
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step synthesis and XPS investigations of chiral NHC–Au(0)/Au(I) nanoparticles†
Adam J.
Young
 a,
Markus
Sauer
b,
Guilherme M. D. M.
Rubio
c,
Akiko
Sato
d,
Annette
Foelske
b,
Christopher J.
Serpell
d,
Jia Min
Chin
*ac and
Michael R.
Reithofer
*c
a,
Markus
Sauer
b,
Guilherme M. D. M.
Rubio
c,
Akiko
Sato
d,
Annette
Foelske
b,
Christopher J.
Serpell
d,
Jia Min
Chin
*ac and
Michael R.
Reithofer
*c
aGray Centre for Advanced Materials, School of Mathematics and Physical Sciences, University of Hull, Cottingham Road, Hull, East Riding of Yorkshire, HU6 7RX, UK. E-mail: j.chin@hull.ac.uk
bVienna University of Technology, Analytical Instrumentation Center, Getreidemarkt 9/BL01A01, A-1060 Wien, Austria
cInstitute of Inorganic Chemistry, University of Vienna, Faculty of Chemistry, Waehringer Strasse 42, 1090 Vienna, Austria. E-mail: michael.reithofer@univie.ac.at
dUniversity of Kent, School of Physical Sciences, Ingram Building Canterbury, Kent, CT2 7NH, UK
First published on 8th April 2019
Abstract
Although N-heterocyclic carbenes (NHCs) have been demonstrated as suitable ligands for the stabilisation of gold nanoparticles (AuNPs) through a variety of methods, the manner in which such AuNPs form is yet to be fully elucidated. We report a simple and fast one-step synthesis of uniform chiral (L/D)-histidin-2-ylidene stabilised gold nanoparticles using the organometallic Au(I) complex as a well defined starting material. The resulting nanoparticles have an average size of 2.35 ± 0.43 nm for the L analog whereas an average size of 2.25 ± 0.39 nm was found for the D analog. X-ray photoelectron spectroscopy analyses reveal the presence of Au(I) and Au(0) in all NHC stabilised AuNPs. In contrast, measured X-ray photoelectron spectra of dodecanethiol protected gold nanoparticles showed only the presence of a Au(0) species. This observation leads us to postulate that AuNPs synthesised from organometallic NHC–Au(I) complexes exhibit a monolayer of Au(I) surrounding a Au(0) core. This work highlights the importance of synthetic method choice for NHC-stabilized AuNPs, as this could determine Au oxidation states and resulting AuNP properties such as catalytic activities and stabilities.
Introduction
Gold nanoparticles (AuNPs) have been exploited for centuries for colouration of glass and pottery, due to the strong interaction of light with electrons on the gold nanoparticle surface, giving rise to surface plasmon resonance (SPR) absorption bands which afford the nanoparticles intense visible colours.1 Recently, interest in such nanoparticles has expanded due to their promise in biomedical applications, such as plasmonic heating for controlled drug release or photothermal therapy. Their large surface area to volume ratio also renders them useful for catalytic applications. Another emerging direction of AuNP research has been towards optoelectronic applications and sensing, alongside the development of reliable methods to tune the sizes, electronic structures, optical properties and stabilities of such nanoparticles.1As AuNP stability is crucial for the aforementioned applications, significant effort has gone into developing ligands with strong Au-binding properties, which are expected to enhance the stabilities of the corresponding AuNPs. N-heterocyclic carbenes (NHCs) in particular, are an attractive ligand class for AuNP stabilization, as the synthetic accessibility2 of NHC ligands allows fine-tuning of their steric and electronic properties according to need. Further, the strong σ-donating and π-accepting properties of NHC ligands results in stable metal–NHC complexes. NHCs have also attracted much attention in the development of novel nanoparticles,3–9 particularly to serve as more strongly bound capping ligands in place of thiols.10–13
It is estimated that the NHC–Au bond strength is about double that of a corresponding Au–thiol bond.14 Recent reports show that NHC-functionalized gold surfaces and NHC–Au nanoparticles outperformed their thiol based counterparts with regards to thermal and chemical stability.10,14,15 However, it is important to note that this is not always the case, and the stability of the resulting NHC-AuNPs depends upon the synthetic procedure employed. For example, Man et al. reported AuNPs stabilized by bidentate NHC ligands, whereby analogous AuNPs were prepared by both bottom-up and top-down methods.16 In the bottom-up approach, molecular NHC-AuBr compounds were reduced with NaBH4 whereas for the top-down approach an exchange reaction of dodecylsulfide ligands with NHC ligands on AuNPs was performed. Although both methods yielded NHC-stabilized AuNPs (NHC-AuNPs), the NPs synthesised via the top-down approach were found to have significantly higher stability against ripening and exogenous thiols. The authors attributed this difference to the smaller size of the bottom-up NPs as well as lower surface coverage by the ligands. MacLeod and Johnson also showed that NPs synthesised from their corresponding NHC–Au(I) complexes undergo ligand exchange reactions when exposed to thiols, releasing an NHC–Au(I) complex and not the free NHC ligand.7 Given the importance of NP stability in AuNP applications, more in-depth understanding of the factors determining AuNP stability, and the differences in AuNPs synthesized via top-down versus bottom-up methods is certainly required.
We have previously reported the synthesis of AuNPs stabilized by histidine-derived NHCs, which showed chiroptical activity.17 However, the AuNPs obtained were size disperse, necessitating a centrifugal size selection process to afford monodispersed nanoparticles. Here, we report that by replacing tBuNH2·BH3 with a stronger reducing agent, NaBH4, and shortening the reaction times, we can obtain smaller nanoparticles with a narrow size dispersion in a one-step manner (Scheme 1). Circular dichroism spectroscopy showed that the resulting AuNPs possess chiroptical activity. To study the Au oxidation state and the NHC-binding in the AuNPs, X-ray photo electron spectroscopy (XPS) was carried out. XPS was also performed on dodecanethiol-stabilized AuNPs (DDTAuNP) for comparison purposes. The findings indicate that for NHC-AuNPs synthesised via the NHC–Au(I) complex, Au exists in both Au(0) and Au(I) oxidation states, which might help to explain the different stabilities of the NPs when they are synthesised by different methods.
 | ||
| Scheme 1 Synthesis of complex 2(L/D) and its reduction with (a) tBuNH3·BH3 in THF, and (b) NaBH4 in DCM/H2O. | ||
Results and discussion
Synthesis of AuNPs
NHC-AuNPs were synthesised starting from their molecular NHC–Au(I) complex, chlorido(1,3-dimethyl-N-Boc-O-methyl-L/D-histidin-2-ylidene)gold(I) [2(L/D)] by dissolving 2(L/D) in DCM. Subsequently an aqueous solution of NaBH4 was added and the resulting biphasic reaction was vigorously stirred for 4 h at room temperature. During the reaction, the organic phase became brown/red, indicating the formation of AuNPs. The NPs 3′-NP(L/D) could be isolated as black powder which was re-dispersible in DCM and THF without any visible aggregation, even after repeated drying. DDTAuNPs were also synthesised according to a previously reported literature procedure, to provide a reference compound for comparative studies.18Characterisation of AuNPs
NMR analysis of NHC-AuNPs confirmed the presence of the capping ligand on the AuNPs, whereas no free imidazolium ligand was detectable in all NHC-AuNP samples, as indicated by the absence of the acidic C2 proton peak at ∼9 ppm. Furthermore, the 13C NMR spectra of samples 3′-NP(L) and 3′-NP(D) show that the carbene NHC peak has shifted from 171.5 ppm in the molecular species 2(L) to 184.5 ppm in the AuNPs. The low field shift of the coordinating carbon demonstrates a change in the chemical environment, suggesting the successful formation of AuNPs. Similar changes in chemical shifts between the molecular and the nanoparticulate species have been recently reported by Johnson et al.7 Further, the absence of a signal arising from the protonated C2 carbon at ∼137.0 ppm shows that no free imidazolium ligand is detectable in solution.7TEM analysis of films obtained via drop casting a solution of 3′-NP(L) and 3′-NP(D) (Fig. 1) in DCM after drying revealed the presence of non-agglomerated, spherical AuNPs with an average diameter (DTEM) of 2.35 ± 0.43 (18.3%) nm and 2.25 ± 0.39 (17.2%) nm respectively, in comparison, the hydrodynamic diameter of 3′-NP(L) and 3′-NP(D) (DDLS) were found to be 4.2 and 6.5 nm respectively (Fig. S11 and S12†). The increased NP diameter as measured by DLS is attributed to the ligand sphere as well as solvent cage, which is typically not observed via TEM. This is further supported by the lack of a plasmon band for the DLS-measured AuNP solutions. This relatively low polydispersity with no additional size selection step lends itself to a facile synthetic method for chiral NHC stabilised AuNPs. The use of a stronger reducing agent and shorter reaction times (4 h) compared to the synthesis of 3-NP(L) and 3-NP(D) (using tBuNH2·BH3 and 24 hours reaction times) led to smaller NPs, which is in keeping with the general observations by others for AuNP synthesis.3,8,19 In contrast, the previously reported 3-NP(L) and 3-NP(D) displayed larger polydispersity,17 necessitating the use of a centrifugal size selection technique taken from Pileni et al. to isolate the ‘larger’ AuNPs.20 Furthermore, the size distribution of the reference DDTAuNPs were found to be (DTEM) 1.98 ± 0.37 (18.7%) nm and a hydrodynamic diameter of (DDLS) 3.1 nm (Fig. S13†).
 | ||
| Fig. 1 TEM images of the respective AuNPs synthesised in this report. | ||
Thermogravimetric analysis (TGA) was performed to estimate the amount of capping ligand present on the surface of the AuNPs, by heating the AuNPs under an N2 atmosphere up to 700 °C. TGA showed that 3′-NP(L) and 3′-NP(D) possessed 49.7% and 49.8% organic component by weight respectively (Fig. S12 and S13† respectively).
The optical properties of 3′-NP(L) and 3′-NP(D) were characterized by UV-Vis as well as by circular dichroism (CD) spectroscopy. 3′-NP(L) and 3′-NP(D) exhibit a brown/red colour when dispersed in DCM (Fig. S6 and S7† respectively) indicating the formation of small AuNPs which do not possess a plasmon resonance band. This was confirmed by UV-Vis analysis where 3′-NP(L) and 3′-NP(D) did not display a surface plasmon resonance (SPR) signal. The reference DDTAuNPs form a brown/red solution in DCM, and UV-Vis spectroscopy showed no SPR signal, as expected from TEM observations of the small DDTAuNPs.
Optical activity of AuNPs
The optical activity of 3′-NP(L) and 3′-NP(D) was analysed by circular dichroism (CD) spectroscopy using DCM as the solvent (Fig. 2). The dichroic effects are visible in the ligand region of the spectrum, which suggests a chiral supramolecular ordering of the histidine-derived ligands on the AuNP surface. Since neither the free ligand (as imidazolinium salt) nor the molecular NHC–Au complex displays a CD effect in the measured region,17 the nanoparticle appears to play a crucial role in ordering the ligand on the surface as the ligand-binding substrate. We attribute the minor deviations from perfect mirroring of spectra between L- and D-variants to differences in NP size which influences the ordering of the ligands on the AuNP surface. | ||
| Fig. 2 CD spectra of synthesised compounds 3′-NP(L) and 3′-NP(D) dispersed in DCM. | ||
Stability studies of AuNPs
The stability of 3′-NP(L) and 3′-NP(D) was tested by dispersing the respective AuNPs in various solvents such as THF, DMF and chloroform. The resulting solutions were either refluxed or heated at 100 °C and stability was monitored through any changes in the UV-Vis spectrum. When AuNPs were heated either in chloroform or DMF, complete decomposition was observed after 1 h by UV-Vis spectroscopy (Fig. S25–S28†). Only samples heated in THF showed a slower degradation with a strong plasmon band being visible after 1 h heating by UV-Vis spectroscopy (Fig. S23 and S24† respectively). Based on these results, THF was chosen to test the stability of 3′-NP(L) and 3′-NP(D) solutions over a 24 h time period at room temperature. The UV-Vis spectra revealed only minor changes in the measured spectra over time. The minor change could be attributed to solvent evaporation or minor degradation of the AuNPs (Fig. S21 and S22† respectively).X-ray photoelectron spectroscopic analysis
XPS was utilized to study the NHC-AuNPs, as it has already been proven to be a powerful tool in the characterization of NHC-coated AuNPs whereby the C 1s and N 1s signals are used to evidence the presence of the NHC ligand on AuNPs.10,16,19–23 In addition to the C 1s and N 1s signals, we also utilized information arising from the Au 4f peaks to investigate Au oxidation states.XPS analysis was carried out on the NPs synthesized in this report (3′-NP(L) and 3′-NP(D)). For comparison against well-established thiol-stabilized AuNPs, we also performed XPS analysis on the reference DDTAuNPs. In addition, XPS measurements were carried out on the 2(D/L) complex to detect possible changes in the NHC ligand structure.
Each signal was deconvoluted using the CASA XPS software package employing Shirley backgrounds and Gaussian–Lorentzian (GL(30)) peak shapes. The C 1s signals of 3′-NP(D/L) (Fig. 3(a)) consist of three components indicative of the histidine-derived NHC ligand. The peaks at (284.7 ± 0.2) eV and (286.3 ± 0.2) eV can be attributed to C–C and C–O/C–N bonds respectively, consistent with previous results.19,20 In addition, an O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O contribution can be detected at (289.2 ± 0.2) eV, confirming the presence of ester and carbamate bonds within the ligand. Similar components and component ratios have been found on the 2(D/L) complex. Thus, the C 1s signals indicate that the structural integrity of the ligands is maintained after reduction.
O contribution can be detected at (289.2 ± 0.2) eV, confirming the presence of ester and carbamate bonds within the ligand. Similar components and component ratios have been found on the 2(D/L) complex. Thus, the C 1s signals indicate that the structural integrity of the ligands is maintained after reduction.
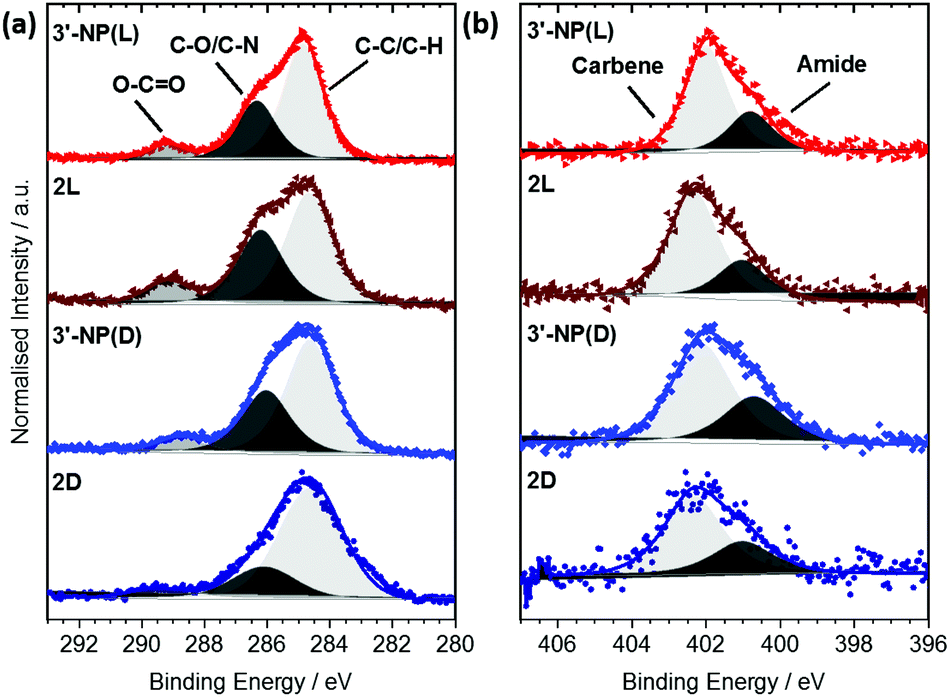 | ||
| Fig. 3 Normalised C 1s (a) and N 1s (b) XP spectra of 3′-NP(D/L) NHC-AuNPs and the 2(D/L) complexes. Raw data are shown as coloured scatter plots. Deconvolutions are shown in grey, resulting envelopes in colour. | ||
The N 1s signals of 3′-NP(D/L) can be deconvoluted into two components at (400.9 ± 0.3) eV and (402.1 ± 0.3) eV (Fig. 3(b)). Hereby the signal at 402.1 eV can be attributed to the carbene type structure whereas the signal at 400.9 eV is attributed to the nitrogen within the amide/carbamate. Binding energy (BE) and relative intensity ratios of the components are in good agreement with the N 1s signals observed in other NHC-stabilized metal nanoparticles.19,24 N 1s signals of the 2(D/L) complex are not significantly different to those of stabilized NPs, again indicating that the NHC ligand is not affected by the reduction process.
XPS data of the Au 4f region of AuNPs have previously been reported by Ling et al.20 as well as by Bridonneau et al.19 and in most cases the authors observed a Au 4f signal composed of two peaks at 84 eV (Au 4f7/2) and 88 eV (Au 4f5/2), consistent with Au(0) (Fig. 4).19,20,25 However, the Au 4f spectra of all NHC-AuNP samples measured in this study not only displayed signals at (84.2 ± 0.3) eV and (87.9 ± 0.3) eV, but also showed significant contributions at (86.2 ± 0.3) eV and (89.9 ± 0.3) eV. The difference in binding energy (ΔBE) between Au(0) and the second component (ΔBE ∼2.0 eV) points to the oxidised Au being mainly Au(I) (ΔBE ∼1.6 eV) and not Au(III) (ΔBE ∼3.3 eV).26
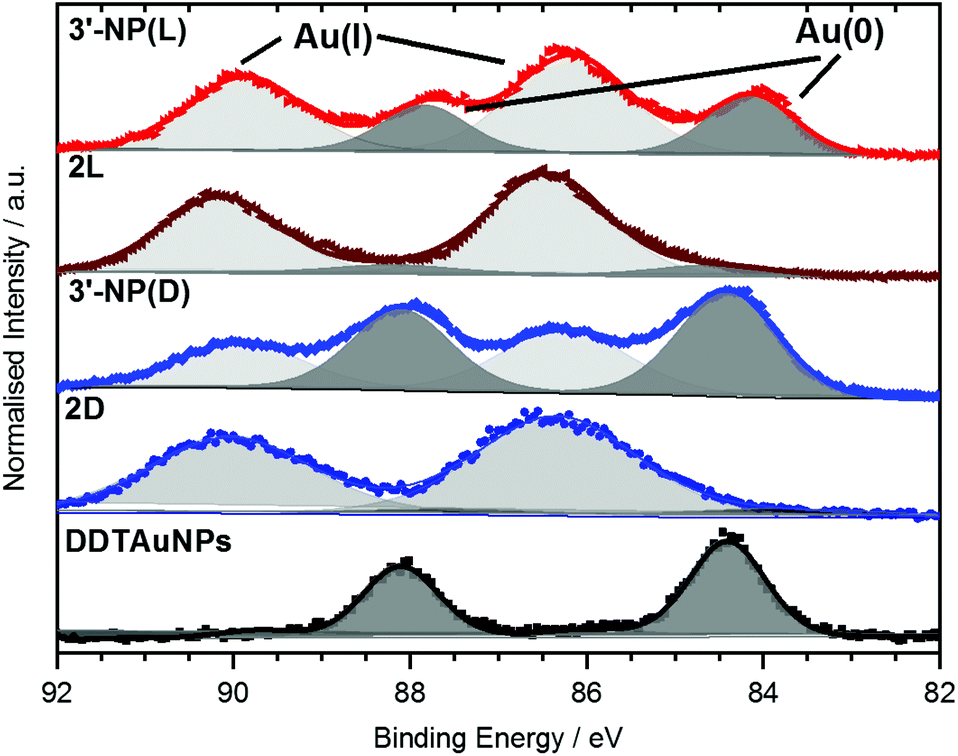 | ||
| Fig. 4 Normalised Au 4f XPS narrow scan spectra of 3′-NP(D/L) NHC-AuNPs and the 2(D/L) complex. Raw data are shown as coloured scatter plots. Deconvolutions are shown in grey, resulting envelopes in colour. | ||
To evaluate if the Au(I) signals arose from unconverted NHC–Au(I)Cl complexes, the Cl 2p signal was measured. Whereas no contributions of Cl 2p were detected for 3′-NP(L), small amounts of Cl were found for 3′-NP(D). Nevertheless, Cl intensity does not scale with the amount of Au(I) detected on those samples, therefore indicating that the detected Cl may not originate from the NHC–Au(I) complex, but more likely from the dichloromethane (DCM) used as solvent for spin coating.
To further elucidate the nature of the Au(I) signal, XPS analysis of the NHC–Au(I) complexes were performed. Au 4f spectra for 2D and 2L showed mainly Au(I) contributions at (86.3 ± 0.2) eV and (90.0 ± 0.2) eV respectively (Fig. 4). These values are shifted by 0.1 eV towards higher BE and therefore are too close to the observed BE values for Au(I) in the NHC-AuNP samples to conclusively rule out the presence of NHC–Au(I)Cl species in the latter. However, 13C NMR analysis of the nanoparticles samples independently indicates that there is no NHC–Au(I)Cl complex present after reduction (Fig. S5†). Therefore, the Au(I) detected in the Au 4f XP spectra is likely present in the AuNPs themselves.
These results are comparable to findings by others, whereby NHC-AuNPs synthesized via a molecular NHC–Au(I) complex were found to possess a layer of Au(I) atoms on top of an Au(0) core, although the exact nature of these Au(I) species still remains unsolved.27 We believe that the presence of Au(I) within our NHC-stabilized AuNPs arises from the synthetic route utilizing reduction of molecular NHC–Au(I) complex to NHC-stabilized AuNPs. Unfortunately, our attempts to prepare analogous NHC-AuNPs from their haloaurate salts (Scheme 2) were unsuccessful.19 Nevertheless, reports by others showed that when NHC-AuNP compounds are synthesised from their imidazolium haloaurate salts, no Au(I) species has been detected by means of XPS.19,20 Furthermore, DDTAuNPs were synthesized via reduction of Au(III) salt and ligand exchange with dodecanethiol for comparison purposes. XPS analysis showed that mainly one Au 4f signal was found (Fig. 4, bottom). This signal can be attributed to Au(0), with indication of only minor amounts of Au(I). Taken together, these results further serve to support our hypothesis that NHC-AuNPs synthesized via NHC–Au(I) complexes may retain Au(I) species in the resulting nanoparticles.
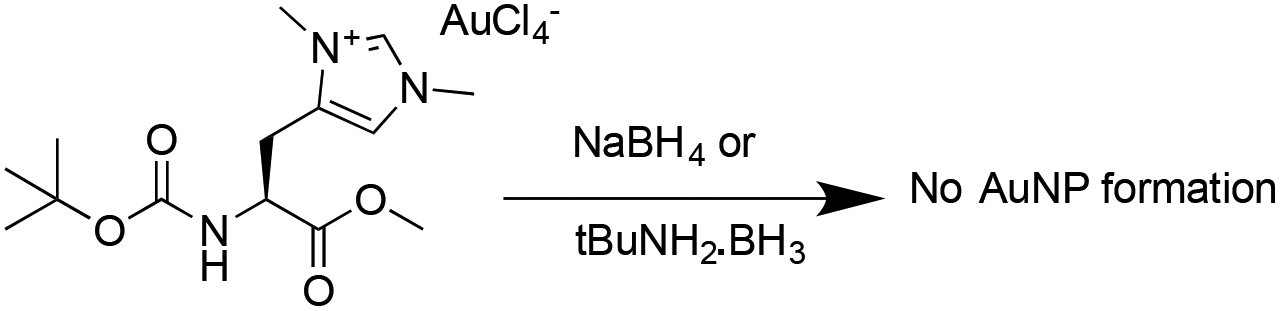 | ||
| Scheme 2 Attempted synthesis of AuNP from haloaurate derivative. | ||
As it is expected that an Au(I) versus an Au(0) surface layer should have a significant influence on the reactivity and stability of such NPs, further studies need to be conducted to understand the reason for Au(I) presence and its nature in the NHC-AuNPs, so as to elucidate design rules for AuNPs with improved functionality. For example, Ye et al. demonstrated that surface binding energies of Au atoms in AuNPs had a noticeable impact on their catalytic activity for lactonization of allene carboxylic acids.27 The importance of Au(I) atoms is further supported as the same reaction has been shown to proceed when a Au(I) NHC complex is exploited.28 With respect to stability, Pileni et al. demonstrated superior oxygen stability of NHC-AuNPs synthesised from NHC–Au(I) complexes when compared to DDTAuNPs.29 Although the oxidation states present in the NHC-AuNPs were not disclosed in the paper, we presume that a mixture of Au(I)/Au(0) oxidation states is likely present in the AuNPs due to the starting materials employed. In contrast, Crudden et al. have shown lower stability of NHC–Au(I) complex-derived AuNPs compared to that of Au(0) derived AuNPs, and Johnson et al. demonstrated release of NHC–Au(I) complexes when they exposed their NHC-AuNPs to thiol ligands.7,16 Therefore, we believe that the method or precursor exploited in the synthesis of NHC-stabilised AuNPs and the nature of the Au oxidation state is fundamentally tied to the overall stability of the resulting AuNPs. As such, future work in our group will focus on synthetic routes to both Au(I)/Au(0) AuNPs and the respective Au(0) AuNP analogues for comparative XPS and stability studies.
Conclusions
We have reported the synthesis of AuNPs stabilised by both L and D enantiomers of histidine derived NHC ligands giving rise to optical activity. This new method of synthesis in conjunction with our previously reported procedure demonstrates that the size of histidine derived NHC-AuNPs is tuneable by simply changing the reducing agent. Bonding environments of the resulting AuNPs were investigated through XPS analysis. The XPS analysis of the NHC stabilised AuNPs shows a combination of Au(I) and Au(0) oxidation states. However, in comparison, a thiol stabilised AuNP only exhibited Au with Au(0) oxidation state. We assume that the NHC derived AuNPs possess a Au(I) monolayer surrounding a Au(0) core. From examining previous reports of NHC-derived AuNPs, we postulate the method in which NHC AuNPs are synthesised determines whether Au(I)/Au(0) hybrids or Au(0) containing AuNPs are obtained. This finding could be of particular interest when considering the use of NHC-stabilised AuNPs in the fields of catalysis and biological applications, due to the potential difference in stability and activity between the two different kinds of AuNPs.Experimental
Synthesis
Cγ), 5.81 (d, J = 8.0 Hz, 1H, NHCO), 4.54 (br, CαHCOO), 3.78 (m, 9H, NCH3 and OCH3), 3.19–2.92 (m, 2H, CβH2), 1.43 (s, 9H, (CH3)3). 13C NMR (125 MHz, CDCl3) δ 184.7 (NCεN), 171.2 (COO), 155.2 (CONH), 129.4 (CγCHη), 120.0 (CηHCγ), 80.9 (C(CH3)3), 53.1 (OCH3), 52.4 (CαHCOO), 38.1 (NCH3), 35.3 (NCH3), 29.9 ((CH3)3), 28.3 (CβH2).
Given the average particle size (2.35 nm), the number of gold atoms in the metal core is estimated to be ∼263 by the method of Leff et al.30
Cγ), 5.90 (d, J = 7.6 Hz, 1H, NHCO), 4.54 (m, CαHCOO), 3.84 (s, 3H, NCH3), 3.82 (s, 3H, NCH3), 3.78 (s, 3H, OCH3), 3.23–2.87 (m, 2H, CβH2), 1.39 (s, 9H, (CH3)3). 13C NMR (125 MHz, CDCl3) δ 184.6 (NCεN), 171.4 (COO), 156.2 (CONH), 130.5 (CγCHη), 121.3 (CηHCγ), 79.1 (C(CH3)3), 53.1 (OCH3), 52.5 (CαHCOO), 38.3 (NCH3), 35.7 (NCH3), 29.8 ((CH3)3), 28.4 (CβH2).
Given the average particle size (2.25 nm), the number of gold atoms in the metal core is estimated to be ∼232 by the method of Leff et al.30
Sample preparation for XPS measurements
Solutions of 2(D), 2(L), 3′-NP(D), 3′-NP(L) and DDTAuNP were dissolved in minimal amount of DCM and spin coated at 2000 rpm onto cleaned Si wafers separately. Si wafers were washed with toluene (twice) and acetone, then dried in an oven at 100 °C before spin coating any samples.XPS measurements
The measurement series was carried out on a SPECS XPS-spectrometer equipped with a monochromatic Al-Kα X-ray (1486.6 eV) source (μFocus 350) and a hemispherical WAL-150 analyser (acceptance angle: 60°). All samples were mounted onto the sample holder using double-sided carbon tape.Pass energies of 100 eV and 30 eV and energy resolutions of 1 eV and 100 meV were used for survey and detail spectra respectively (beam energy and spot size: 70 W onto 400 μm; source angle: 30° to sample surface normal; analyser angle: 51° to sample surface normal; base pressure: 8 × 10−10 mbar; pressure during measurements: 3 × 10−9 mbar). Low energy electrons from a broad spot flood gun (SPECS FG 22, 5 eV, 25 μA) were used to minimise charging effects during XPS measurements.
Data analysis was performed using CASA XPS software, employing transmission corrections (as per the instrument vendor's specifications), Shirley/Tougaard backgrounds31,32 and Scofield sensitivity factors.33 For charge correction the SiO2 species occurring in the Si 2p spectra of the substrates was used and set to 103.6 eV BE. Deconvolution of the spectra was carried out using least-square fitting (Marquardt) with Gaussian–Lorentzian (GL(30)) peak shapes.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. R. R. thanks the University of Vienna for a start-up grant. A. J. Y. thanks the University of Hull for the provision of a University Scholarship. C. J. S. thanks the University of Kent for financial support. A. S. thanks the Leverhulme Trust for financial support (RPG-2017-188).Notes and references
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer, Berlin, Heidelberg, 1995, vol. 25 Search PubMed.
- S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef.
- K. Salorinne, R. W. Y. Man, C.-H. Li, M. Taki, M. Nambo and C. M. Crudden, Angew. Chem., Int. Ed., 2017, 56, 6198–6202 CrossRef CAS PubMed.
- A. Ferry, K. Schaepe, P. Tegeder, C. Richter, K. M. Chepiga, B. J. Ravoo and F. Glorius, ACS Catal., 2015, 5, 5414–5420 CrossRef CAS.
- P. Lara, L. M. Martínez-Prieto, M. Roselló-Merino, C. Richter, F. Glorius, S. Conejero, K. Philippot and B. Chaudret, Nano-Struct. Nano-Objects, 2016, 6, 39–45 CrossRef CAS.
- C. J. Serpell, J. Cookson, A. L. Thompson, C. M. Brown and P. D. Beer, Dalton Trans., 2013, 1385–1393 RSC.
- M. J. MacLeod and J. A. Johnson, J. Am. Chem. Soc., 2015, 137, 7974–7977 CrossRef CAS PubMed.
- S. Roland, X. Ling and M.-P. Pileni, Langmuir, 2016, 32, 7683–7696 CrossRef CAS PubMed.
- J. Vignolle and T. D. Tilley, Chem. Commun., 2009, 21, 7230–7232 RSC.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef CAS PubMed.
- G. Wang, A. Rühling, S. Amirjalayer, M. Knor, J. B. Ernst, C. Richter, H.-J. Gao, A. Timmer, H.-Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, Nat. Chem., 2016, 9, 152–156 CrossRef PubMed.
- J. Crespo, Y. Guari, A. Ibarra, J. Larionova, T. Lasanta, D. Laurencin, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos and S. Richeter, Dalton Trans., 2014, 15713–15718 RSC.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- P. Pyykk and N. Runeberg, Chem. – Asian J., 2006, 1, 623–628 CrossRef PubMed.
- X. Ling, N. Schaeffer, S. Roland and M.-P. Pileni, Langmuir, 2015, 31, 12873–12882 CrossRef CAS PubMed.
- R. W. Y. Man, C.-H. Li, M. W. A. MacLean, O. V. Zenkina, M. T. Zamora, L. N. Saunders, A. Rousina-Webb, M. Nambo and C. M. Crudden, J. Am. Chem. Soc., 2017, 140, 1576–1579 CrossRef PubMed.
- A. J. Young, C. J. Serpell, J. M. Chin and M. R. Reithofer, Chem. Commun., 2017, 53, 12426–12429 RSC.
- A. Sharma, B. P. Singh and A. K. Gathania, Indian J. Pure Appl. Phys., 2014, 52, 93–100 CAS.
- N. Bridonneau, L. Hippolyte, D. Mercier, D. Portehault, M. Desage-El Murr, P. Marcus, L. Fensterbank, C. Chanéac and F. Ribot, Dalton Trans., 2018, 6850–6859 RSC.
- X. Ling, S. Roland and M. P. Pileni, Chem. Mater., 2015, 27, 414–423 CrossRef CAS.
- M. R. Narouz, C.-H. Li, A. Nazemi and C. M. Crudden, Langmuir, 2017, 33, 14211–14219 CrossRef CAS PubMed.
- H. Lu, Z. Zhou, O. V. Prezhdo and R. L. Brutchey, J. Am. Chem. Soc., 2016, 138, 14844–14847 CrossRef CAS PubMed.
- E. C. Hurst, K. Wilson, I. J. S. Fairlamb and V. Chechik, New J. Chem., 2009, 33, 1837–1840 RSC.
- L. M. Martínez-Prieto, L. Rakers, A. M. López-Vinasco, I. Cano, Y. Coppel, K. Philippot, F. Glorius, B. Chaudret and P. W. N. M. van Leeuwen, Chem. – Eur. J., 2017, 23, 12779–12786 CrossRef PubMed.
- A. Bakker, A. Timmer, E. Kolodzeiski, M. Freitag, H. Y. Gao, H. Mönig, S. Amirjalayer, F. Glorius and H. Fuchs, J. Am. Chem. Soc., 2018, 140, 11889–11892 CrossRef CAS PubMed.
- J. P. Sylvestre, S. Poulin, A. V. Kabashin, E. Sacher, M. Meunier and J. H. T. Luong, J. Phys. Chem. B, 2004, 108, 16864–16869 CrossRef CAS.
- R. Ye, A. V. Zhukhovitskiy, R. V. Kazantsev, S. C. Fakra, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2018, 140, 4144–4149 CrossRef CAS PubMed.
- X. Z. Shu, S. C. Nguyen, Y. He, F. Oba, Q. Zhang, C. Canlas, G. A. Somorjai, A. P. Alivisatos and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7083–7086 CrossRef CAS PubMed.
- X. Ling, N. Schaeffer, S. Roland and M. P. Pileni, Langmuir, 2015, 31, 12873–12882 CrossRef CAS PubMed.
- D. V. Leff, P. C. Ohara, J. R. Heath and W. M. Gelbart, J. Phys. Chem., 1995, 99, 7036–7041 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- S. Tougaard, Surf. Interface Anal., 1997, 25, 137–154 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr00905a |
| This journal is © The Royal Society of Chemistry 2019 |