2010, 49, 8435–8438 CrossRef CAS.
- G. M. Day, T. G. Cooper, A. J. Cruz-Cabeza, K. E. Hejczyk, H. L. Ammon, S. X. M. Boerrigter, J. S. Tan, R. G. Della Valle, E. Venuti, J. Jose, S. R. Gadre, G. R. Desiraju, T. S. Thakur, B. P. Van Eijck, J. C. Facelli, V. E. Bazterra, M. B. Ferraro, D. W. M. Hofmann, M. A. Neumann, F. J. J. Leusen, J. Kendrick, S. L. Price, A. J. Misquitta, P. G. Karamertzanis, G. W. A. Welch, H. A. Scheraga, Y. A. Arnautova, M. U. Schmidt, J. Van De Streek, A. K. Wolf and B. Schweizer, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 107–125 CrossRef CAS.
- A. Otero-de-la-roza, B. H. Cao, I. K. Price, J. E. Hein and E. R. Johnson, Angew. Chem., Int. Ed., 2014, 53, 7879–7882 CrossRef CAS.
- I. Baglai, M. Leeman, K. Wurst, B. Kaptein, R. M. Kellogg and W. L. Noorduin, Chem. Commun., 2018, 54, 10832–10834 RSC.
- M. van der Meijden, M. Leeman, E. Gelens, W. Noorduin, H. Meekes, W. van Enckevort, B. Kaptein, E. Vlieg and R. Kellogg, Org. Process Res. Dev., 2009, 13, 1195–1198 CrossRef CAS.
- P. Wilmink, C. Rougeot, K. Wurst, M. Sanselme, M. Van Der Meijden, W. Saletra, G. Coquerel and R. M. Kellogg, Org. Process Res. Dev., 2015, 19, 302–308 CrossRef CAS.
- A. Galland, V. Dupray, B. Berton, S. Morin-Grognet, M. Sanselme, H. Atmani and G. Coquerel, Cryst. Growth Des., 2009, 9, 2713–2718 CrossRef CAS.
- F. Simon, S. Clevers, V. Dupray and G. Coquerel, Chem. Eng. Technol., 2015, 38, 971–983 CrossRef CAS.
- The acidity of the α-proton to the carbonyl group in 3a,b will be significantly less than in phenylglycinamide derivatives owing the extra stabilization by the phenyl group present in the latter.12 A previously deracemized phenylglycinamide imine has a pKa in the order of 17–18 in MeOH.10 We estimate from literature values that the pKa values of imines 3a,b will be 3–4 units higher.23,24 The pKa of DBU in MeOH is 12.
- M. J. O. Donnell, W. D. Bennett, W. A. Bruder, W. N. Jacobsen, K. Knuth, B. Leclef, R. L. Pelt, F. G. Bordwell, S. R. Mrozack and T. A. Cripe, J. Am. Chem. Soc., 1988, 110, 8520–8525 CrossRef.
- M. J. O'Donnell, J. D. Keeton, V. V. Khau and J. C. Bollinger, Can. J. Chem., 2006, 84, 1301–1312 CrossRef.
- W. L. Noorduin, H. Meekes, W. J. P. Van Enckevort, A. Millemaggi, M. Leeman, B. Kaptein, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 6445–6447 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02660b |
| This journal is © The Royal Society of Chemistry 2019 |