
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple fabrication of layered halide perovskite platelets and enhanced photoluminescence from mechanically exfoliated flakes†
Balaji
Dhanabalan
ac,
Andrea
Castelli
 a,
Milan
Palei
b,
Davide
Spirito
b,
Liberato
Manna
a,
Roman
Krahne
*b and
Milena
Arciniegas
*a
a,
Milan
Palei
b,
Davide
Spirito
b,
Liberato
Manna
a,
Roman
Krahne
*b and
Milena
Arciniegas
*a
aNanochemistry Department, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy. E-mail: Milena.Arciniegas@iit.it
bOptoelectronics Group, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy. E-mail: Roman.Krahne@iit.it
cDipartimento di Chimica e Chimica Industriale, Università degli Studi di Genova, Via Dodecaneso, 31, 16146, Genova, Italy
First published on 10th April 2019
Abstract
Rapid progress on the fabrication of lead halide perovskite crystals has led to highly promising performance in optoelectronic devices, particularly from three-dimensional crystals. Recently, these efforts have been extended to layered perovskite structures, motivated in part by their good environmental stability. Furthermore, layered perovskites represent a nanocrystal system with micron-size extensions and strong confinement in one dimension that is highly appealing for studying fundamental photophysics. Here, we report a facile route for the growth of single-layered perovskite platelets, which is demonstrated using four different organic cations acting as spacers, providing a layer interdistance from approx. 1.3 nm to 2.4 nm. The resulting ensembles of platelets exhibit a strong emission band in the deep blue spectral region characterized by two emission peaks and a photoluminescence quantum yield (PLQY) up to 15%. Thin 2D layered perovskite flakes can be readily obtained by mechanical exfoliation, and their emission shows a PLQY as high as 42%, which can be related to reduced reabsorption in the exfoliated crystals. Furthermore, the low energy peak that was observed in the double peak emission from the platelet ensembles is suppressed in the exfoliated flakes. Therefore, the exfoliated flakes manifest a more colour-pure blue emission with strongly increased radiative rate as compared to the dried platelet aggregates obtained directly from the synthesis. The straightforward fabrication strategy that employs solely polar solvents with low environmental impact provides a highly appealing route towards two-dimensional perovskite systems with bright and stable emission in the blue spectral range.
Introduction
In recent years, lead halide perovskites have attracted much attention due to their exceptional electronic and optical properties and low-cost fabrication and solution processability.1–5 In addition to the typically cubic, three-dimensional nanocrystals, structures with lower dimensionality such as perovskite nanosheets, nanowires, and the so-called 0D perovskites have been fabricated through simple modifications of wet chemistry fabrication protocols.6–9 Among the low-dimensional perovskites, two-dimensional (2D) layered crystals have been the focus of several studies, given their bright emission, possible white fluorescence, and, in particular, their environmental stability.10–14 Moreover, 2D perovskites have exciton binding energies of about 300 meV that are larger than those of their 3D counterparts, which is advantageous for potential applications in the field of optoelectronics.15–17Layered perovskite crystals are made of a sequence of inorganic layers formed by corner-sharing [PbX6]4− octahedra, which are chemically passivated by organic moieties with smaller dielectric constant and higher exciton binding energies than the inorganic layer.18,19 The use of different organic moieties in the fabrication of 2D layered perovskites influences the layer arrangements, and it can also induce structural distortions, and thereby alter the electronic structure of the resulting material.20–22 Thus, the choice of the organic moieties can be seen as a strategy to tune the photoluminescence (PL) of layered perovskites at the nano- and macro-scale. Until now, extensive research into green- and red-emitting low-dimensional perovskites has been reported, achieving relatively high PLQY,23–26 while the synthesis of blue- and white-emitting layered crystals with desired performance is still a challenge. For instance, the maximum PLQY reported for microscopic white-emitting layered perovskites remains around 9%,27–29 and in the case of blue-emitting low-dimensional crystals, a PLQY of 26% from atomically thin structures9 and 54% from microdisks in solution have been reached when using multiple solvents,30 although significantly higher PLQY values have been reported for other types of white- and blue-emitting perovskites.31,32 In line with this, the current synthetic protocols used for high-quality layered perovskite materials commonly involve a pre-preparation process of synthesizing ammonium halide salts, long reaction times, and the use of multiple solvents such as dimethylformamide, dimethyl sulfoxide, N-methylformamide, hexane, toluene, and acetone, making the purification and drying of the layered structures difficult.27,28,30,33
In this work, we report a facile strategy for the fabrication of layered perovskite platelets using different types of amines, which serve as spacers between layers of octahedra. These amines are dissolved in acetone, which is a much more eco-friendly polar solvent compared to the ones typically used. Except for the decylamine-based crystals that show a nearly white emission and a broad long tail up to 700 nm, all the as-synthesized structures exhibit a deep blue emission associated with two emission peaks stemming from single layers. In a single crystal, multiple layers are stacked and their spacing depends on the organic molecule species, from about 1.3 nm (for butylamine) to 2.4 nm (for decylamine). From our syntheses, we obtain large aggregates of such crystals that in the following we will call ‘ensembles’. From such ensembles, we measured relatively high PLQYs of approx. 8% and 15% for the layered crystals prepared using butylamine and phenethylamine, respectively. The PLQY is strongly enhanced when exfoliating flakes with few monolayers from these ensembles and reaches 18% and 42%, correspondingly. Our work provides a novel route for a low-cost, flexible, and rapid processability of layered perovskites toward device fabrication, by combining a straightforward synthesis using more ecofriendly solvents, with a facile exfoliation process that produces thin flakes with bright blue emission.
Results and discussion
Fabrication and structural characterization
Hybrid ammonium lead halide layered perovskite crystals were synthesized through a facile protocol in which only acetone was used as solvent to prepare a mixture of PbBr2 and HBr. Two different aliphatic primary amines – butylamine and decylamine – and two aromatic ones – benzylamine and phenethylamine – were selected to investigate their role as an organic spacer on the optical properties of the final crystals. The selected amine was directly injected into the prepared mixture, as illustrated in Fig. 1a (see the Experimental section for details). With this simple protocol, we obtained layered structures with the general formula RAm2MAn−1PbnBr3n+1, where RAm is the general ammonium ion resulting from the protonation of the selected primary amine, MA corresponds to methylammonium ions and n is the number of layers in the structures. The prepared material is formed of stacked monolayers (n = 1) because no MA source nor other ions (such as formamidinium or Cs+) that can promote a 3D crystal growth were employed in the synthesis.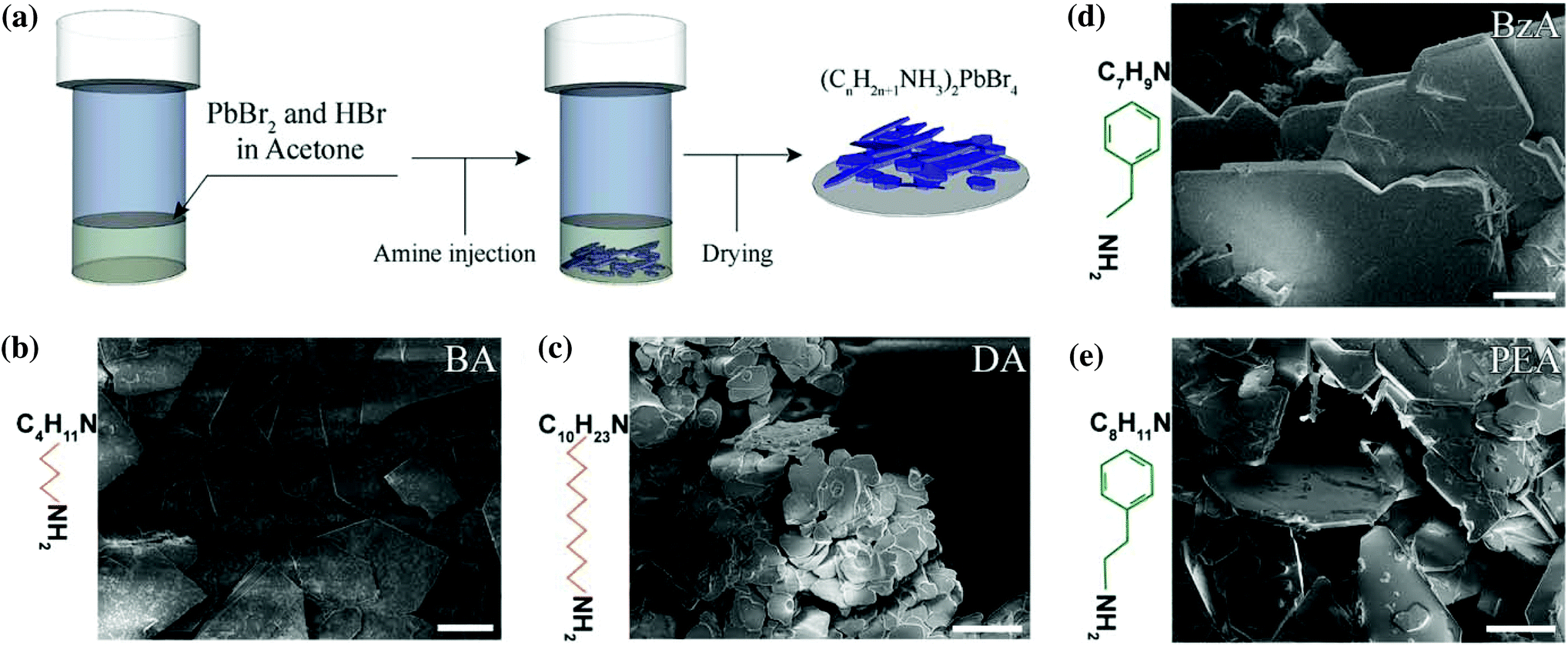 | ||
| Fig. 1 (a) Schematic diagram of the facile fabrication process of the layered perovskite by injecting a primary amine to a mixture of PbBr2 in HBr and acetone. (b–e) SEM images of the produced platelets when using short and long aliphatic molecules, such as butylammonium (BA) (b) and decylammonium (DA) (c), and aromatic ones as benzylammonium (BzA) (d) and phenethylammonium (PEA) (e). Scale bars: 100 μm. | ||
Thin microscopic-sized crystals with a platelet shape were observed immediately after the addition of the selected amine and they precipitated at the bottom of the vial in a few seconds (Movie S1†). The ensembles of platelets were collected and dried before further analysis was performed.
Fig. 1b–e report a collection of Scanning Electron Microscopy (SEM) images of the resulting crystals showing the formation of two-dimensional structures with a platelet-like morphology from all the amines used in the experiments. A macroscopic view of the crystal morphologies is provided in Fig. S1.†
More extended platelets with sharper facets are observed from butylammonium (BA)- (Fig. 1b), benzylammonium (BzA)- (Fig. 1d), and phenethylammonium (PEA)-based crystals (Fig. 1e), with lateral sizes of a few hundreds of micrometers, whereas more rounded structures are observed from the decylammonium (DA)-based ones (Fig. 1c), with a reduced lateral size of less than 60 μm. Such differences in the size of the as-synthesized perovskites when using decylamine in their synthesis might be ascribed to a hindered diffusion of the precursors to the growing flakes. That is, the long and hydrophobic DA tails that surround the initial flakes might act as a diffusion barrier for the migration of Pb2+ and Br− ions to favour the growth of larger structures from the precursor solution.
X-ray diffraction (XRD) patterns acquired from the ensembles are shown in Fig. 2a. Equally spaced and sharp diffraction peaks arising from (0 0 2l) reflections are observed in all the structures that were prepared with the different organic cations. This is an indication of the single-phase and highly oriented nature of the crystals, which is in line with a previous work from our group on layered perovskite flakes obtained through a more complex process.28 The periodicity, d, obtained from the diffraction peaks as a function of the type of amine used in the experiments is shown in Fig. 2b. That is, 13.83 ± 0.06 Å and 24.12 ± 0.14 Å for BA- and DA-based crystals, respectively; and 16.71 ± 0.15 Å and 16.68 ± 0.07 Å for BzA- and PEA-based crystals. In the case of the aliphatic amines, the crystals synthesized by employing a longer chain are characterized by a significantly larger periodicity than those prepared with a shorter one, as expected (Fig. 2b). However, in the case of aromatic amines, the resulting layered structures exhibit a similar periodicity d, independent of the nominal size of their organic cations. We confirmed this behavior using octylamine and dopamine in our fabrication protocol as additional aliphatic and aromatic primary amines, respectively. The collected XRD patterns from these structures are shown in Fig. S2a.† The calculated d values are 21.11 ± 0.11 Å and 17.14 ± 0.06 Å for octylamine- and dopamine-based crystals, respectively. The lack of differences on the periodicity when using different lengths of aromatic amines indicates the ability of such amines to acquire a relaxed structural state when packing around the octahedral layers, in particular for PEA molecules that can adopt a rotational organization in similar structures to favour a π–π stacking.33 Considering the obtained d values, the 6 Å thickness of a lead halide octahedral layer, and the length of the organic spacers, the estimated periodicity corresponds to one inorganic monolayer (n = 1) for all the amines. Therefore, all the resulting crystals are made of single [PbBr4]2− octahedral layers coated on the top and bottom by the selected cation. The [PbBr4]2− octahedral layer passivation of the cations leads to an interdistance of ca. 0.8 nm and 1.8 nm for (BA)2PbBr4 and (DA)2PbBr4, respectively, and to 1.07 nm for the aromatic amines (BzA)2PbBr4 and (PEA)2PbBr4.
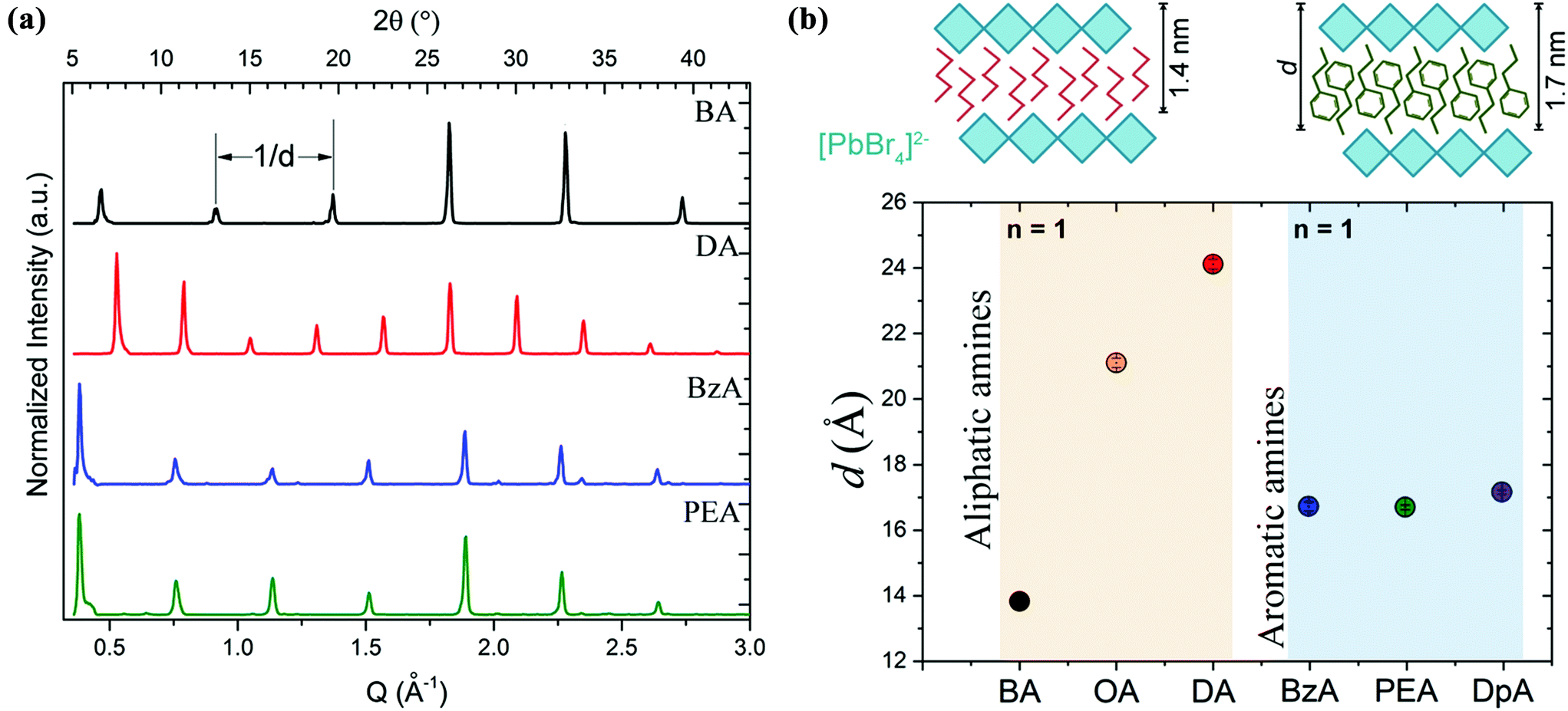 | ||
| Fig. 2 (a) X-ray diffraction patterns collected from the as-synthesized platelets prepared from different types of amines showing periodic diffraction peaks that correspond to a layer stacking with an interdistance, d, related to the ordered stacking of [PbBr4]2− layers separated by organic spacers. (b) The calculated interdistance d as a function of the type of cation of the fabricated layered perovskites, including octylammonium (OA) and dopammonium (DpA). The sketches in (b) highlight the layered structure formed with two different types of cations (BA in red and PEA in green), while preserving similar d values. | ||
Optical characterization of the layered perovskites ensembles
In contrast to the 3D crystals of APbBr3 perovskites (with A = Cs, methylammonium or formammidinium) that exhibit green emission,7 the ensembles of layered platelets produced in this work show a bluish-white emission under UV light (Fig. S3†). This blue-shifted emission is in line with a strong confinement of charge carriers inside the inorganic layer of octahedra, as it is observed in other colloidal quantum well systems such as CdSe nanoplatelets.34–36Fig. 3a displays a set of absorption and emission spectra collected from the selected layered perovskites. The PL profiles acquired from (OA)2PbBr4 and (DpA)2PbBr4 are shown in Fig. S2b.†Table 1 summarizes the emission and absorbance peak positions retrieved by fitting the spectra with Lorentzian functions; in the case of (BzA)2PbBr4 crystals, a sum of a Gaussian and a Voigt function was needed to fit the PL profile (Fig. S4†). All the absorption spectra show two peaks in the 390–429 nm region (3.16 eV–2.89 eV) that can be related to confined charge carrier states. Both the high- and low-energy peaks result from single-layer (n = 1) perovskites, as corroborated by the XRD analysis. | ||
| Fig. 3 (a) Absorption (thin lines) and photoluminescence (thick lines) spectra acquired from the different perovskite platelet ensembles. The dotted lines indicate the emission peaks located at 412 nm (P1) and 426 nm (P2) from the BA-based structures that are similar to those observed from PEA-based ones. The PLQY is also included in the panel. (b) CIE chromaticity coordinates of the as-synthesized layered perovskites showing that, except for the DA-based crystals, the structures exhibit a deep-blue colour. (c) Decay traces of the normalized PL as a function of time for the four different single-layer perovskites acquired at the high-energy peak, P1. | ||
| Perovskite ensembles | Absorption peaks, nm (eV) | Emission peaks, nm (eV) | ||
|---|---|---|---|---|
| P1 | P2 | P1 | P2 | |
| (BA)2PbBr4 | 392 (3.16) | 412 (3.01) | 411 (3.02) | 429 (2.89) |
| (DA)2PbBr4 | 377 (3.29) | 399 (3.11) | 396 (3.13) | 406 (3.05) |
| (BzA)2PbBr4 | 389 (3.19) | 412 (3.01) | 410 (3.02) | 419 (2.97) |
| (PEA)2PbBr4 | 396 (3.13) | 414 (3.00) | 412 (3.01) | 426 (2.91) |
In addition, the peaks obtained from the DA-based crystals are slightly blue-shifted with respect to those of the other crystals. Since the periodicity d is significantly larger for the DA-based platelets, this blue shift could be correlated to a stronger confinement of the excitons in the 2D layer. The PL spectra display two red-shifted emission peaks (P1 and P2 at high and low energies, respectively), when compared to the absorption; this includes the (BzA)2PbBr4 crystals, which show an emission peak P1 that contributes less to the PL distribution, as indicated by our fitting analysis reported in Fig. S4.†
Furthermore, all platelets exhibit a broad PL tail that extends up to the red region of the visible spectrum, which can be attributed to emission from defect states formed within the layers of the perovskite crystal that is typically observed from white-emitting layered perovskites.28,37,38 This tail is most pronounced for the (DA)2PbBr4 crystals. Interestingly, the PLQY obtained from the ensembles is much higher when the two emission peaks are well defined and well separated, as is the case for the BA- and PEA-based crystals (Fig. 3a, top and bottom spectra), which exhibit PLQY values of 8% and 15%, respectively, with a standard deviation below 2% (see Table S1†). We note also that for these platelets, the low energy peak is stronger than the high-energy one in their PL spectra (Fig. 3a). For the emission from such monolayer (n = 1) perovskite structures, the interlayer coupling within a single platelet can be expected to play a minor role, which is supported by the similarity of the optical spectra from the (BA)2PbBr4 and (PEA)2PbBr4 crystals that manifest slightly different periodicity. One other very interesting aspect is that the high-energy emission peak P1 overlaps with the low energy absorption band for all crystals. This overlap is particularly evident for the BA- and PEA-based crystals, where the peaks are well defined, and the P1 emission peak is found at higher energy than the maximum of the low energy absorption peak (see dashed lines in Fig. 3a as a guide to the eye). Fig. 3b shows the corresponding Commission Internationale de l'Eclairage (CIE) coordinates for the layered perovskites studied in this work, indicating that their emission colours are located in the deep-blue region, except for (DA)2PbBr4 crystals that display a near-white emission caused by their strong low energy tail. The PL decay traces reported in Fig. 3c show a much faster decay for the (DA)2PbBr4 and (BzA)2PbBr4 ensembles of platelets than for the (BA)2PbBr4 and (PEA)2PbBr4 ones, which correlates with their low PLQY. Correspondingly, the fastest lifetime component, τ1, has a much larger weight in the curve fitting, as compared to the bright emitting (BA)2PbBr4 and (PEA)2PbBr4 perovskites, while the dominant longer lifetime component, τ2, is reduced, as reported in Table S1.† That, together with the PLQY, could indicate that τ1 is mostly due to non-radiative decay, while the lifetime of the radiative decay is dominated by τ2, and indeed its weight is much stronger for the bright emitting ensembles. The average lifetimes that could be extracted from the fitting (see Fig. S7 in the ESI† for details) reflect the faster decay of the less bright perovskite crystals and yield 3.44 ns and 2.33 ns for the (DA)2PbBr4 and (BzA)2PbBr4 ensembles, respectively, and 5.43 ns and 9.24 ns for the (BA)2PbBr4 and (PEA)2PbBr4 ones, respectively. Therefore, we infer that the organic moieties play a key role in the photophysics of the layered perovskite crystals, and that the DA and BzA molecules appear to lead to a less efficient passivation of the octahedral layers, and thus to more defects that induce a more pronounced non-radiative decay.
Exfoliation of perovskite flakes
The ensembles of platelets were mechanically exfoliated using a non-emissive 3 M Scotch tape and the exfoliated flakes were transferred to a SiO2/Si substrate (see the Experimental section for details). In the following, we focus on the brighter (BA)2PbBr4 and (PEA)2PbBr4 crystals, where optical investigation of the exfoliated flakes was possible.Fig. 4a and b show optical images of the exfoliated flakes, evidencing different colour contrasts due to the thin film interference effect. The colour resulting from these interferences can be associated with the number of monolayers in the exfoliated flake. The white-framed regions in Fig. 4a and b contain the flakes with the smallest number of monolayers, x. To determine x, we used atomic force microscopy (AFM) operating in intermittent contact mode. The topographic AFM images are displayed in Fig. 4c and d, and the height profiles along the pink and blue dotted lines are shown in Fig. 4e and f. As discussed earlier, the layers in the (BA)2PbBr4 and (PEA)2PbBr4 crystals are separated by about 1.4 nm and 1.7 nm, respectively. Thus, the steps in the height profiles in Fig. 4e and f of the thin (blue) and thick (pink) exfoliated flakes correspond to 6 and 12 layers for (BA)2PbBr4 flakes, and to 5 and 13 layers for (PEA)2PbBr4.
 | ||
| Fig. 4 (a–b) Optical micrographs of the mechanically exfoliated (BA)2PbBr4 (a) and (PEA)2PbBr4 (b) flakes transferred to a SiO2/Si substrate. Optical interferences between the flake surface and the substrate lead to different colours that are related to different thicknesses. Scale bars: 15 μm. (c–d) Intermittent contact mode AFM images collected from the white-framed regions in (a) and (b), respectively. Scale bars: 1 μm. (e–f) AFM height profiles acquired along the blue and pink dotted lines in (c) and (d) showing a step height (in blue) of 8.2 nm that corresponds to 6 monolayers, and a 20.3 nm (12 monolayers) step (in pink) for exfoliated (BA)2PbBr4 flakes (e). Similar steps in height are found in exfoliated (PEA)2PbBr4 flakes (f). The pink arrow in (d) indicates a thick region containing approximately 13 monolayers (with a height of 22.4 nm). | ||
Next, we collected the absorption and emission spectra from the exfoliated flakes, which are displayed in Fig. 5a for (PEA)2PbBr4 crystals and in Fig. S5† for (BA)2PbBr4 ones. Table S2† lists the absorbance and emission peak positions before and after exfoliation of the selected crystals. The absorbance manifests a double-peak structure as observed from the ensembles, and its profile does not depend significantly on the region from which the spectra were collected, i.e. from regions with predominantly thick or thin flakes. Compared to the ensemble absorbance spectra, the low energy peak of the flake absorbance has a strongly reduced intensity and is blue shifted by a few nanometers. The emission spectrum exhibits only one sharp and well-defined peak that occurs at the energy of the P1 emission peak recorded from the ensembles. Therefore, the low energy emission (P2) that was observed from the ensembles is either strongly suppressed or not present. Furthermore, we measured the PLQY of the exfoliated flakes and compared the values to those of the ensembles (see the Methods section for details). We found that the exfoliated crystals show a much higher PLQY, by a factor of 2–3, than that of their ensembles (see comparative analysis in Fig. S6†), with average PLQY values of 18% and 42% for (BA)2PbBr4 and (PEA)2PbBr4, respectively.
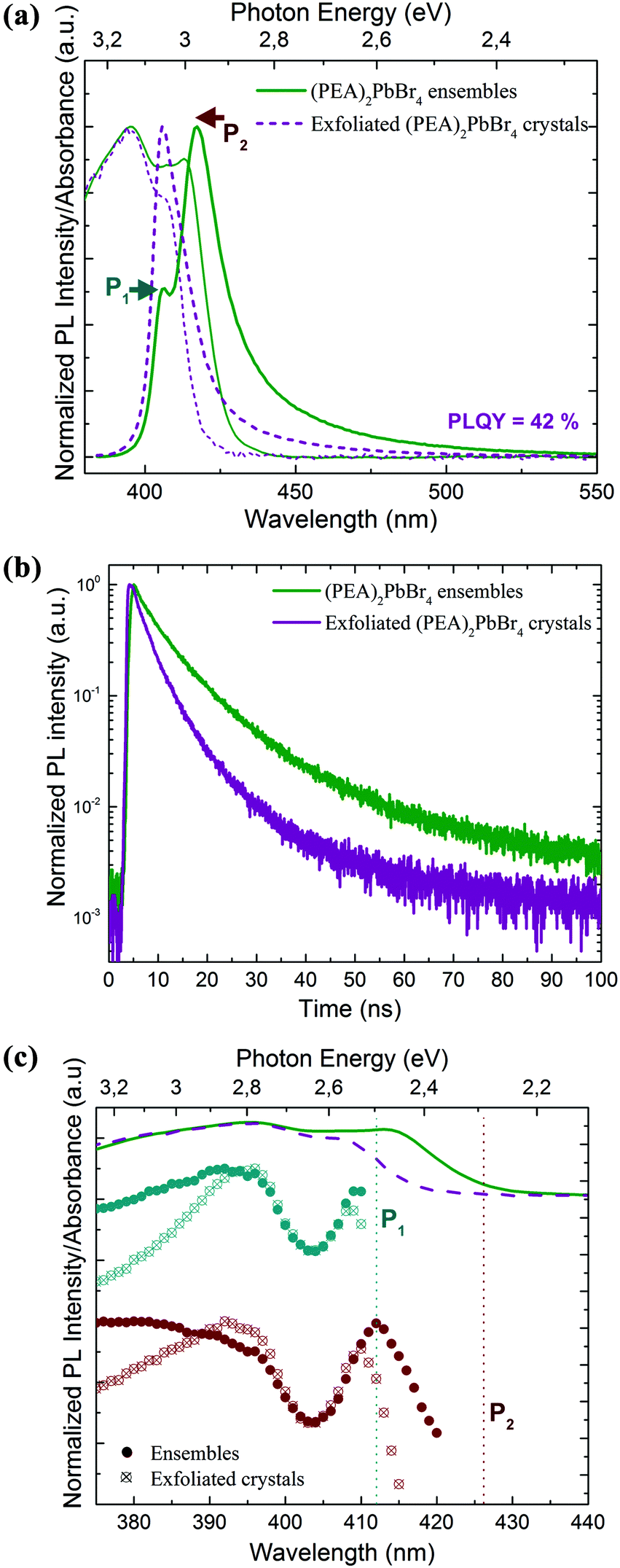 | ||
| Fig. 5 (a) Normalized PL and absorbance spectra acquired before and after exfoliation of (PEA)2PbBr4 crystals showing a sharp and single emission peak (P1) with respect to their ensembles. The coloured arrows indicate the position of the P1 and P2 emission peaks. (b) Normalized PL intensities as a function of time acquired at the high-energy peak from ensembles and exfoliated flakes. (c) PL excitation spectra acquired from ensemble (solid dots) and exfoliated (empty dots) (PEA)2PbBr4 flakes recorded at 412 nm (dark cyan, P1) and 426 nm (red, P2). The absorption spectra for the ensembles (green solid line) and exfoliated (violet dashed line) flakes are included on the top of the plots. The vertical lines highlight the position of the P1 and P2 emission peaks. The spectra are shifted vertically for clarity. | ||
These higher PLQY values in the exfoliated samples with respect to their ensemble counterparts can be explained by a reduced reabsorption and the presence of fewer impurities in the exfoliated crystals.33,39 The obtained PLQY values are comparable to those reported in the literature for similar layered structures emitting in the same energy range.28,33,40
The PL time traces in Fig. 5b and Fig. S5b† are characterized by a much faster decay for the exfoliated flakes (see Fig. S7†), which, together with the increase in PLQY, implies that the radiative rate in the flakes is strongly enhanced compared to their ensemble structures.
With the average lifetime from the PL decay fitting and the PLQY values of the selected crystals, we can calculate the average radiative and non-radiative rates of the emission using eqn (1) and (2):41,42
 | (1) |
 | (2) |
Towards the implementation of the 2D layered perovskite materials in devices such as light-emitting diodes, their stability with respect to elevated temperatures (that might be needed for processing and/or might be experienced by the material under operation conditions) and under light exposure, is of interest. We measured the PL intensity of (BA)2PbBr4 and (PEA)2PbBr4 ensembles and exfoliated flakes before and after heat treatment at 80 °C for 1 hour, and under illumination with UV light at an excitation power of 1.2 mW cm−2, as reported in Fig. S9 and S10.† In both cases, we observe a significant reduction of the emission intensity, but stable emission colour.
Conclusions
We demonstrated the straightforward fabrication of 2D layered lead halide perovskites with bright emission in the deep blue spectral region by facile injection of primary amines (with alkylic and aromatic tails) into a solution of precursors dissolved in acetone. Single flakes consisting of stacked few monolayers of [PbBr4]2− octahedra, spaced by a double layer of organic counterions, can be obtained via mechanical exfoliation. The resulting crystals show further improved optical properties with a PLQY up to 42%, manifesting a single emission peak at a wavelength of around 400 nm. Interestingly, we observe two well-separated energy levels close to the band edge, and from the exfoliated flakes, only the emission of the higher energy level can be detected, while in 2D platelet ensembles, the emission from the low energy peak dominates. This behaviour indicates a bright (high energy) state and a dark (low energy) state that are separated by around 130 meV, where the emission of the dark state can only be activated by reabsorption of the bright state emission. Our work opens a perspective on the implementation of layered perovskites in devices, where ultra-flat deep blue emitters are needed, and towards model systems for excitons and light–matter interaction in 2D systems at room temperature.Experimental
Materials
PbBr2 (98%), hydrobromic acid (HBr, 48% m m−1 in water), butylamine 99%, octylamine 99%, decylamine 99%, benzylamine 99%, phenethylamine 99%, and dopamine hydrochloride were purchased from Sigma-Aldrich and used without further purification.Growth of layered perovskite ensembles
Starting solutions were prepared by placing a stoichiometric amount of PbBr2 (95 mg) in a vial and adding 60 μl of HBr and 1 mL of acetone. A clear and transparent solution was obtained by vigorously shaking the mixture. Then, a stoichiometric amount of the selected primary amine (for aliphatic ones: butylamine, octylamine, and decylamine; for aromatic ones: benzylamine, phenethylamine, and dopamine) was added to the mixture. The reaction run over few minutes under strong magnetic stirring. The crystals were formed within a few minutes and they were collected and dried overnight on filter paper. All the experiments were conducted at room temperature.Exfoliation of the layered perovskite ensembles
As-synthesized crystals were mechanically exfoliated using a non-emissive, one-sided 3M Scotch® tape. The crystals were placed on the tape and the exfoliation was carried out by gentle pushing of a clear part of the tape onto the ensembles. The exfoliation process was repeated on the freshly exposed layers several time. The tape with exfoliated layered perovskite flakes was attached to a glass substrate for further analysis.Structural and morphological characterization
Optical images were collected using a Zeta-20 non-contact optical profiler (Zeta Instruments, USA) from ensembles on glass substrate and exfoliated crystals on SiO2-coated Si substrate. Scanning electron microscopy analysis was conducted on an FEI Nova 600 NanoLab instrument from the perovskite ensembles by depositing them directly on the Si substrates. Atomic force microscopy images and topological analysis of thin layers of exfoliated crystals were performed using an MFP-3D AFM system (Asylum Research, Goleta, CA, USA), operating in an intermittent contact mode. The samples for AFM imaging were prepared by transferring the exfoliated structures from the tape to the SiO2/Si substrate (285 nm) of 5 mm × 5 mm. The scan size was set as 5 μm × 5 μm for (PEA)2PbBr4 samples and 10 μm × 10 μm for (BA)2PbBr4 samples with a resolution of 256 × 256 pixels.Powder X-ray diffraction patterns were acquired on a PANalytical Empyrean X-ray diffractometer equipped with a 1.8 kW CuKα ceramic X-ray tube and a PIXcel3D 2 × 2 area detector, operating at 45 kV and 40 mA. Layered perovskite ensembles were placed on a zero-diffraction Si substrate for the XRD analysis.
Optical characterization
The absorption spectra were collected from dried crystals using a Varian Cary 5000 ultraviolet–visible–near-infrared (UV–vis–NIR) spectrophotometer equipped with an external diffuse reflectance accessory, and operating in absorption geometry. The PL, PLE spectra and PLQY values on both ensembles and exfoliated crystals were acquired using an Edinburgh Instruments fluorescence spectrometer (FLS920) equipped with a xenon lamp with a monochromator for steady-state PL excitation. The PL spectra of ensembles and exfoliated crystals were collected with an excitation wavelength of 350 nm. The PLE spectra on the samples were recorded by fixing the emission wavelength at P1 and P2 with a spectral resolution of 0.5 nm for (PEA)2PbBr4 ensembles and of 1 nm and 0.2 nm for (BA)2PbBr4 ensembles. In the case of the exfoliated samples, the spectral resolution was 1 nm and 0.5 nm, respectively. The PLE spectra were collected with a 0.5 s dwell time. The PLQY values were obtained from five samples from different synthesis batches by exciting them at a wavelength of 375 nm with an excitation power density of 1.5 mW cm−2, using a calibrated integrating sphere. Three measurements were performed on each sample of ensembles and exfoliated crystals with a standard experimental error below 5%. Time-resolved PL measurements were carried out with a time-correlated single-photon counting (TCSPC) unit coupled to a pulsed diode laser. The samples were excited at 375 nm with 50 ps pulses at a repetition rate of 0.05–1 MHz and a spectral collection window of 10 nm. The samples for all of these studies were prepared by transferring the tape with the ensemble and exfoliated 2D layered perovskite crystals to a clean glass substrate. The PL decay curves were acquired at the high- and low-energy peaks P1 and P2. Micro-PL experiments were performed by adapting an Olympus BX41 microscope. A bifurcated optical fibre was connected to a 349 nm laser and a Horiba iHR320 spectrometer; the common end of the fibre was used to excite the sample and collect the spectra via a system of lenses, mounted on a port of the microscope. UV-compatible objectives were used. The spot size was of the order of a few microns. A red-pass filter with an edge at 400 nm was inserted at the entrance of the spectrometer. The thermal stability study was carried out by placing the samples on glass substrates on a pre-heated hot plate. The temperature was set to 80 °C and kept for 60 min. For optical stability analysis, the samples were continuously illuminated by a UV laser working at 349 nm with a power density of 1.2 mW cm−2 for 60 min and their corresponding PL was collected subsequently.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MA and RK acknowledge financial support by the EU Horizon2020 MSCA Rise project “COMPASS-691185”. MA and RK thank Ivan Infante for the fruitful discussion on the photophysics of 2D layered perovskites and Vincenzo Caligiuri for support on the PLQY measurements. BD and MA thank Marco Salerno and the Materials Characterization Facility at the Istituto Italiano di Tecnologia for technical support on the AFM measurements.References
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC
.
- K. Wang, S. Wang, S. Xiao and Q. Song, Adv. Opt. Mater., 2018, 6, 1800278 CrossRef
- M. P. Arciniegas, A. Castelli, S. Piazza, S. Dogan, L. Ceseracciu, R. Krahne, M. Duocastella and L. Manna, Adv. Funct. Mater., 2017, 27, 1701613 CrossRef
- H. Huang, Q. Xue, B. Chen, Y. Xiong, J. Schneider, C. Zhi, H. Zhong and A. L. Rogach, Angew. Chem., 2017, 129, 9699–9704 CrossRef
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed
- R. Quintero-Bermudez, A. Gold-Parker, A. H. Proppe, R. Munir, Z. Yang, S. O. Kelley, A. Amassian, M. F. Toney and E. H. Sargent, Nat. Mater., 2018, 17, 900–907 CrossRef CAS PubMed
- J. Shamsi, A. L. Abdelhady, S. Accornero, M. Arciniegas, L. Goldoni, A. R. S. Kandada, A. Petrozza and L. Manna, ACS Energy Lett., 2016, 1, 1042–1048 CrossRef CAS PubMed
- F. Palazon, C. Urso, L. De Trizio, Q. Akkerman, S. Marras, F. Locardi, I. Nelli, M. Ferretti, M. Prato and L. Manna, ACS Energy Lett., 2017, 2, 2445–2448 CrossRef CAS PubMed
- L. Dou, A. B. Wong, Y. Yu, M. Lai, N. Kornienko, S. W. Eaton, A. Fu, C. G. Bischak, J. Ma and T. Ding, Science, 2015, 349, 1518–1521 CrossRef CAS PubMed
- S. Wang, Y. Yao, J. Kong, S. Zhao, Z. Sun, Z. Wu, L. Li and J. Luo, Chem. Commun., 2018, 54, 4053–4056 RSC
- J. Shamsi, P. Rastogi, V. Caligiuri, A. L. Abdelhady, D. Spirito, L. Manna and R. Krahne, ACS Nano, 2017, 11, 10206–10213 CrossRef CAS PubMed
- L. Lv, Y. Xu, H. Fang, W. Luo, F. Xu, L. Liu, B. Wang, X. Zhang, D. Yang, W. Hu and A. Dong, Nanoscale, 2016, 8, 13589–13596 RSC
- L. Pedesseau, D. Sapori, B. Traore, R. Robles, H.-H. Fang, M. A. Loi, H. Tsai, W. Nie, J.-C. Blancon, A. Neukirch, S. Tretiak, A. D. Mohite, C. Katan, J. Even and M. Kepenekian, ACS Nano, 2016, 10, 9776–9786 CrossRef CAS PubMed
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 7843–7850 CrossRef CAS PubMed
- L. Etgar, Energy Environ. Sci., 2018, 11, 234–242 RSC
- D. B. Straus and C. R. Kagan, J. Phys. Chem. Lett., 2018, 9, 1434–1447 CrossRef CAS PubMed
- N. Kitazawa, M. Aono and Y. Watanabe, Thin Solid Films, 2010, 518, 3199–3203 CrossRef CAS
- N. Kawano, M. Koshimizu, Y. Sun, N. Yahaba, Y. Fujimoto, T. Yanagida and K. Asai, J. Phys. Chem. C, 2014, 118, 9101–9106 CrossRef CAS
- Y. Chen, Y. Sun, J. Peng, J. Tang, K. Zheng and Z. Liang, Adv. Mater., 2018, 30, 1703487 CrossRef PubMed
- D. N. Congreve, M. C. Weidman, M. Seitz, W. Paritmongkol, N. S. Dahod and W. A. Tisdale, ACS Photonics, 2017, 4, 476–481 CrossRef CAS
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed
- Q. Zhang, L. Chu, F. Zhou, W. Ji and G. Eda, Adv. Mater., 2018, 30, 1704055 CrossRef PubMed
- C. Zhou, H. Lin, Y. Tian, Z. Yuan, R. Clark, B. Chen, L. J. van de Burgt, J. C. Wang, Y. Zhou and K. Hanson, Chem. Sci., 2018, 9, 586–593 RSC
- C. Zhou, Y. Tian, Z. Yuan, H. Lin, B. Chen, R. Clark, T. Dilbeck, Y. Zhou, J. Hurley and J. Neu, ACS Appl. Mater. Interfaces, 2017, 9, 44579–44583 CrossRef CAS PubMed
- D. Solis-Ibarra, I. Smith and H. Karunadasa, Chem. Sci., 2015, 6, 4054–4059 RSC
- M. D. Smith, A. Jaffe, E. R. Dohner, A. M. Lindenberg and H. I. Karunadasa, Chem. Sci., 2017, 8, 4497–4504 RSC
- L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS PubMed
- A. Castelli, G. Biffi, L. Ceseracciu, D. Spirito, M. Prato, D. Altamura, C. Giannini, S. Artyukhin, R. Krahne and L. Manna, Adv. Mater., 2018, 1805608 Search PubMed
- M. D. Smith, B. A. Connor and H. I. Karunadasa, Chem. Rev., 2019, 119, 3104–3139 CrossRef CAS PubMed
- Z. Yuan, Y. Shu, Y. Tian, Y. Xin and B. Ma, Chem. Commun., 2015, 51, 16385–16388 RSC
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed
- C. Zhou, H. Lin, M. Worku, J. Neu, Y. Zhou, Y. Tian, S. Lee, P. Djurovich, T. Siegrist and B. Ma, J. Am. Chem. Soc., 2018, 140, 13181–13184 CrossRef CAS PubMed
- X. Gong, O. Voznyy, A. Jain, W. Liu, R. Sabatini, Z. Piontkowski, G. Walters, G. Bappi, S. Nokhrin, O. Bushuyev, M. Yuan, C. Riccardo, D. McCamant, S. O. Kelley and E. H. Sargent, Nat. Mater., 2018, 17, 550–556 CrossRef CAS PubMed
- S. Ithurria, M. D. Tessier, B. Mahler, R. P. S. M. Lobo, B. Dubertret and Al. L. Efros, Nat. Mater., 2011, 10, 936 CrossRef CAS PubMed
- A. Polovitsyn, Z. Dang, J. L. Movilla, B. Martín-García, A. H. Khan, G. H. V. Bertrand, R. Brescia and I. Moreels, Chem. Mater., 2017, 29, 5671–5680 CrossRef CAS
- J. Q. Grim, S. Christodoulou, F. Di Stasio, R. Krahne, R. Cingolani, L. Manna and I. Moreels, Nat. Nanotechnol., 2014, 9, 891 CrossRef CAS PubMed
- M. D. Smith and H. I. Karunadasa, Acc. Chem. Res., 2018, 51, 619–627 CrossRef CAS PubMed
- Z. Wu, C. Ji, Z. Sun, S. Wang, S. Zhao, W. Zhang, L. Li and J. Luo, J. Mater. Chem. C, 2018, 6, 1171–1175 RSC
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef CAS
- D. Liang, Y. Peng, Y. Fu, M. J. Shearer, J. Zhang, J. Zhai, Y. Zhang, R. J. Hamers, T. L. Andrew and S. Jin, ACS Nano, 2016, 10, 6897–6904 CrossRef CAS PubMed
- B. Omogo, J. F. Aldana and C. D. Heyes, J. Phys. Chem. C, 2013, 117, 2317–2327 CrossRef CAS PubMed
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer US, New York, 2006 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr00638a |
| This journal is © The Royal Society of Chemistry 2019 |