
Urchin-like NiO–NiCo2O4 heterostructure microsphere catalysts for enhanced rechargeable non-aqueous Li–O2 batteries†
Wen
Zhao
a,
Xiaomin
Li
a,
Rui
Yin
a,
Lei
Qian
*a,
Xiaoshuai
Huang
a,
Hu
Liu
 bc,
Jiaoxia
Zhang
bd,
Jun
Wang
*a,
Tao
Ding
*e and
Zhanhu
Guo
*b
bc,
Jiaoxia
Zhang
bd,
Jun
Wang
*a,
Tao
Ding
*e and
Zhanhu
Guo
*b
aKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials (Ministry of Education), Shandong University, 17923 Jingshi Road, Jinan 250061, China. E-mail: qleric@sdu.edu.cn; jw707@sdu.edu.cn
bIntegrated Composites Lab (ICL), Department of Chemical & Biomolecular Engineering, University of Tennessee, Knoxville, TN 37996, USA. E-mail: zguo10@utk.edu
cKey Laboratory of Materials Processing and Mold (Zhengzhou University), Ministry of Education, National Engineering Research Center for Advanced Polymer Processing Technology, Zhengzhou University, Zhengzhou, 450002, China
dSchool of Material Science and Engineering, Jiangsu University of Science and Technology, Zhenjiang, Jiangsu 212003, China
eCollege of Chemistry and Chemical Engineering, Henan University, Kaifeng 475004, P. R. China. E-mail: dingtao@henu.edu.cn
First published on 26th November 2018
Abstract
Urchin-like NiO–NiCo2O4 microspheres with heterostructures were successfully synthesized through a facile hydrothermal method, followed by thermal treatment. The unique structure of NiO–NiCo2O4 with the synergetic effect between NiCo2O4 and NiO, and the heterostructure favour the catalytic activity towards Li–O2 batteries. NiCo2O4 is helpful for boosting both the oxygen reduction reaction and oxygen evolution reaction for the Li–O2 batteries and NiO is likely to promote the decomposition of certain by-products. The special urchin-like morphology facilitates the continuous oxygen flow and accommodates Li2O2. Moreover, benefitting from the heterostructure, NiO–NiCo2O4 microspheres are able to promote the transport of Li ions and electrons to further improve battery performance. Li–O2 batteries utilizing a NiO–NiCo2O4 microsphere electrode show a much higher specific capacity and a lower overpotential than those with a Super P electrode. Moreover, they exhibit an enhanced cycling stability. The electrode can be continuously discharged and charged without obvious terminal voltage variation for 80 cycles, as the discharge capacity is restricted at 600 mA h g−1, suggesting that NiO–NiCo2O4 is a promising catalyst for Li–O2 batteries.
1. Introduction
The increasing demand for future energy storage systems has driven rapid growth of battery research.1,2 In order to meet the target for applications in electric vehicles and grid storage, various battery systems, such as lithium–ion, lithium–sulphur and metal–air batteries, have been studied.3–7 Recently, Li–O2 batteries have attracted enormous attention due to their extremely high theoretical energy density and potential applications in transportation, portable electronics and grid energy storage.8–10 The discharge and charge processes in Li–O2 batteries are related to the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER), respectively.11–13 The practical applications of Li–O2 batteries have been restricted by many problems, including high discharge/charge overpotential caused by sluggish reaction kinetics,14–16 electrolyte decomposition under high voltage, and safe issues resulting from the active Li anode.17–19 Developing bifunctional electrocatalysts to efficiently catalyse both the ORR and OER is one of the keys to achieve high performance Li–O2 batteries. The electrocatalysts should be able to facilitate the rapid decomposition of discharge products to improve the capacity, round-trip efficiency and cycling stability performance of Li–O2 batteries.20–22Although noble metals and their alloys have been proved to be efficient bifunctional catalysts,23–25 high electrocatalytic performance transition metal oxides, such as Co3O4,26–29 MnO2,30–32 MnCo2O4,33–35 and NiCo2O4,36–38 are more suitable for practical uses because of their low cost and abundant reserves. Among them, NiCo2O4 is one of the most promising materials due to its large amount of catalytic active sites from two redox couples of Co3+/Co2+ and Ni3+/Ni2+.20,39 Besides, compared with NiO and Co3O4, NiCo2O4 possesses enhanced electrical conductivity.40,41 It has also been reported that the microstructure of catalysts significantly influenced their catalytic activity.39,42–44 In order to obtain a better Li–O2 battery performance, NiCo2O4 cathode catalysts with various microstructures have been prepared and studied, such as nanotubes, nanoflakes, nanosheets, nanowire arrays and microspheres.40,45–48 Recently, urchin-like NiCo2O4 microspheres have attracted great interest, owing to their high specific surface area, short diffusion path for ions or electrons and enough channels for mass transport, which are in favour of higher electrocatalytic performance for Li–O2 batteries.39,49 However, Li–O2 batteries based on single phase metal oxides still suffer from poor capacities, high overpotential and unsatisfactory cycling stability which limited their practical applications. Therefore, many efforts have been made to design novel hybrid electrodes in order to achieve synergetic effects between two components. For instance, it has been reported that one-dimensional RuO2/Mn2O3 fibers,50 RuO2-δ-MnO2 nanosheets,32 mesoporous ZnO/ZnFe2O4 nanocages,51 Fe2O3@MnO2 composites,52 and NiO–RuO2 nanoparticles53 were prepared and they exhibited favourable battery performance because of the synergetic effects between two components.
Compared with mixture or homogeneous doping bulk, heterostructures constructed from coupling nanocrystals with different band gaps have great potential applications in photocatalysis, sensors and energy storage because of their interface effects which offer unprecedented properties.54 Benefiting from the existing internal electric field, heterostructures are able to enhance the surface reaction kinetics as well as to facilitate charge transport.55,56 Thus, from the perspective of enhancing the electronic conductivity and ion diffusion capability, the design and fabrication of a complex heterostructure by employing two different materials might be a feasible strategy to obtain better electrocatalytic performance in Li–O2 batteries. It was also found that NiO played an important role in decomposing the by-products containing carbonate/carboxylate species.57,58 The NiO catalyst can promote the formation of thinner passivation layers to avoid the hindrance of charge transfer from the cumulative passivation layers.58 Therefore, the NiO catalyst can facilitate efficient oxidation of carbonate/carboxylate species, as well as promote the peroxide oxidation. Mixed metal oxides containing NiO were designed to improve their electrocatalytic performance.59 For example, Tan et al.60 reported a nanostructured RuO2/NiO cathode which enabled the lithium–air batteries to be truly operated in ambient air at 500 mA h g−1 for 200 cycles. Liu et al.61 developed a method for synthesizing ultrafine NiO/CoO catalysts which retained a capacity of >1000 mA h g−1 after 100 cycles. Many previous studies have successfully employed the NiO–NiCo2O4 composite as the electrode for supercapacitors, and the composites exhibited good catalytic activity towards the ORR and OER.62–66 Nevertheless, there are far few reports on NiO–NiCo2O4 composites used as cathode catalysts for Li–O2 batteries. They are expected to effectively facilitate the formation and decomposition of Li2O2 with less by-product accumulation during cycling.
Herein, inspired by the unique advantages of heterostructures and the benefits of NiO–NiCo2O4 composites, we proposed and constructed NiO–NiCo2O4 microspheres with a heterostructure through a facile hydrothermal method following annealing treatment. This unique structure was able to promote fast transport of ions and electrons to obtain enhanced battery performance. The capacity, reversibility and rate performance of the batteries were investigated by electrocatalytic testing and the results demonstrated that the NiO–NiCo2O4 microspheres with the heterostructure were favourable for the catalytic activity of nonaqueous Li–O2 batteries.
2. Experimental
2.1 Preparation of NiO–NiCo2O4 microspheres with the heterostructure
In a typical hydrothermal preparation process, 1.25 mmol NiCl2·6H2O, 2.5 mmol CoCl3·6H2O and 4.5 mmol urea were dissolved in 25 mL deionized water at room temperature and the solution was ultrasonically treated for 30 minutes. It was then transferred to a 50 mL stainless steel autoclave with a Teflon liner and was heated to 120 °C and maintained for 8 hours. After being cooled down to room temperature, the product was collected by centrifugation and washed with deionized water and ethanol several times, followed by drying at 60 °C under vacuum for 12 h. Finally, the obtained product was calcined at different temperatures for 3 h with a heating rate of 2 °C min−1 in air to obtain urchin-like heterostructure NiO–NiCo2O4 microspheres. Different NiO–NiCo2O4 microspheres were fabricated by applying calcination temperatures of 400, 500 and 600 °C and they were designated as NCO-400, NCO-500 and NCO-600, respectively.2.2 Materials characterization
X-ray diffraction (XRD) patterns were recorded on a D8 Advance X-ray diffractometer using Cu Kα radiation (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) was carried out on a ThermoFisher K-Alpha instrument. The XPS data were analysed by CasaXPS software and all the results were calibrated by using C 1s = 284.6 eV. The morphology and microstructure of the synthesized materials were examined using a field emission scanning electron microscope (FESEM: SU-70) equipped with an energy dispersive X-ray (EDX) analyser and a transmission electron microscope (TEM: JEOL-JEM2100, operated at 200 kV).2.3 Electrochemical investigation
The electrochemical performance of the Li–O2 batteries was analysed using a 2032-type coin cell with holes in its top cover. To prepare the electrodes tested in Li–O2 cells, 40 wt% catalyst, 40 wt% Super P carbon and 20 wt% poly(tetrafluoroethylene) were mixed in isopropanol to prepare a slurry, which was homogeneously dispersed on a carbon paper (TGP-H-060, Toray) and dried at 120 °C under vacuum for 12 h. All the cells were assembled in a glovebox under an Ar atmosphere, in which both oxygen and water contents were less than 0.1 ppm. Li–O2 batteries consist of a Li foil anode, a glass fibre separator soaked with the electrolyte containing 1 M lithium bis(trifluoromethanesulfonyl)imide in triethylene glycol dimethyl ether (LiTFSI/TEGDME) and a catalyst coated oxygen cathode. Galvanostatic discharge/charge tests were carried out in an O2-purged chamber on a multichannel battery testing system (LAND CT 2001A) in the potential range of 2.35–4.35 V at current densities of 100, 200, 300, 400 and 500 mA g−1, respectively. The specific capacities were calculated based on the amount of the active material in the cathodes. Cyclic voltammetry (CV) was performed on an electrochemical workstation (RST5002F) at a scan rate of 0.1 mV s−1 in the potential range of 2.35–4.35 V (vs. Li/Li+). Electrochemical impedance spectroscopy (EIS) was tested on the same electrochemical workstation using a sine wave of 0.01 V amplitude over a frequency range of 100 kHz–0.01 Hz.3. Results and discussion
The synthetic procedure of NiO–NiCo2O4 microspheres with a heterostructure, shown in Fig. 1, was carried out through a two-step route. Firstly, the NiO–NiCo2O4 precursor was prepared by a hydrothermal reaction. Afterwards, the precursor was calcined to achieve NiO–NiCo2O4 heterostructure microspheres. The crystal structure and composition of the as-prepared materials were characterized by XRD analysis. The XRD pattern (Fig. S1a†) of the precursor revealed that it was composed of cobalt–nickel bimetallic carbonate hydroxide. Fig. 2a shows the XRD pattern of the as-prepared NiO–NiCo2O4 microspheres with different calcination temperatures. All the peaks matched well with the cubic NiCo2O4 (JCPDS no.: 20-0781) and cubic NiO (JCPDS no.: 65-5745), which clearly demonstrated that the synthesized products were composed of NiCo2O4 and NiO. No presence of the NiO peak was observed in the XRD result of NCO-400, while the cubic phase of NiO peaks appeared in the XRD result of NCO-500 and NCO-600. This was consistent with the observation of the NiO phase when the firing temperature was raised to 500 °C.67 The cubic NiO phase was initially formed in the sample annealed at 500 °C and co-existed with NiCo2O4 as surface coverage. It was possibly because the spinel NiCo2O4 was decomposed with the loss of NiO.67–69 However, the specific mechanism still needs further study to confirm.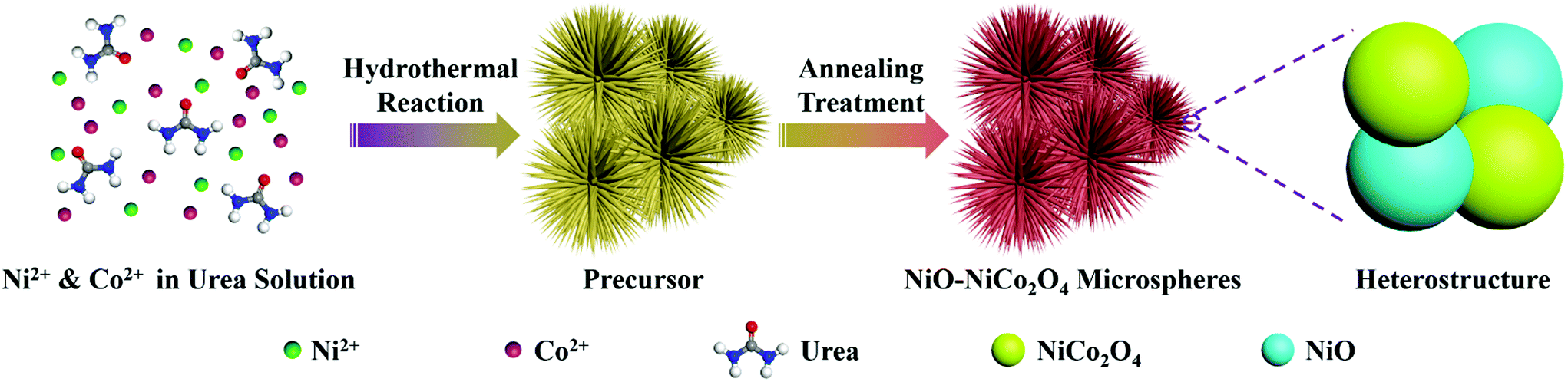 | ||
| Fig. 1 Schematic illustration of the formation of NiO–NiCo2O4 microspheres through a two-step method. | ||
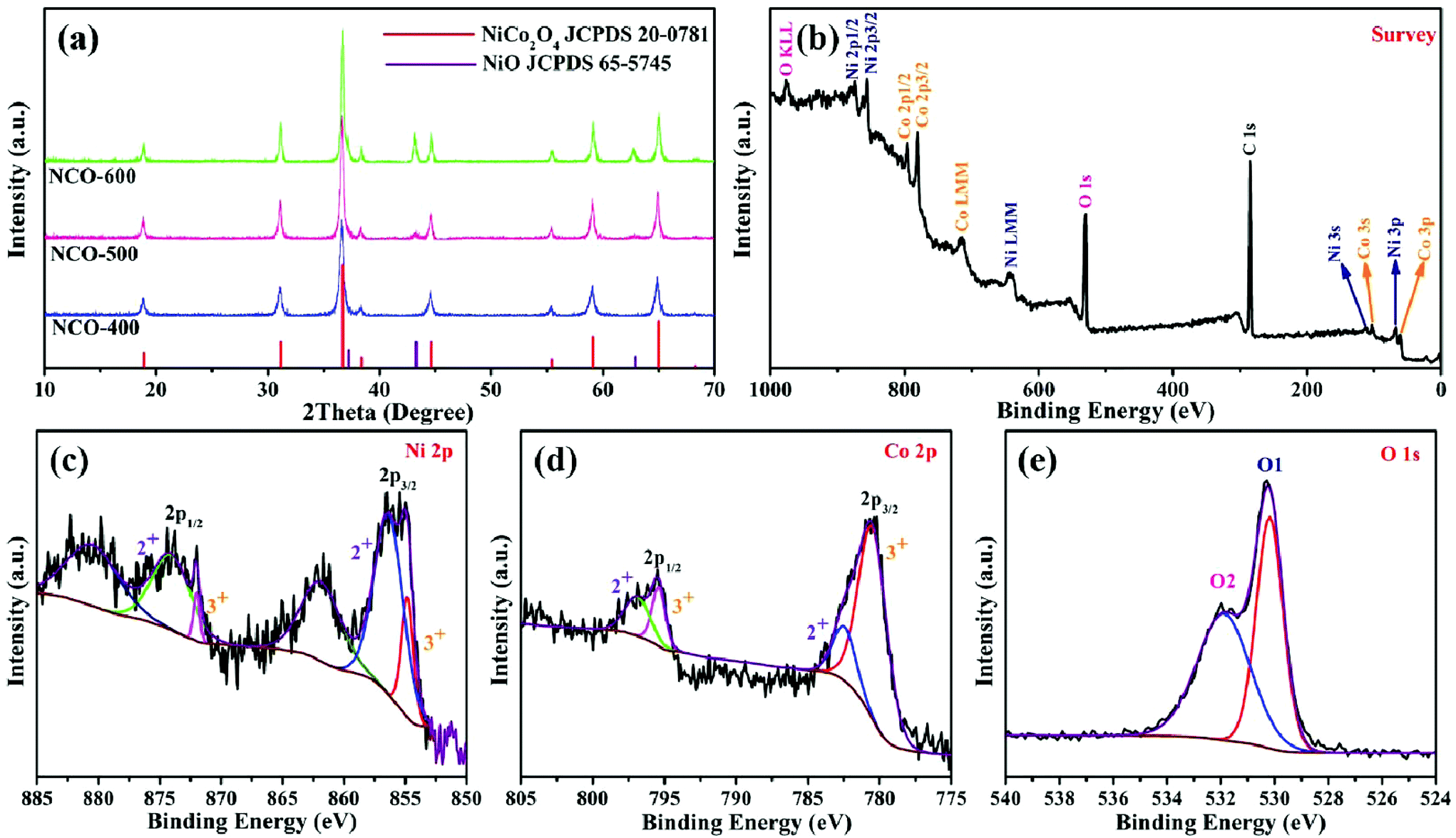 | ||
| Fig. 2 (a) XRD patterns of NCO-400, NCO-500 and NCO-600; XPS spectra of NCO-500: (b) survey, (c) Ni 2p, (d) Co 2p and (e) O 1s. | ||
XPS measurement was carried out to further understand the chemical composition and the elemental oxidation state of NiO–NiCo2O4 samples. Fig. 2b shows the survey spectrum of NCO-500, which indicates the presence of C, O, Co and Ni elements. In Fig. 2c of the high-resolution spectrum of Ni 2p, the peaks were well-fitted with two spin–orbit doublets and two shakeup satellite peaks. The fitting peaks with binding energies of 856.5 and 874.1 eV were indexed to Ni2+ signals, while the peaks with those of 854.8 and 872.0 eV were indexed to Ni3+ signals. The satellite peaks at 861.9 and 880.7 eV were shakeup peaks of Ni.70,71 In a similar way, Co 2p can be fitted to two spin–orbit doublets (Fig. 2d). The fitting peaks at 782.5 and 797.0 eV were indexed to Co2+ signals, while the peaks at 780.4 and 795.4 eV were indexed to Co3+ signals.71,72 These results indicated that in NCO-500, both Ni and Co were partially oxidized and reduced, respectively, to balance the formation of oxygen vacancies.73,74 The spectrum of O 1s (Fig. 2e) shows two oxygen contributions. The fitting peak at 530.1 eV is assigned to the metal–oxygen bond, and the peak at 531.8 eV is usually associated with a high number of defect sites.75,76
The morphology and microstructure of the NiO–NiCo2O4 samples were characterized by FESEM and TEM. The images in Fig. S1b† show the morphology of the NiO–NiCo2O4 precursor. It generally presented a spherical urchin shape structure with a uniform diameter of about 5 μm. The FESEM images of the as-prepared NCO-500 in Fig. 3a and c show that the structure did not change after calcination. It was observed from Fig. 3a that each urchin-like NCO-500 sphere displayed a uniform diameter of 5 μm with numerous small nanorods radially-grown from the center. The elements in the NCO-500 sample were characterized by EDS and the results are shown in Fig. 3b, which confirmed the presence of Ni, Co and O elements. Element mapping results in Fig. 3d, e and f show the uniform distribution of Ni, Co and O elements in an NCO-500 sphere. The FESEM images of NCO-400 and NCO-600 with the according element mapping results are shown in Fig. S2 and S3,† respectively, and the morphology is similar to that of NCO-500.
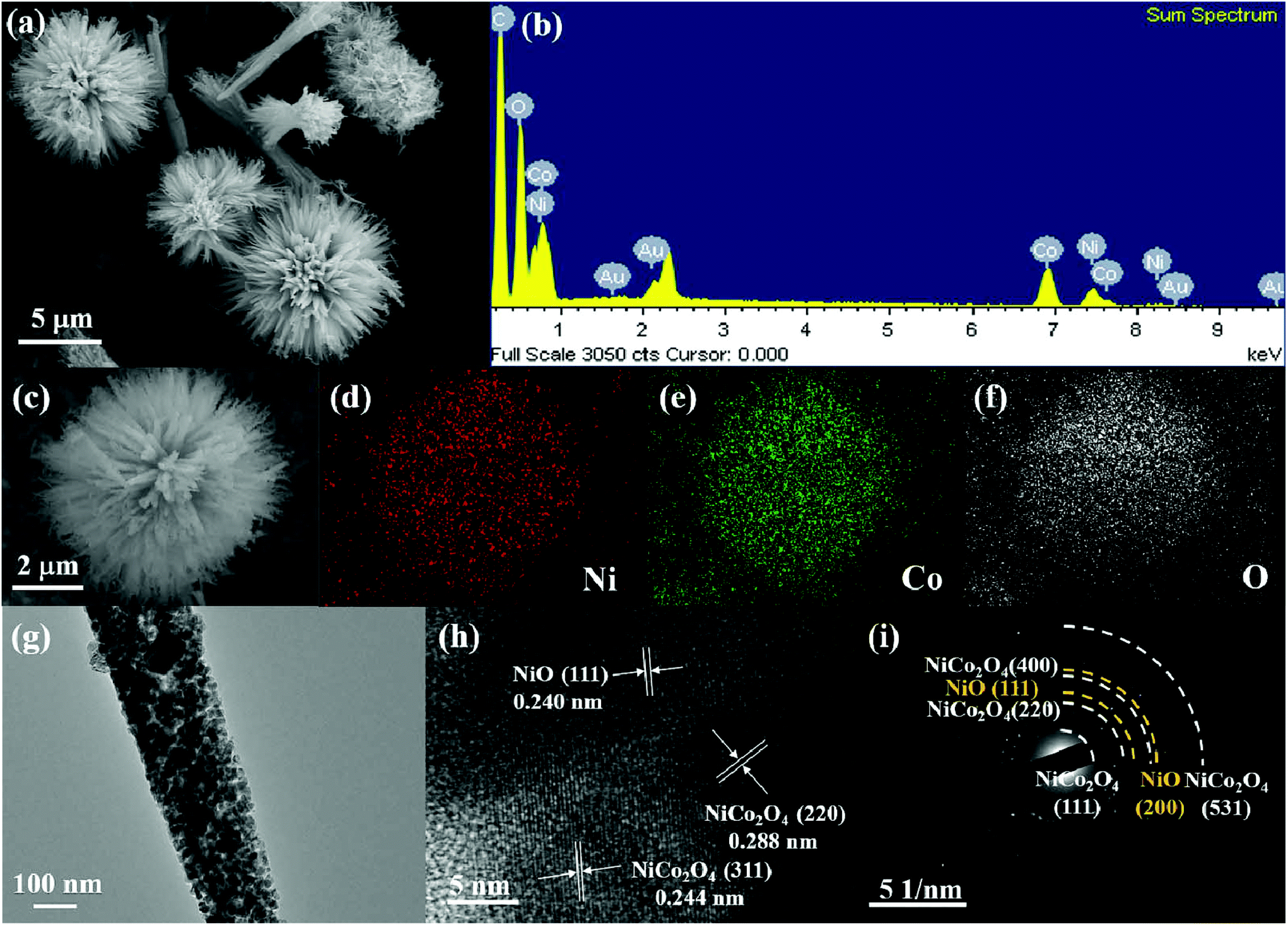 | ||
| Fig. 3 FESEM images of NCO-500 at (a) low and (c) high magnifications; (b) EDS result of NCO-500; element mapping images of NCO-500: (d) Ni, (e) Co and (f) O; (g) TEM images of NCO-500; (h) high resolution TEM image; (i) selected area electron diffraction of NCO-500. | ||
NCO-500 was tested for TEM to further examine its detailed morphology and component structure. Fig. 3g shows the nanorods which constituted the NiO–NiCo2O4 urchin-like microspheres. The HRTEM image in Fig. 3h clearly presents the lattice fringes with spacings of about 0.288, 0.244 and 0.240 nm, corresponding to the (220) and (311) planes of NiCo2O4 and the (111) planes of NiO, respectively, which are consistent with the results of the XRD pattern. The corresponding SAED pattern in Fig. 3h shows well-defined rings indexed to the NiO and spinel NiCo2O4 phases, indicating the polycrystalline characteristic of NiO–NiCo2O4 urchin-like microspheres.
Different CV curves of the Li–O2 cells containing the NiO–NiCo2O4 microspheres and pure SP electrodes in the LiTFSI/TEGDME electrolyte were studied, as shown in Fig. S4.† Compared with the pure SP electrode, the NiO–NiCo2O4 electrodes exhibited considerably larger cathodic and anodic current values and higher cathodic potentials, indicating higher bifunctional electrocatalytic activity towards the ORR and OER in non-aqueous Li–O2 batteries. Particularly, the NCO-500 electrode showed the highest current value among all samples, indicating its excellent catalytic activity. As a result, it demonstrated that the NiO–NiCo2O4 electrode may have remarkable electrochemical properties towards both the formation and decomposition of discharge products during discharge and charge processes.
The Li–O2 battery performances of NiO–NiCo2O4 heterostructure microspheres and pure SP electrodes were further studied by galvanostatically discharge and charge tests. Fig. 4a shows the initial full discharge/charge profiles for the Li–O2 batteries with NCO-400, NCO-500, NCO-600 and pure SP electrodes at a current density of 100 mA g−1 from 2.35 to 4.35 V vs. Li+/Li. The NiO–NiCo2O4 based Li–O2 battery showed a lower overpotential and a much higher specific capacity than that of a pure SP based Li–O2 battery. The discharge/charge capacities of the pure SP electrode were 1750/1619 mA h g−1. The electrodes with NiO–NiCo2O4 exhibited dramatically improved discharge/charge capacities. In brief, the NCO-400, NCO-500 and NCO-600 electrodes delivered discharge/charge capacities of 7092/7563, 9231/8349 and 5429/4622 mA h g−1, respectively. In order to eliminate the capacity influence of the carbon paper, a Li–O2 cell using only the carbon paper as a cathode material was also assembled. It was observed from Fig. S5† that the discharge/charge capacities of the carbon paper were quite limited, indicating that the contribution of the carbon paper to the total capacity was negligible. Therefore, it is reasonable to use carbon paper as the current collector in the testing of Li–O2 batteries.
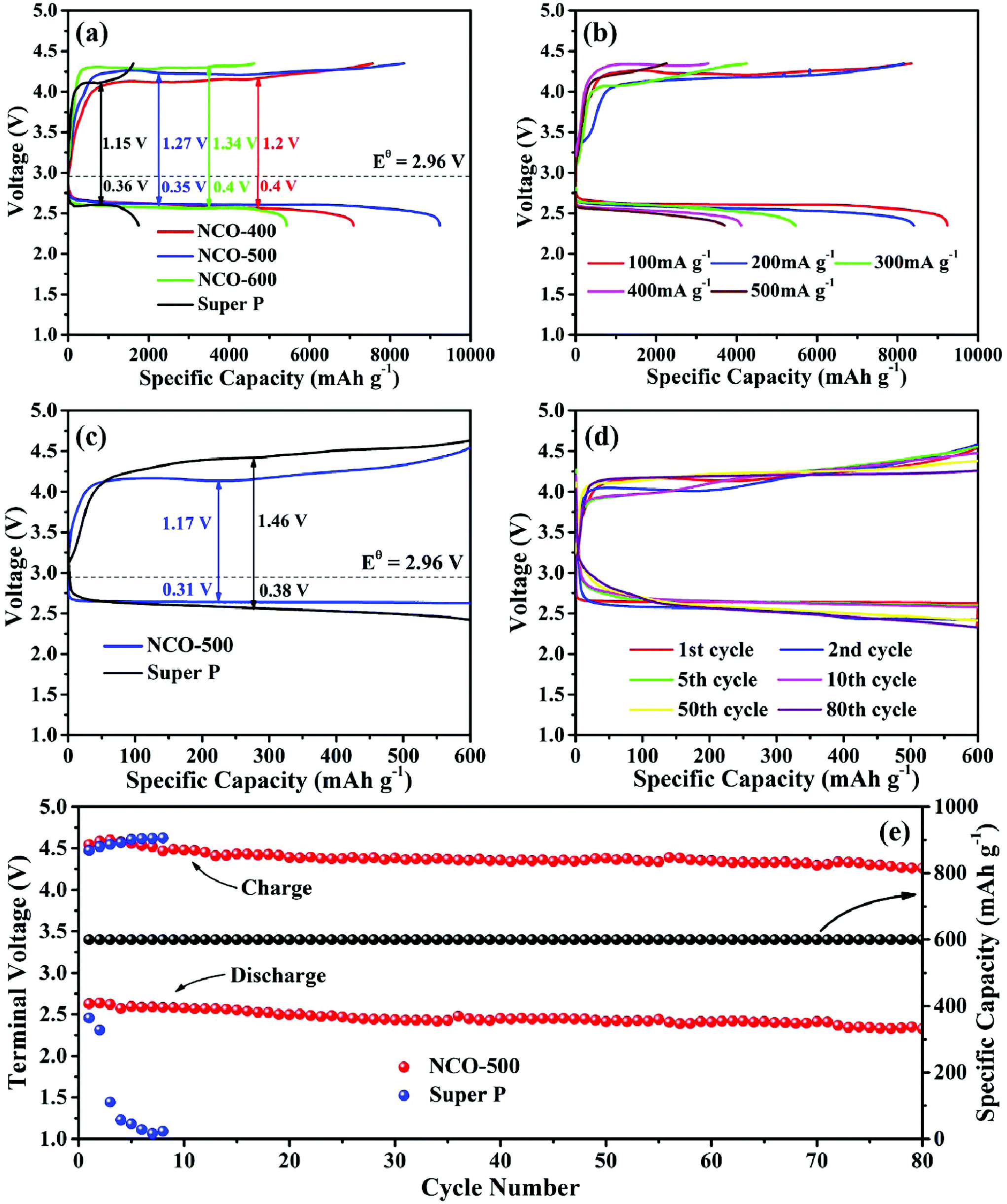 | ||
| Fig. 4 The initial discharge/charge profiles of (a) NCO-400, NCO-500, NCO-600 and pure SP electrodes at 100 mA g−1, (b) the NCO-500 electrode at different current densities; (c) initial discharge/charge plots of the NCO-500 and SP electrodes under a capacity limit of 600 mA h g−1 at 100 mA g−1; (d) typical discharge/charge profiles of NCO-500 electrode under a capacity limit of 600 mA h g−1 at 100 mA g−1; (e) cycling performances of the NCO-500 and SP electrodes under a capacity limit of 600 mA h g−1 at 100 mA g−1. | ||
In Fig. 4b, as the current densities varied from 200, 300, 400 to 500 mA g−1, the NCO-500 electrode also showed a relatively high discharge/charge performance of 8406/8162, 5461/4257, 4113/3292 and 3711/2254 mA h g−1, respectively, indicating that the NCO-500 composite delivered a good rate capability. Fig. 4c exhibits the typical initial discharge and charge profiles of the NCO-500 electrode and the SP electrode with a fixed capacity of 600 mA h g−1 at a current density of 100 mA g−1. The NCO-500 electrode showed a much lower discharge and charge overpotential. Fig. 4d exhibits the selected discharge/charge profiles for the NCO-500 electrode at a current density of 100 mA g−1 with a fixed specific capacity of 600 mA h g−1 and the specific capacity showed no loss up to 80 cycles. The cycling stability of the NCO-500 and SP based electrodes was also manifested by the discharge/charge terminal voltage at the current density of 100 mA g−1 with a fixed capacity of 600 mA h g−1, and the NCO-500 electrode demonstrated a favourable cycling performance for the Li–O2 batteries. As shown in Fig. 4e, the discharge/charge terminal voltage for the NCO-500 electrode remained stable over 80 cycles (2.32 and 4.25 V respectively), while for the SP electrode, its discharge terminal voltage dropped quickly after the first 2 cycles.
Table S1† summarizes the Li–O2 battery performance of the NCO-500 electrode in comparison with some representative NiO-based and NiCo2O4-based electrodes reported in the literature. It clearly demonstrated that the NCO-500 composite electrode showed a better performance than most of the other electrodes under similar testing conditions, especially in terms of capacity and cycling stability, making the NCO-500 composite a promising material for advanced Li–O2 batteries. Compared with other NiO-based and NiCo2O4-based materials, NCO-500 combined the benefits of NiO and NiCo2O4. The synergetic effect between NiCo2O4 and NiO favors enhancing the catalytic activity towards Li–O2 batteries. NiCo2O4 can help boost both the ORR and OER, while NiO is likely to promote the decomposition of certain by-products. Moreover, the unique urchin-like heterostructure also provides advantages. The special morphology facilitates the continuous oxygen flow and the heterostructure promotes the transport of Li ions and electrons to further improve the battery performance.
EIS analysis was also conducted on different samples in the frequency range from 0.01 Hz to 10 kHz, as shown in Fig. 5a. Each curve consists of two parts, a small semicircle at the high-frequency region attributes to the charge-transfer resistance and a slope line at the low-frequency region is associated with the ion diffusion process within the electrode.76–78 From the Nyquist plots, it was found that the NCO-500 electrode delivered the lowest charge transfer resistance among all the four samples, indicating that it enabled an efficient charge transport on the electrode/electrolyte interface and ensured the electrochemical activity during cycling. The EIS test of the NCO-500 electrode at different discharge/recharge stages (Fig. 5b) was also performed to further identify the discharge and recharge characteristics. After the first discharge, a larger charge-transfer resistance was observed compared with that of the fresh electrode. This is because of the formation and accumulation of Li2O2 on the electrode, which was hypothesized to prevent the transfer of electrons and lead to high electrical resistivity.79,80 After being recharged, the charge-transfer resistance of the NCO-500 electrode slightly changed compared with that of the fresh electrode, suggesting that the formation and decomposition of the reaction product is reversible. The charge-transfer resistance experienced almost no change after the 80th cycle, mainly because less irreversible by-products existed on the electrode, confirming its remarkable catalytic properties.81,82
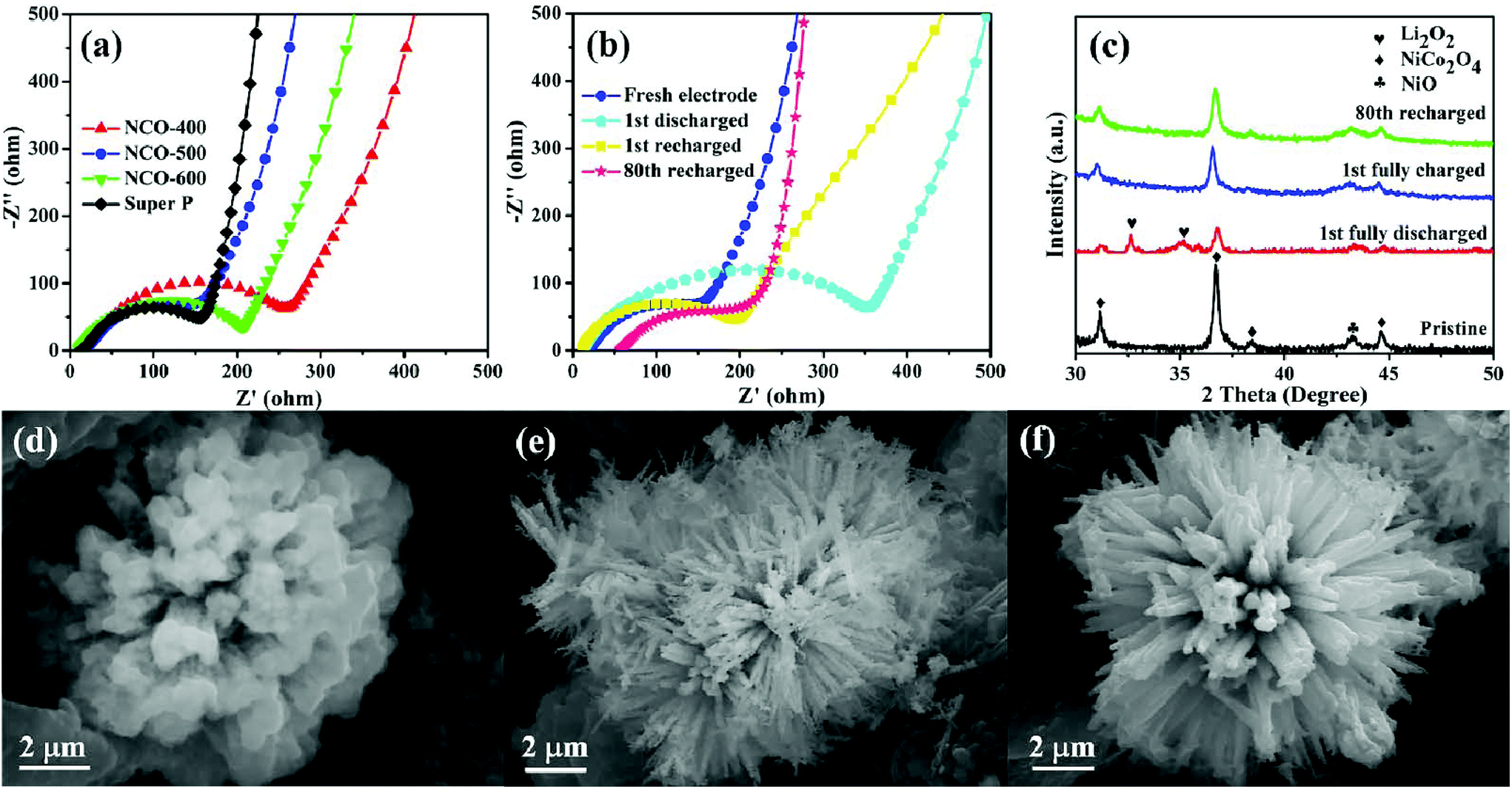 | ||
| Fig. 5 (a) EIS plots of NCO-400, NCO-500, NCO-600 and SP electrodes; (b) EIS plots of NCO-500 electrodes at fresh, 1st cycle discharged, 1st cycle recharged, and 80th cycle recharged states; (c) XRD patterns of the NCO-500 electrode at fresh, 1st cycle discharged, 1st cycle recharged, and 80th cycle recharged states; FESEM images of NCO-500 electrodes at (d) the 1st cycle discharged to 2.35 V, (e) the 1st cycle recharged to 4.35 V and (f) the 80th cycle recharged stages. | ||
To have a further understanding of the changes in the composition and morphology of the NCO-500 electrode at different discharge/recharge stages, ex situ XRD and FESEM investigations were conducted. Fig. 5c shows the XRD patterns of the NCO-500 electrode obtained before discharge, after discharge to 2.35 V, after recharge to 4.35 V and after 80th cycle, respectively. Compared with the XRD pattern of the fresh electrode, the characteristic Li2O2 peaks were distinctly observed in the electrode after the 1st full discharge to 2.35 V. These two peaks were assigned to the (100) and (101) of Li2O2, and this indicated that Li2O2 is the major crystalline discharge product.16,21,83 After being recharged to 4.35 V and after the 80th fixed-capacity cycle, the peaks of Li2O2 were not found in the XRD pattern. The result demonstrates that the NCO-500 electrode exhibited an excellent cycling stability and can efficiently catalyse the formation and decomposition of Li2O2 during cycling. As shown in Fig. 5d, the urchin-like sphere was wrapped up with a Li2O2 film after the cell was fully discharged to 2.35 V. Whether Li2O2 was formed as particles or as films was controlled by the solubility of LiO2.84 The particle-shaped Li2O2 grew via a solution mechanism due to the high soluble LiO2, while the film-shaped Li2O2 grew through a surface growth model of low soluble LiO2. In this case, film-like Li2O2 was formed via a surface growth pathway on the urchin-like NiO–NiCo2O4 cathode, providing a large contact area between the discharge product and cathode surfaces.85 As a benefit, the electron transfer during the charge process was facilitated, contributing to a reduced charge overpotential and good rechargeability.86,87 After being recharged to 4.35 V, the film disappeared, and the urchin-like structure of the NCO-500 electrode appeared again as shown in Fig. 5e. The morphology of the electrode also retained after 80th cycle at a fixed capacity of 600 mA h g−1, as shown in Fig. 5f.
Fig. 6 shows the schematic illustration for the formation and decomposition of film-like Li2O2 during the discharge and recharge process in Li–O2 batteries with the NiO–NiCo2O4 electrode. The urchin-like morphology promoted the diffusion of oxygen and electron and provided sufficient space for loading Li2O2 to mitigate volumetric variation during cycling. The heterostructure microspheres effectively prevented the diffusion of discharge products and accelerated electron transfer for Li2O2 conversion reactions. The synergetic effect among NiCo2O4, NiO and the heterostructure also contributed to the superior electrocatalytic ability of NiO–NiCo2O4 electrodes.57 Therefore, this NiO–NiCo2O4 heterostructure exhibited excellent electrocatalytic performance in Li–O2 batteries.
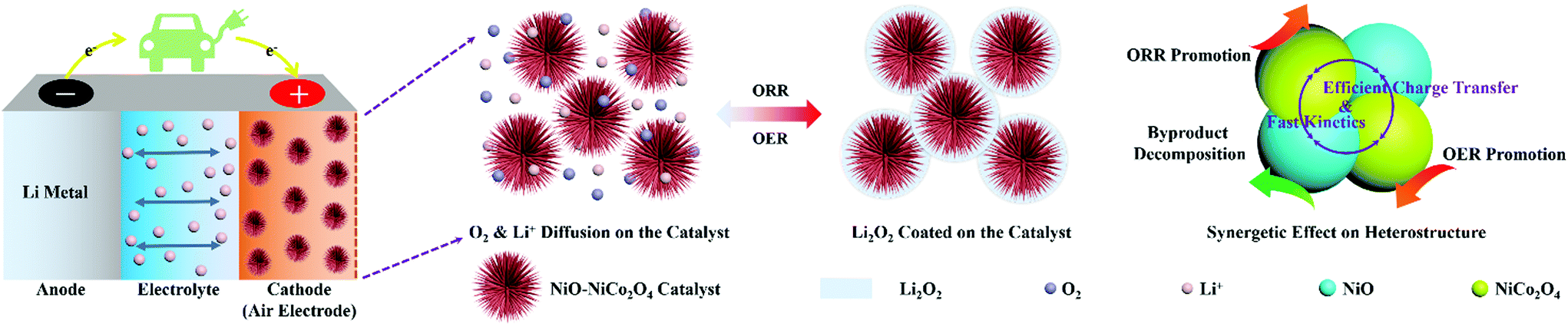 | ||
| Fig. 6 Schematic illustration for the reaction mechanism during discharge and recharge process. | ||
4. Conclusions
In summary, urchin-like NiO–NiCo2O4 microspheres with heterostructures were successfully synthesized through a facile hydrothermal-assisted method and presented favourable catalytic activity towards both the ORR and the OER for nonaqueous Li–O2 batteries. The assembled batteries employing NCO-500 as a catalyst delivered a high discharge/charge capacity of 9231/8349 mA h g−1 at the current density of 100 mA g−1. Besides, NCO-500 also exhibited good rate capability and cycling stability. Even when the current density increased to 500 mA g−1, discharge/charge capacities of 3711/2254 mA h g−1 were still achieved. When discharge/charge capacities are limited to 600 mA h g−1, Li–O2 batteries containing the NCO-500 electrode could be continuously cycled for 80 cycles without obvious terminal voltage variation and exhibited no capacity loss at the current density of 100 mA g−1. This superior electrocatalytic performance resulted from the unique heterostructure of NiO–NiCo2O4. It can facilitate continuous oxygen flow and charge transport during cycle as well as provide enough reaction sites for Li2O2 deposition and decomposition. Furthermore, according to the reported literature,57 the introduction of the NiO phase is likely to promote the decomposition of certain by-products. This study suggests that the NiO–NiCo2O4 microspheres with heterostructures are promising cathode catalyst materials for Li–O2 batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 51672162), the China Postdoctoral Science Foundation (2017M622198), the Natural Science Foundation of Shandong Province (ZR2017BEM018), the Open Project Program of Key Laboratory for Analytical Science of Food Safety and Biology, Ministry of Education (FS18010) and the Fundamental Research Funds of Shandong University (No. 2017JC035).Notes and references
- (a) X. M. Lou, C. F. Lin, Q. Luo, J. B. Zhao, B. Wang, J. B. Li, Q. Shao, X. K. Guo, N. Wang and Z. H. Guo, ChemElectroChem, 2017, 4, 3171–3180 CrossRef CAS; (b) M. Idrees, S. Batool, J. Kong, Q. Zhuang, H. Liu, Q. Shao, N. Lu, Y. Feng, E. K. Wujcik, Q. Gao, T. Ding, R. Wei and Z. Guo, Electrochim. Acta, 2019, 296, 925–937 CrossRef CAS; (c) Y. Guo, Y. Li, X. Lou, J. Guan, Y. Li, X. Mai, H. Liu, C. X. Zhao, N. Wang, C. Yan, G. Gao, H. Yuan, J. Dai, R. Su and Z. Guo, J. Mater. Sci., 2018, 53, 13790–13800 CrossRef CAS; (d) J. Guan, H. Xiao, X. Lou, Y. Guo, X. Luo, Y. Li, C. Yan, X. Yan, G. Gao, H. Yuan, J. Dai, R. Su and Z. Guo, ES Energy. Environ., 2018, 1, 80–88 Search PubMed.
- C. F. Lin, L. Hu, C. B. Cheng, K. Sun, X. K. Guo, Q. Shao, J. Li, N. Wang and Z. H. Guo, Electrochim. Acta, 2018, 260, 65–72 CrossRef CAS.
- R. J. Li, C. F. Lin, N. Wang, L. J. Luo, Y. J. Chen, J. B. Li and Z. H. Guo, Adv. Compos. Hybrid Mater., 2018, 1, 440–459 CrossRef.
- J. Guan, H. Y. Xiao, X. Y. Lou, Y. G. Guo, X. M. Luo, Y. S. Li, C. Yan, X. R. Yan, G. L. Gao, H. Yuan, J. Da, R. J. Su, W. X. Gu and Z. H. Guo, Eng. Sci., 2018, 1, 80–88 Search PubMed.
- X. C. Zhao, P. Yang, L. J. Yang, Y. Cheng, H. Y. Chen, H. Liu, G. Wang, V. Murugadoss, S. Angaiah and Z. H. Guo, ES Mater. Manuf., 2018, 1, 67–71 Search PubMed.
- S. L. Zhai, L. Wei, H. E. Karahan, Y. Q. Wang, C. J. Wang, A. Montoya, Q. Shao, X. Wang and Y. Chen, Carbon, 2018, 132, 698–708 CrossRef CAS.
- L. K. Yang, X. Wang, X. M. Mai, T. Wang, C. Wang, X. Li, V. Murugadoss, Q. Shao, S. Angaiah and Z. H. Guo, J. Colloid Interface Sci., 2019, 534, 459–468 CrossRef CAS PubMed.
- Y. Li, L. L. Zou, J. Li, K. Guo, X. W. Dong, X. W. Li, X. Z. Xue, H. F. Zhang and H. Yang, Electrochim. Acta, 2014, 129, 14–20 CrossRef CAS.
- G. X. Liu, L. Zhang, S. Q. Wang, L. X. Ding and H. H. Wang, J. Mater. Chem. A, 2017, 5, 14530–14536 RSC.
- J. Christensen, P. Albertus, R. S. Sanchez Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1–R30 CrossRef CAS.
- W. B. Luo, X. W. Gao, D. Q. Shi, S. L. Chou, J. Z. Wang and H. K. Liu, Small, 2016, 12, 3031–3038 CrossRef CAS PubMed.
- K. M. Liao, X. B. Wang, Y. Sun, D. M. Tang, M. Han, P. He, X. F. Jiang, T. Zhang and H. S. Zhou, Energy Environ. Sci., 2015, 8, 1992–1997 RSC.
- Y. Zhang, L. Qian, W. Zhao, X. M. Li, X. S. Huang, X. M. Mai, Z. K. Wang, Q. Shao, X. R. Yan and Z. H. Guo, J. Electrochem. Soc., 2018, 165, H510–H516 CrossRef CAS.
- Z. Y. Guo, D. D. Zhou, X. L. Dong, Z. J. Qiu, Y. G. Wang and Y. Y. Xia, Adv. Mater., 2013, 25, 5668–5672 CrossRef CAS PubMed.
- H. G. Jung, J. Hassoun, J. B. Park, Y. K. Sun and B. Scrosati, Nat. Chem., 2012, 4, 579–585 CrossRef CAS PubMed.
- Z. L. Jian, P. Liu, F. J. Li, P. He, X. W. Guo, M. W. Chen and H. S. Zhou, Angew. Chem., Int. Ed., 2014, 53, 442–446 CrossRef CAS PubMed.
- Z. Y. Guo, G. N. Zhu, Z. J. Qiu, Y. G. Wang and Y. Y. Xia, Electrochem. Commun., 2012, 25, 26–29 CrossRef CAS.
- J. J. Xu, Z. L. Wang, D. Xu, L. L. Zhang and X. B. Zhang, Nat. Commun., 2013, 4, 2438 CrossRef PubMed.
- M. Leskes, N. E. Drewett, L. J. Hardwick, P. G. Bruce, G. R. Goward and C. P. Grey, Angew. Chem., Int. Ed., 2012, 51, 8560–8563 CrossRef CAS PubMed.
- B. Sun, J. Q. Zhang, P. Munroe, H. J. Ahn and G. X. Wang, Electrochem. Commun., 2013, 31, 88–91 CrossRef CAS.
- F. Li, D. M. Tang, Y. Chen, D. Golberg, H. Kitaura, T. Zhang, A. Yamada and H. Zhou, Nano Lett., 2013, 13, 4702–4707 CrossRef CAS PubMed.
- X. F. Hu, X. P. Han, Y. X. Hu, F. Y. Cheng and J. Chen, Nanoscale, 2014, 6, 3522–3525 RSC.
- D. Oh, J. Qi, Y. C. Lu, Y. Zhang, Y. Shao Horn and A. M. Belcher, Nat. Commun., 2013, 4, 2756 CrossRef PubMed.
- F. J. Li, D. M. Tang, Z. L. Jian, D. Q. Liu, D. Golberg, A. Yamada and H. S. Zhou, Adv. Mater., 2014, 26, 4659–4664 CrossRef CAS PubMed.
- J. Lu, L. Cheng, K. C. Lau, E. Tyo, X. Y. Luo, J. G. Wen, D. Miller, R. S. Assary, H. H. Wang, P. Redfern, H. M. Wu, J. B. Park, Y. K. Sun, S. Vajda, K. Amine and L. A. Curtiss, Nat. Commun., 2014, 5, 4895 CrossRef CAS PubMed.
- W. H. Ryu, T. H. Yoon, S. H. Song, S. Jeon, Y. J. Park and I. D. Kim, Nano Lett., 2013, 13, 4190–4197 CrossRef CAS PubMed.
- L. L. Huang, Y. J. Mao, G. Q. Wang, X. K. Xia, J. Xie, S. C. Zhang, G. H. Du, G. S. Cao and X. B. Zhao, NewJ. Chem., 2016, 40, 6812–6818 RSC.
- J. K. Wang, R. Gao, D. Zhou, Z. J. Chen, Z. H. Wu, G. Schumacher, Z. B. Hu and X. F. Liu, ACS Catal., 2017, 7, 6533–6541 CrossRef CAS.
- L. T. Yan, H. Z. Wang, D. Huang and H. M. Luo, Eng. Sci., 2018, 1, 4–20 CrossRef.
- Y. Cao, Z. K. Wei, J. He, J. Zang, Q. Zhang, M. S. Zheng and Q. F. Dong, Energy Environ. Sci., 2012, 5, 9765–9768 RSC.
- S. Y. Liu, Y. G. Zhu, J. Xie, Y. Huo, H. Y. Yang, T. J. Zhu, G. S. Cao, X. B. Zhao and S. C. Zhang, Adv. Energy Mater., 2014, 4, 1301960 CrossRef.
- G. Q. Wang, L. L. Huang, W. Huang, J. Xie, G. H. Du, S. C. Zhang, P. Y. Zhu, G. S. Cao and X. B. Zhao, Nanoscale, 2015, 7, 20614–20624 RSC.
- S. C. Ma, L. Q. Sun, L. N. Cong, X. G. Gao, C. Yao, X. Guo, L. H. Tai, P. Mei, Y. P. Zeng, H. M. Xie and R. S. Wang, J. Phys. Chem. C, 2013, 117, 25890–25897 CrossRef CAS.
- H. L. Wang, Y. Yang, Y. Y. Liang, G. Y. Zheng, Y. G. Li, Y. Cui and H. J. Dai, Energy Environ. Sci., 2012, 5, 7931–7935 RSC.
- L. Zou, J. F. Cheng, Y. X. Jiang, Y. P. Gong, B. Chi, J. Pu and L. Jian, RSC Adv., 2016, 6, 31248–31255 RSC.
- S. J. Peng, Y. X. Hu, L. L. Li, X. P. Han, F. Y. Cheng, M. Srinivasan, Q. Y. Yan, S. Ramakrishna and J. Chen, Nano Energy, 2015, 13, 718–726 CrossRef CAS.
- C. Shen, Z. Y. Wen, F. Wang, K. Rui, Y. Lu and X. W. Wu, J. Power Sources, 2015, 294, 593–601 CrossRef CAS.
- D. A. Agyeman, M. Park and Y. M. Kang, J. Mater. Chem. A, 2017, 5, 22234–22241 RSC.
- J. Wang, T. Qiu, X. Chen, Y. L. Lu and W. S. Yang, J. Power Sources, 2014, 268, 341–348 CrossRef CAS.
- L. Y. Li, L. F. Shen, P. Nie, G. Pang, J. Wang, H. S. Li, S. Y. Dong and X. G. Zhang, J. Mater. Chem. A, 2015, 3, 24309–24314 RSC.
- B. Kirubasankar, V. Murugadoss, J. Lin, T. Ding, M. Y. Dong, H. Liu, J. X. Zhang, T. X. Li, N. Wang, Z. H. Guo and S. Angaiah, Nanoscale, 2018, 10, 20414–20425 RSC.
- B. Song, T. T. Wang, H. G. Sun, Q. Shao, J. K. Zhao, K. K. Song, L. H. Hao, L. Wang and Z. H. Guo, Dalton Trans., 2017, 46, 15769–15777 RSC.
- T. M. Su, Q. Shao, Z. Z. Qin, Z. H. Guo and Z. L. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- Y. Ma, M. L. Ma, X. Q. Yin, Q. Shao, N. Lu, Y. N. Feng, Y. Lu, E. K. Wujcik, X. M. Mai, C. Wang and Z. H. Guo, Polymer, 2018, 156, 128–135 CrossRef CAS.
- L. J. Wang, T. Zhu, Z. Y. Lyu, J. Zhang, L. L. Gong, S. N. Xiao, J. Liu, W. H. Dong, X. H. Cui, G. W. Ho and W. Chen, RSC Adv., 2016, 6, 98867–98873 RSC.
- L. X. Zhang, S. L. Zhang, K. J. Zhang, G. J. Xu, X. He, S. M. Dong, Z. H. Liu, C. S. Huang, L. Gu and G. L. Cui, Chem. Commun., 2013, 49, 3540–3542 RSC.
- B. Sun, X. D. Huang, S. Q. Chen, Y. F. Zhao, J. Q. Zhang, P. Munroe and G. X. Wang, J. Mater. Chem. A, 2014, 2, 12053–12059 RSC.
- W. M. Liu, T. T. Gao, Y. Yang, Q. Sun and Z. W. Fu, Phys. Chem. Chem. Phys., 2013, 15, 15806–15810 RSC.
- X. Y. Yu, X. Z. Yao, T. Luo, Y. Jia, J. H. Liu and X. J. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 3689–3695 CrossRef CAS PubMed.
- K. R. Yoon, G. Y. Lee, J. W. Jung, N. H. Kim, S. O. Kim and I. D. Kim, Nano Lett., 2016, 16, 2076–2083 CrossRef CAS PubMed.
- W. Yin, Y. Shen, F. Zou, X. L. Hu, B. Chi and Y. H. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 4947–4954 CrossRef CAS PubMed.
- X. F. Hu, F. Y. Cheng, N. Zhang, X. P. Han and J. Chen, Small, 2015, 11, 5545–5550 CrossRef CAS PubMed.
- P. Tan, W. Shyy, M. C. Wu, Y. Y. Huang and T. S. Zhao, J. Power Sources, 2016, 326, 303–312 CrossRef CAS.
- (a) Y. Zheng, T. F. Zhou, C. F. Zhang, J. F. Mao, H. K. Liu and Z. P. Guo, Angew. Chem., Int. Ed., 2016, 55, 3408–3413 CrossRef CAS PubMed; (b) Z. Zhao, R. Guan, J. Zhang, Z. Zhao and P. Bai, Acta Metall. Sin. (Engl. Lett.), 2017, 30, 66–72 CrossRef CAS; (c) Z. Zhao, P. Bai1, R. Guan, V. Murugadoss, H. Liu, X. Wang and Z. Guo, Mater. Sci. Eng., A, 2018, 734, 200–209 CrossRef CAS; (d) Y. Zhao, L. Qi and Y. Jin, et al. , J. Alloys Compd., 2015, 647, 1104–1110 CrossRef CAS; (e) Y. Zhao, S. Deng, H. Liu, J. Zhang, Z. Guo and H. Hou, Comput. Mater. Sci., 2018, 154, 365–370 CrossRef CAS; (f) C. Wang, Z. He, X. Xie, X. Mai, Y. Li, T. Li, M. Zhao, C. Yan, H. Liu, E. Wujcik and Z. Guo, Macromol. Mater. Eng., 2018, 3, 1700462 CrossRef; (g) Z. Wang, H. Zhu, N. Cao, R. Du, Y. Liu and G. Zhao, Mater. Lett., 2017, 186, 274–278 CrossRef CAS; (h) C. Wang, B. Mo, Z. He, Q. Shao, D. Pan, E. Wujick, J. Guo, X. Xie, X. Xie and Z. Guo, J. Membr. Sci., 2018, 556, 118–125 CrossRef CAS.
- (a) W. W. Zhou, C. W. Cheng, J. P. Liu, Y. Y. Tay, J. Jiang, X. T. Jia, J. X. Zhang, H. Gong, H. H. Hng, T. Yu and H. J. Fan, Adv. Funct. Mater., 2011, 21, 2439–2445 CrossRef CAS; (b) L. Zhang, M. Qin, W. Yu, Q. Zhang, H. Xie, Z. Sun, Q. Shao, X. Guo, L. Hao, Y. Zheng and Z. Guo, J. Electrochem. Soc., 2017, 164, H1086–H1090 CrossRef CAS; (c) L. Zhang, W. Yu, C. Han, J. Guo, Q. Zhang, H. Xie, Q. Shao, Z. Sun and Z. Guo, J. Electrochem. Soc., 2017, 164, H651–H656 CrossRef CAS; (d) C. Wang, B. Mo, Z. He, C. X. Zhao, L. Zhang, Q. Shao, X. Guo, E. Wujcik and Z. Guo, Polymer, 2018, 138, 363–368 CrossRef CAS.
- (a) L. Y. Hu, C. L. Dai, H. Liu, Y. Li, B. L. Shen, Y. M. Chen, S. J. Bao and M. W. Xu, Adv. Energy Mater., 2018, 1800709 CrossRef; (b) D. Jiang, V. Murugadoss, Y. Wang, J. Lin, T. Ding, Z. Wang, Q. Shao, C. Wang, H. Liu, N. Lu, R. Wei, S. Angaiah and Z. Guo, Polym. Rev., 2018 DOI:10.1080/15583724.2018.1546737 , in press; (c) W. Du, X. Wang, J. Zhan, X. Sun, L. Kang, F. Jiang, X. Zhang, Q. Shao, M. Dong, H. Liu, V. Murugadoss and Z. Guo, Electrochim. Acta, 2019, 296, 907–915 CrossRef CAS; (d) L. Wang, H. Qiu, C. Liang, P. Song, Y. Han, Y. Han, J. Gu, J. Kong, D. Pan and Z. Guo, Carbon, 2019, 141, 506–514 CrossRef CAS; (e) H. Liu, Q. Li, S. Zhang, R. Yin, X. Liu, Y. He, K. Dai, C. Shan, J. Guo, C. Liu, C. Shen, X. Wang, N. Wang, Z. Wang, R. Wei and Z. Guo, J. Mater. Chem. C, 2018, 6, 12121–12141 RSC.
- M. Hong, H. C. Choi and H. R. Byon, Chem. Mater., 2015, 27, 2234–2241 CrossRef CAS.
- S. F. Tong, M. B. Zheng, Y. Lu, Z. X. Lin, J. Li, X. P. Zhang, Y. Shi, P. He and H. S. Zhou, J. Mater. Chem. A, 2015, 3, 16177–16182 RSC.
- J. Y. Tian, Q. Shao, X. J. Dong, J. L. Zheng, D. Pan, X. Y. Zhang, H. L. Cao, L. H. Hao, J. R. Liu, X. M. Mai and Z. H. Guo, Electrochim. Acta, 2018, 261, 236–245 CrossRef CAS.
- P. Tan, Z. H. Wei, W. Shyy, T. S. Zhao and X. B. Zhu, Energy Environ. Sci., 2016, 9, 1783–1793 RSC.
- B. Liu, P. Yan, W. Xu, J. Zheng, Y. He, L. Luo, M. E. Bowden, C. M. Wang and J. G. Zhang, Nano Lett., 2016, 16, 4932–4939 CrossRef CAS PubMed.
- A. Shanmugavani and R. K. Selvan, Electrochim. Acta, 2016, 189, 283–294 CrossRef CAS.
- C. Wei, Y. Huang, M. H. Chen, J. Yan, W. Yao and X. F. Chen, J. Colloid Interface Sci., 2017, 504, 1–11 CrossRef CAS PubMed.
- J. Zhao, Z. J. Li, M. Zhang, A. Meng and Q. D. Li, ACS Sustainable Chem. Eng., 2016, 4, 3598–3608 CrossRef CAS.
- X. J. Liu, J. F. Liu and X. M. Sun, J. Mater. Chem. A, 2015, 3, 13900–13905 RSC.
- F. T. Ran, X. B. Yang and L. Shao, Adv. Compos. Hybrid Mater., 2018, 1, 32–55 CrossRef.
- D. P. Lapham and A. C. C. Tseung, J. Mater. Sci., 2004, 39, 251–264 CrossRef CAS.
- L. Wei, K. Lozano and Y. B. Mao, Eng. Sci., 2018, 3, 62–66 Search PubMed.
- Y. N. Feng, E. Witkoske, E. S. Bell, Y. Wang, A. Tzempelikos, I. T. Ferguson and N. Lu, ES Mater. Manuf., 2018, 1, 13–20 Search PubMed.
- Q. Q. Tang, M. M. Chen, L. Wang and G. C. Wang, J. Power Sources, 2015, 273, 654–662 CrossRef CAS.
- C. Mahala and M. Basu, ACS Omega, 2017, 2, 7559–7567 CrossRef CAS.
- J. F. Marco, J. R. Gancedo, M. Gracia, J. L. Gautier, E. I. Ríos, H. M. Palmer, C. Greaves and F. J. Berry, J. Mater. Chem., 2001, 11, 3087–3093 RSC.
- D. L. Liu, C. Zhang, Y. F. Yu, Y. M. Shi, Y. Yu, Z. Q. Niu and B. Zhang, Nano Res., 2017, 11, 603–613 CrossRef.
- C. Chang, L. Zhang, C. W. Hsu, X. F. Chuah and S. Y. Lu, ACS Appl. Mater. Interfaces, 2018, 10, 417–426 CrossRef CAS PubMed.
- X. F. Lu, D. J. Wu, R. Z. Li, Q. Li, S. H. Ye, Y. X. Tong and G. R. Li, J. Mater. Chem. A, 2014, 2, 4706–4713 RSC.
- J. Zhang, F. Liu, J. P. Cheng and X. B. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 17630–17640 CrossRef CAS PubMed.
- J. M. Xu, J. S. Wu, L. L. Luo, X. Q. Chen, H. B. Qin, V. Dravid, S. B. Mi and C. L. Jia, J. Power Sources, 2015, 274, 816–822 CrossRef CAS.
- E. Jokar, A. I. Zad and S. Shahrokhian, J. Solid State Electrochem., 2014, 19, 269–274 CrossRef.
- S. Lau and L. A. Archer, Nano Lett., 2015, 15, 5995–6002 CrossRef CAS PubMed.
- L. L. Liu, H. P. Guo, Y. Y. Hou, J. Wang, L. J. Fu, J. Chen, H. K. Liu, J. Z. Wang and Y. P. Wu, J. Mater. Chem. A, 2017, 5, 14673–14681 RSC.
- B. D. McCloskey, R. Scheffler, A. Speidel, D. S. Bethune, R. M. Shelby and A. C. Luntz, J. Am. Chem. Soc., 2011, 133, 18038–18041 CrossRef CAS PubMed.
- S. A. Freunberger, Y. H. Chen, N. E. Drewett, L. J. Hardwick, F. Barde and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- L. L. Liu, J. Wang, Y. Hou, J. Chen, H. K. Liu, J. Z. Wang and Y. P. Wu, Small, 2016, 12, 602–611 CrossRef CAS PubMed.
- Z. Sadighi, J. P. Liu, F. Ciucci and J. K. Kim, Nanoscale, 2018, 10, 15588–15599 RSC.
- F. L. Meng, Z. W. Chang, J. J. Xu, X. B. Zhang and J. M. Yan, Mater. Horiz., 2018, 5, 298–302 RSC.
- X. Z. Ren, M. J. Huang, S. Luo, Y. L. Li, L. B. Deng, H. W. Mi, L. N. Sun and P. X. Zhang, J. Mater. Chem. A, 2018, 6, 10856–10867 RSC.
- Y. Xing, Y. Yang, R. J. Chen, M. C. Luo, N. Chen, Y. S. Ye, J. Qian, L. Li, F. Wu and S. J. Guo, Small, 2018, 14, 1704366 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr08457b |
| This journal is © The Royal Society of Chemistry 2019 |