
A general and rapid room-temperature synthesis approach for metal sulphide nanocrystals with tunable properties†
Yang
Liu
 a,
Maixian
Liu
b,
Deqiang
Yin
a,
Dewei
Zhu
a and
Mark T.
Swihart
*a
a,
Maixian
Liu
b,
Deqiang
Yin
a,
Dewei
Zhu
a and
Mark T.
Swihart
*a
aDepartment of Chemical and Biological Engineering, University at Buffalo, The State University of New York, Buffalo, New York 14260, USA. E-mail: swihart@buffalo.edu
bDepartment of Pharmaceutical Science, University at Buffalo, The State University of New York, Buffalo, New York 14260, USA
First published on 3rd December 2018
Abstract
Colloidal metal sulphide (MS) nanocrystals (NCs) have recently attracted considerable attention because of their tunable properties that can be exploited in various physical, chemical and biological applications. In this work, we present a novel and general method for synthesis of monodispersed binary (CuS, Ag2S, CdS, PbS, and SnS), ternary (Ag–In–S, Cu–In–S and Cu–Sn–S) and quaternary (Cu–Zn–Sn–S) MS NCs. The synthesis is conducted at room temperature, with an immediate crystallization process and up to 60 seconds of growth time, enabling rapid synthesis without external heating. For some of the ternary and quaternary NCs produced with relatively low crystallinity, we then carried out a “colloidal annealing” process to improve their crystallinity without changing their composition. Moreover, we show that the morphology and optical properties of the NCs can be tuned by varying the concentration of precursors and reaction time. The shape evolution and photoluminescence of particular MS NCs were also studied. These results not only provide insights into the growth mechanisms of MS NCs, but also yield a generalized, low cost, and potentially scalable method to fabricate them.
Introduction
Colloidal semiconductor NCs have attracted broad attention due to their size, shape, and composition-dependent electrical, optical, magnetic and mechanical properties.1,2 Metal sulphides, an important family of semiconductors, have been applied in many fields including bioimaging,3,4 photocatalysis,5,6 optoelectronic devices,7 and photovoltaic applications8,9 that exploit their diverse and tunable features. For example, plasmonic copper sulphide NCs have shown great promise for use in photothermal therapy10,11 and photoacoustic imaging.12,13 Developing high-yield and scalable synthesis of MS NCs with precisely tunable optical properties is desirable for both fundamental studies and practical applications.14 Over the last decade, a variety of MS NCs have been prepared by many synthesis routes, including one-pot thermal decomposition of single precursors,15,16 injecting organo-sulphur precursors into metal-organo complexes,17,18 and thermolysis of ligands in the presence of metal-organo complexes.4,19We previously showed that covellite CuS NPls can be prepared by mixing ammonium sulphide (AS) solution with Cu-OAm complex at ambient conditions.20 Moreover, providing additional AS can produce larger CuS NPls with a corresponding red-shift of their near-infrared localized surfaced plasmon resonance (NIR LSPR) absorbance. In this contribution, we expand this room-temperature synthetic method to many different metal sulphide (MS) NCs. Although several reports focused on generic methods to synthesize binary MS NCs have been published,21–23 preparing ternary and quaternary MS NCs using a simple and generalized synthetic route remains a formidable challenge. Therefore, developing a general solution method for the direct fabrication of a variety of binary, ternary and quaternary MS NCs is still of great significance.
Traditional approaches to synthesis of colloidal metal sulphide NCs usually suffer from difficulty in achieving and controlling a desired level of precursor reactivity.24 Utilizing a highly-reactive sulphur precursor is one means of increasing the conversion of less reactive cations. Robinson's group previously reported a generic method for scalable synthesis of monodisperse metal sulphide NCs by using OAm-AS (dried using molecular sieves) and aqueous AS solutions as the sulphur precursors.22 However, many of the reactions were carried out at elevated temperatures. Postsynthetic modification of MS NCs using AS has not been studied, and doing so is necessary to understand the effect of additional AS on determining the shape of NCs. Therefore, we aimed to develop a rapid room-temperature method to synthesize MS NCs with well-controlled morphology by combining OAm, metal precursors, AS, and toluene (as a solvent and diluent).
Here, we present a novel and general method to synthesize a variety of binary, ternary and quaternary MS NCs. Compared to traditional colloidal synthesis, which requires external heating, this approach enables a rapid, low temperature synthesis by directly mixing metal-OAm complexes with an AS solution. The MS crystallization process occurs immediately, as indicated by a rapid colour change upon introducing the AS solution. This is illustrated Fig. 1, in which the vials in the top row contain metal precursors in OAm and those in the bottom row are diluted dispersions of MS NCs in hexane with a cation concentration of ∼2.5 mM. Furthermore, these MS NCs behaved differently when additional AS was added after the initial particle formation. We observed additional growth upon introducing more AS to the CuS, Ag2S, and CdS NCs. However, further growth of PbS and SnS NCs upon AS addition was minimal. Ternary and quaternary NCs can be prepared by the same method, but in some cases with low crystallinity. This general method provides an opportunity to study the properties of distinct MS NCs using a consistent fabrication process.
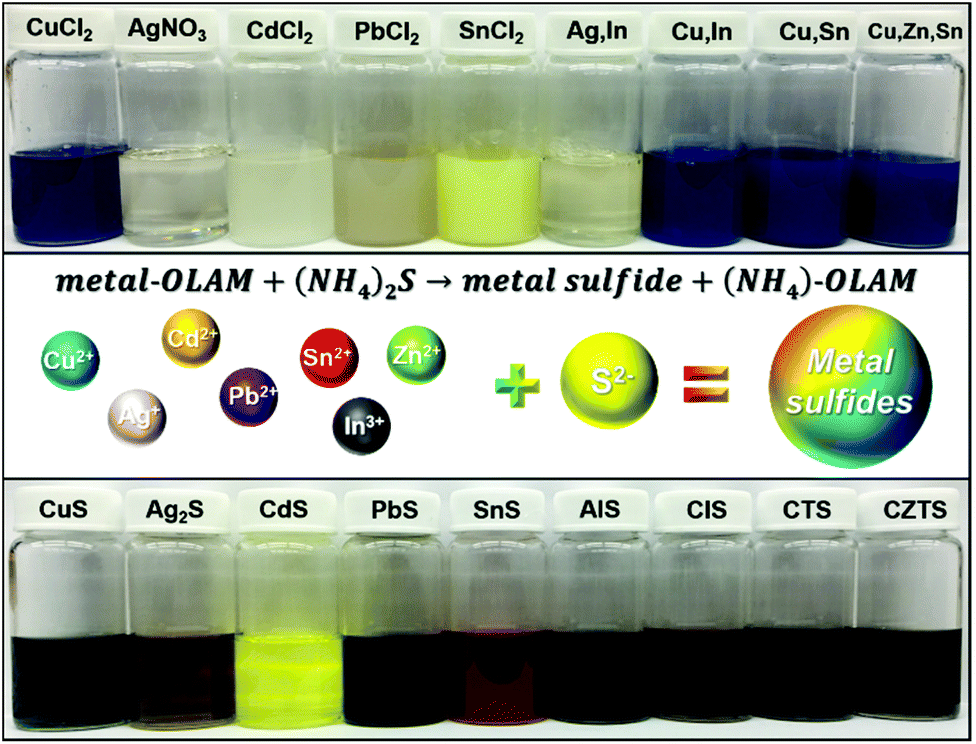 | ||
| Fig. 1 The vials on top are the metal precursors in OAm and vials in the bottom are the corresponding MS NCs dispersed in hexane with cation concentration of ∼2.5 mM. | ||
Experimental section
Chemicals
Oleylamine (70%, OAm), oleic acid (technical grade, OA 90%), silver nitrate (AgNO3), tin(II) chloride (SnCl2), tin(IV) chloride (SnCl4), cadmium sulphide (CdCl2), lead chloride (PbCl2), indium acetate (In(ac)3), zinc chloride (ZnCl2) and hexane were purchased from Sigma-Aldrich. CuCl2·2H2O, toluene, and ammonium sulphide (21.2 wt% in water) were purchased from Fisher Scientific. All chemicals were used as received.Preparation of cation precursor
The cation precursor can be generally prepared by sonicating the metal salt(s) with 5 mL of OAm and 5 mL of toluene for about 3–5 min in a 20 mL vial. All cation precursors formed uniform solutions or suspensions of various colours without precipitation. In contrast to the other metal salts, PbCl2 in OAm required over 30 min to dissolve or disperse. Heating can be used to accelerate dissolution of PbCl2, and subsequent cooling of the solution to room temperature does not lead to precipitation.Synthesis of metal sulphide NCs
The general method of synthesizing MS NCs is to inject AS solution into the solution or suspension of cation precursor(s) at room temperature. The volume of OAm and toluene remains the same in all cases, while reaction parameters including the volume of AS solution and quantity of metal salts were varied as listed in Table S1.† To purify the MS NCs, 10 mL ethanol was added to destabilize the NC dispersion, followed by centrifugation at 5000 rpm for 1 min. Then the NCs were redispersed in hexane and washed with ethanol to remove excess sulphur and ligands. The MS NCs could be redispersed in organic solvents including chloroform, hexane and toluene for further characterization.Synthesis of PbS NPs
The synthesis of PbS NPs was based on Cademartiri's method with some modifications.25 First, 9.6 mmol PbCl2 was dissolved in 16 mL OAm and the mixture was heated to 120 °C under argon and held at this temperature for 30 min to remove dissolved oxygen. Simultaneously, 1 mmol sulphur powder was dissolved in 8 mL OAm. The 8 mL OAm-S was then injected into the Pb-OAm complex and the reaction vessel was kept at 120 °C for 15 min before quenching by adding 20 mL of ethanol. To remove impurities, the dispersion was centrifuged and the precipitate was redispersed in 3 mL hexane to which 6 mL of OA was added before centrifuging a second time. The precipitate was redispersed in hexane then destabilized with ethanol before centrifuging a third time. The precipitate was redispersed in 5 mL toluene for further characterization and for studying PbS shape evolution.Shape evolution of PbS NCs
To investigate shape evolution of PbS NCs, 0.5 mL of PbS NPs in toluene was added to a mixture of 5 mL OAm and 5 mL toluene at room temperature. Then, 0.5 mL of AS solution was added and the reaction vessel was stirred for 5 min. To purify the PbS NCs, 10 mL ethanol was added into the solution followed by centrifugation at 5000 rpm for 1 min. The precipitate was collected and redispersed in toluene.Post-synthetic heat treatment of MS NCs
To improve the crystallinity of the ternary and quaternary MS NCs, a dispersion of each type of MS NC was prepared by adding dry NC (one batch) into 3 mL of OAm and sonicating. 7 mL of OAm was held under flowing argon at ambient temperature for 30 min before heating to 200 °C. Next, 3 mL of NC dispersion was injected into the mixture, and the solution was held at this temperature for 10 min. To collect the NPls, 10 mL of ethanol was added, followed by centrifuging at 5000 rpm for 1 min.Characterization
Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2010 or an FEI Tecnai F20 G2, in both cases at an accelerating voltage of 200 kV. TEM grids were prepared by dropping a dilute NC dispersion onto a carbon-coated copper or nickel TEM grid and allowing the sample to dry under air. Size distributions were obtained from TEM images by measuring at least 150 NCs from multiple TEM images from different areas of the grid, using Nanomeasurer v. 1.2. STEM imaging was carried out on a FEI Tecnai F20 200 kV STEM with a Fischione HAADF STEM detector using samples prepared on Ni TEM grids. Powder X-ray diffraction (XRD) measurements were carried out using a Rigaku Ultima IV diffractometer with a Cu Kα X-ray source. Samples were prepared by drop-casting concentrated NC dispersions onto glass slides. UV-vis-NIR spectra of NC dispersions in hexane were taken on a Shimadzu UV-3600. PL emission spectra were recorded by a Fluorolog-3.11 Jobin Yvon-Horiba spectrofluorometer. Elemental analysis of NCs by energy-dispersive X-ray spectroscopy (EDX) was obtained using an Oxford Instruments X-Max 20 mm2 EDX detector within a Zeiss Auriga scanning electron microscope (SEM). Inductively coupled plasma optical emission spectrometry (ICP-OES), using an Agilent 730 spectrometer, was employed to quantify the amount of Ag in the product NPs to determine the reactant conversion and product yield of large scale synthesis. For ICP-OES analysis, the dried Ag2S NPs were digested in aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) HCl/HNO3) for 48 h.
:1 (v/v) HCl/HNO3) for 48 h.
Results and discussion
Reaction kinetics
All MS NCs were prepared by injecting aqueous AS into a solution of cation precursor(s) (metal-OAm complex(es)) in toluene at ambient conditions. Organo-sulphur precursors are often prepared by dissolving sulphur powder in organic ligands (neat or diluted in solvent). Other commercially-available sulphur-containing organic compounds (e.g. di-tert-butyl disulphide, dodecanethiol), which thermally decompose to release a reactive sulphur component at elevated temperature can also be used.26–29 In contrast to traditional organic and some other inorganic sulphur sources (e.g. Na2S), AS can not only be readily dissolved in organic solvents containing ligands but can also release highly reactive S2− at low temperature.22 Common commercially available AS solution (∼20 wt% AS in water), contains ∼3.1 mmol of S2− per mL solution. The high reactivity of AS, and the use of excess AS drive rapid formation of MS NCs. In the present case, the reaction rate is very high because an excess of highly-reactive AS was used as the sulfur precursor. The rapid reaction provides limited possibilities for controlling the reaction kinetics to change the product composition or phase. In the case of CuS NPls, as shown in Fig. 1, the dark blue colour of the Cu-OAm solution shows the presence of Cu2+. After adding AS solution, the colour of the solution changed to dark green instantly, indicating formation of covellite CuS. Similarly, homogeneous solutions were obtained by mixing other metal salts with OAm and toluene via sonication. When AS was introduced, the various solutions exhibited different colours such as bright yellow, dark brown and reddish brown, due to formation of NCs with specific absorbances at visible wavelengths. The scale of this process for producing metal sulphide NCs can readily be increased. As an example, Fig. S1† demonstrates results of a scaled up synthesis of Ag2S NPs at a batch size of ∼4 g. Based on ICP-OES analysis, the yield of the large scale synthesis was 95.1% of the Ag provided in the precursor, suggesting this synthesis is scalable with high yield.Although many kinds of MS NCs have been prepared by this method, we note the limit of this approach for some cations. Tin has two air-stable oxidation states which are tin(II) and tin(IV).30,31 A creamy yellow homogenous solution was obtained by sonicating SnCl2 with OAm and toluene at room temperature (Fig. 1, fifth from the left top). However, as shown in Fig. S2 in the ESI,† a notable quantity of powder remained in the bottom of the vial after sonicating SnCl4 with OAm. This can be understood based on the fact that Sn4+ has greater hardness than Sn2+, in the context of hard–soft acid–base theory, so that Sn4+ is expected to interact more strongly with Cl−, rather than –NH2. Ternary and quaternary MS NCs can also be synthesized by this method, however, some of them exhibit relatively low crystallinity, such as CuInS2 and Cu2ZnSnS4. Although TEM images, HRTEM images and XRD patterns reveal that these NCs are small particles with low diffraction intensity (Fig. S2†), the EDX elemental analysis in Table S2† shows that all the product NCs (including binary, ternary and quaternary NCs without heat treatment) have compositions close to the expected stoichiometric ratios. These MS NCs are usually synthesized using high temperature to improve cation reactivity towards S2−. These low-crystalline NCs can be potentially be used in specific applications that involve annealing. For example, most processes for producing CIS and CZTS photovoltaics from nanocrystals involve annealing nanocrystalline films in a sulphur or selenium-containing atmosphere.32–35 To improve the quality and crystallinity of these MS NCs, we carried out post-synthetic heat treatment on them (see Experimental section for details). As shown in Fig. S3,† the corresponding XRD patterns and HRTEM images indicate that quality of the NCs after the “colloidal annealing” was much improved, while the overall size increased. We attribute the size increase to additional growth or slight aggregation during high temperature treatment. In contrast to the noisy XRD patterns of the as-prepared ternary and quaternary NCs, clearer XRD patterns corresponding to the most thermodynamically stable phases are observed after colloidal annealing. These crystal phases all have crystal structures in which the atoms are packed in an fcc-like lattice (zincblende-like structures), rather than wurtzite-like phases with hcp packing, which generally require higher formation energy. We note that the elemental ratios of these alloy NCs were almost unchanged after the colloidal annealing process. This reflects the fact that our room-temperature method is able to produce NCs with the expected stoichiometric ratios, such that their crystallinity can be improved by thermal treatment without changing their composition.
Morphology of MS NCs
The MS NCs synthesized using this general room temperature method are typically quite monodisperse. Here, we present typical TEM and HRTEM image of MS NCs, with corresponding fast Fourier transforms (FFT). Fig. 2a shows a TEM image of 19.2 ± 1.5 nm covellite CuS nanoplatelets (NPls). Size-distribution histograms for all samples are presented in Fig. S4.† The side-view high-resolution TEM (HRTEM) image of a covellite CuS NPl with lattice fringes in the inset of Fig. 2a shows the product CuS is single crystalline, with interplanar spacings of 0.19 nm, 0.33 nm and 0.27 nm, corresponding to the (110), (100), and (006) planes of covellite, respectively (Fig. 2a and Fig. S5a†). The TEM and HRTEM images show that the 〈100〉 and 〈010〉 directions lie within the plane of the CuS NPls, while the 〈001〉 direction corresponds to the NPl thickness. To test the sensitivity of our method to the oxidation state of the starting cation precursor, we replaced CuCl2 by CuCl in the reaction to produce CuS NPls while keeping other factors fixed. The results indicated that, upon mixing with OAm and toluene at ambient conditions, the Cu(I) was oxidized to Cu(II), then followed the same reaction mechanism that produced CuS NPls using CuCl2 (Fig. S5b and c†).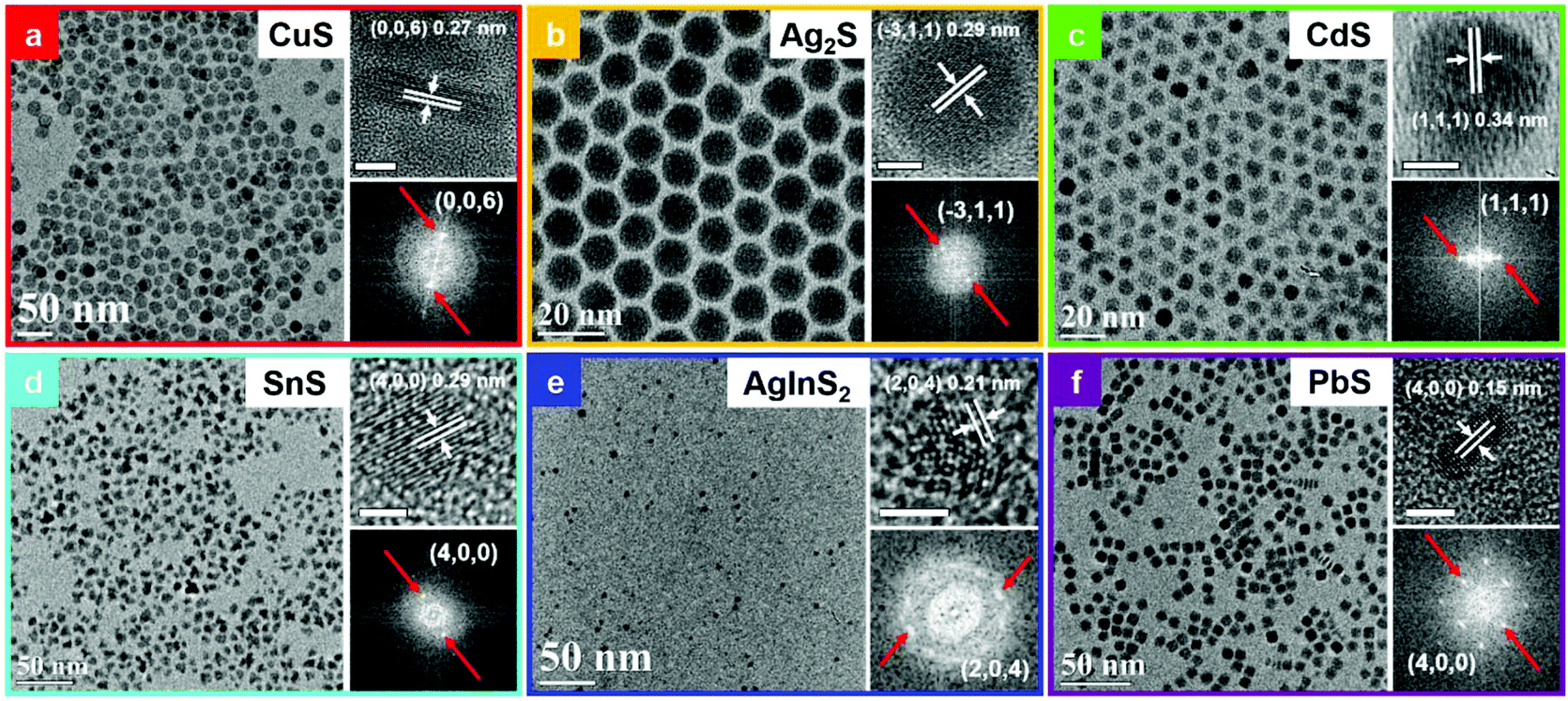 | ||
| Fig. 2 TEM images, HRTEM images and corresponding FFT of the HRTEM images of MS NCs, as labelled. All unlabelled scale bars are 5 nm. | ||
Fig. 2b shows a typical TEM image of spherical Ag2S NCs of 11.6 ± 0.5 nm diameter. HRTEM of Ag2S nanoparticles (NPs) shows an interplanar spacing of 0.29 nm, which matches the (![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 11) lattice spacing of the monoclinic Ag2S phase. The TEM image in Fig. 2c shows that the CdS NCs produced by this method have a truncated triangular morphology, due to the fast growth rate of (001) planes in wurtzite CdS as well as Ostwald ripening.36 The HRTEM image in the inset indicates that CdS NCs exhibit an interplanar spacing of 0.34 nm, which matches the spacing of (111) planes in wurtzite CdS. TEM and HRTEM images of SnS NCs are shown in Fig. 2d. The interplanar spacing of 0.29 nm matches the (400) planes of a cubic phase of SnS, as discussed further below. A typical TEM image of AgInS2 quantum dots (QDs) is displayed in Fig. 2e. The lattice fringe spacing of 0.21 nm in the corresponding HRTEM image matches the (204) planes of AgInS2.
11) lattice spacing of the monoclinic Ag2S phase. The TEM image in Fig. 2c shows that the CdS NCs produced by this method have a truncated triangular morphology, due to the fast growth rate of (001) planes in wurtzite CdS as well as Ostwald ripening.36 The HRTEM image in the inset indicates that CdS NCs exhibit an interplanar spacing of 0.34 nm, which matches the spacing of (111) planes in wurtzite CdS. TEM and HRTEM images of SnS NCs are shown in Fig. 2d. The interplanar spacing of 0.29 nm matches the (400) planes of a cubic phase of SnS, as discussed further below. A typical TEM image of AgInS2 quantum dots (QDs) is displayed in Fig. 2e. The lattice fringe spacing of 0.21 nm in the corresponding HRTEM image matches the (204) planes of AgInS2.
PbS NPls exhibit a square plate-like shape with dimension of 7.7 ± 0.6 × 2.7 ± 0.3 nm, as shown in Fig. 2f. This is, to the best of our knowledge, the first report of monodisperse square PbS NPls. The side-view HRTEM image of PbS NPl indicates an interplanar spacing of 0.15 nm, corresponds to (400) planes in the galena PbS phase. According to previous reports, PbS nanocubes can be synthesized at precursor ratios of Pb:S < 1, while at higher Pb:S precursor ratios, the PbS NCs adopt a cuboctahedral shape.37,38 The Pb:S ratio in this rapid reaction is ∼0.1, leading to the formation of the truncated cuboid morphology. To investigate the formation of PbS NPls, we then studied the Pb-OAm precursor, which appears to be a dispersion, rather than a true solution. The Pb-OAm complex was centrifuged to collect Pb-OAm species, then the Pb-OAm was redispersed in toluene to form a suspension. TEM imaging of the diluted suspension revealed that the Pb-OAm is in chain-like assemblies in which Pb-based chelates were linked by OAm (Fig. S6a†). PbS nanocubes have isotropic growth rates on six facets of a cube, producing a cubic shape. However, the chain-like template of Pb-OAm suppresses growth on a pair of equivalent facets, due to passivation by OAm. Thus, we hypothesize that when S2− ions react with Pb2+ on the surface of growing nanoparticles, the other two pairs of equivalent facets possess higher reaction rates, leading to anisotropic growth in the shape of a square plate (Fig. S6b†). Our results are also supported by observations from a prior report in which pseudo-hexagonal PbS NPls were produced within lamellar amine/oleate-bilayer mesophase templates.39
Crystal phase and optical properties of MS NCs
The study of crystal phase and optical properties of these MS NCs further helps us to understand the capabilities and potential of this synthetic method. Fig. 3(a–f) present XRD patterns of MS NCs prepared using this method. Fig. 3a shows that the crystal phase of the NPls is covellite CuS (ICDD PDF 04-007-1392). Several phases of SnS with similar diffraction patterns are known, but the XRD pattern of Fig. 3(b) most closely matches the recently-reported π-cubic phase of SnS, which has a large unit cell containing 32 Sn and 32 S atoms and a lattice constant of a = 11.6233 Å.40,41 The crystal phases of other MS NCs were confirmed by the close correspondence of their XRD patterns to standard references, as illustrated in Fig. 3. The XRD study clearly reveals that these MS NCs exhibit substantial crystallinity when synthesized at room temperature within a short growth time.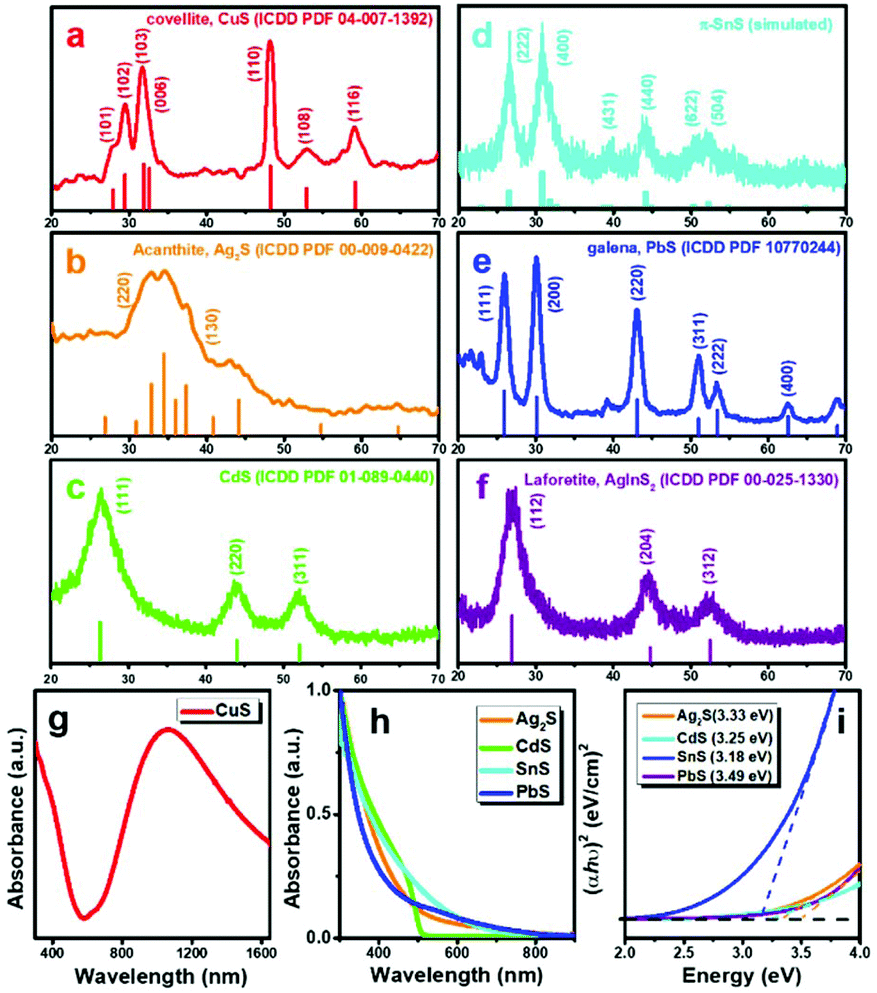 | ||
| Fig. 3 (a)–(f) XRD patterns of MS NCs, (g) UV-vis-NIR spectrum of covellite CuS NPls. (h) UV-vis absorbance spectra of MS NCs. (i) Calculated direct band gaps of MS NCs. | ||
The UV-vis-NIR absorbance spectrum shows that the ∼19 nm CuS NPls have strong absorbance at NIR wavelengths, peaking at ∼1100 nm. This LSPR absorbance arises because CuS is p-type semiconductor with intrinsically high carrier concentration.42 The absorbance spectra and corresponding calculated band gaps of other binary MS NCs are presented in Fig. 3(h and i). The differences in absorbance and band gaps of these MS NCs arise from their morphology, size, crystal structure, and electronic structure. Ternary AgInS2 QDs are of great interest for applications such as bioimaging and biosensing, due to their low toxicity and wide emission spectrum as a luminescent semiconductor.43,44 The photoluminescence (PL) spectrum of AgInS2 QDs shown in Fig. S7,† exhibits a broad emission peak at ∼700 nm with a PL quantum yield (QY) of 2.0% (Fig. S7,† Rhodamine B [QY = 31% in water] was used as a reference).45
Tuning size and properties of MS NCs
Most of the MS NCs are known for their tunable size- and shape-dependent properties.46 The ability to control the size and morphology of MS NCs is of great interest. In our previous report, we discussed the growth mechanism of CuS NPls by adjusting the ratio of Cu:S and AS injection manner.20 Covellite NPls exhibit LSPR absorbance that depends upon their aspect ratio (diameter over thickness), with their dominant in-plane dipolar resonance red-shifting as the aspect ratio increases.20,47 Similarly, we find that small Ag2S NPs can be prepared by reducing the amount of AS solution (10 μl) and the reaction time (10 s) for Ag2S synthesis. As shown in Fig. 4a, this method produced monodispersed Ag2S NPs of 4.1 ± 0.5 nm diameter. Fig. 4(b and c) reveal that the Ag2S NPs have a lattice spacing of 0.22 nm, corresponding to (130) planes of acanthite Ag2S. When the radius of a NC approaches the exciton Bohr radius, the motions of electron–hole pairs are confined. For some semiconductors such as Ag2S, CdS and ZnS, they can be excited by specific incident light producing emission light over a wide spectral range due to quantum confinement. The Bohr radius in Ag2S is 2.2 nm.48 However, we could not detect any PL from these small Ag2S NPs, most likely due to surface defects that provide non-radiative recombination pathways.
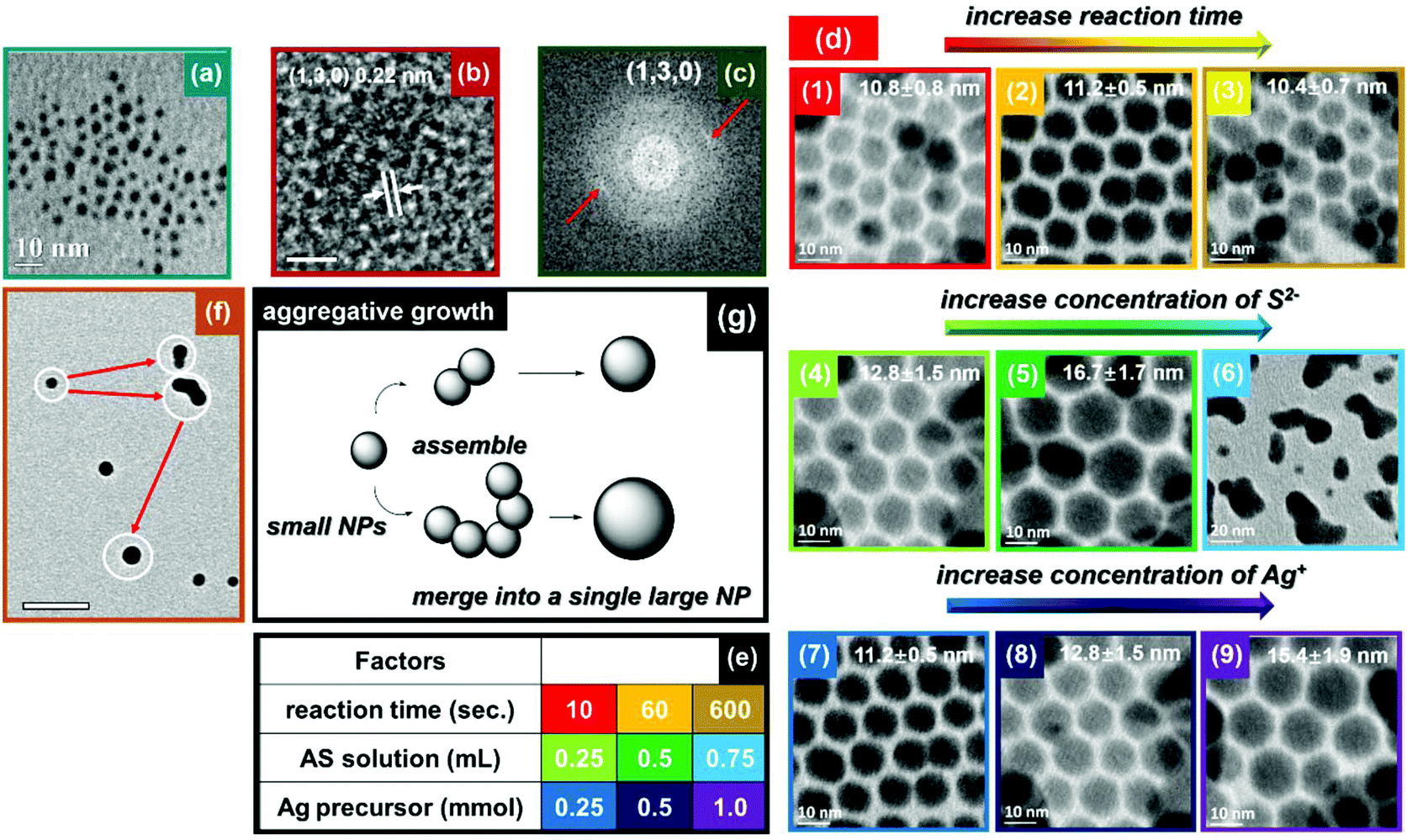 | ||
| Fig. 4 (a) TEM image, (b) HRTEM image and (c) corresponding FFT of 4.1 nm Ag2S NPs. (d) Integrated study on growth of Ag2S NPs: panels (1–3), (4–6) and (7–9) focus on factors of reaction time, concentration of S2−, and concentration of Ag+, respectively. (e) The parameters employed in the integrated study on Ag2S growth mechanism. (f) TEM image illustrating aggregative growth of Ag2S NPs. (g) Schematic illustration of aggregative growth of Ag2S NPs. The scale bars in panel (b) and (f) are 2 nm and 50 nm, respectively. | ||
Ag2S is a potential candidate for NIR QDs for in vivo photoacoustic imaging and diagnostics, due to its negligible toxicity and ideal band gap.49,50 Note that photoacoustic imaging only requires strong absorbance of light, and not PL, so NPs with surface defects that reduce PL may not be a problem. In order to study the growth mechanism of Ag2S NPs, we carried out more systematic synthesis of them. The room-temperature growth of Ag2S NCs depends upon at least three factors: reaction time, concentration of S2−, and concentration of Ag+. As shown in Fig. 4d, the results related to reaction time indicate the size of the Ag2S NPs changes very little with increasing reaction time from 10 s to 600 s (first row). This implies that reactions were accomplished rapidly. On the other hand, continuously increasing the concentration of Ag+ and S2− can facilitate NP growth. In addition, we observed aggregation of Ag2S NPs when employing a larger excess of S2− (second row). This is consistent with our previous discussion that excess S2− can partially remove ligands from the surface of the NP, leading to additional growth and ultimately aggregation of metal sulphide NPs. In order to assess this growth mechanism, we carefully analyzed TEM images of Ag2S NPs during growth. As shown in Fig. 4f, instead of Ostwald ripening, the growth of Ag2S NPs follows an aggregative growth process. The newly-nucleated small NPs possess high surface energy, which prompts two or more NPs to coalesce upon contact. The formation of dumbbell-like structures clearly illustrates this sintering of small NPs. Finally, dumbbell-like structures experience a shape evolution to spheres in order to minimize their surface energy. Fig. 4g provides a schematic illustration of the mechanism of the aggregative growth.
CdS QDs have attracted much attention due to their direct band gap at visible wavelengths, high PL quantum yield, and long-term photostability.51–54 Thus, CdS QDs have been explored widely for use in light emitting diodes, sensors, lasers and biomedical imaging and labeling. Here, we produced CdS NCs with reduced size to test their photoluminescence. The small CdS QDs were prepared by adding 10 μl of AS solution into Cd-OAm complex containing 0.1 mmol of Cd2+. Knowing that CdS QDs can emit light over a wide range of wavelengths by tailoring their size, we employed different amounts of AS solution to obtain CdS QDs with tunable emission wavelengths. The UV-vis and PL spectra of CdS QDs prepared with different S2− concentrations are recorded in Fig. 5(a and b). In the UV-vis spectra, a red shift in absorbance from 370 to 430 nm was observed due to reduced quantum confinement with increasing amount of AS solution (from 10 to 200 μl). The PL emission peaks of CdS QDs in hexane solution were tuned from 500–675 nm, red-shifting with increasing amount of S2−, due to increasing size of the CdS QDs. Large Stokes shifts are often ascribed to polydispersity, interactions between surface atoms and ligands, and the presence of trap states.55–57 In this case, we attribute the large Stokes shift to the presence of defects that are inevitably formed during low temperature synthesis. We note that the shift increased (from 0.87 eV to 1.05 eV), with increasing size of the CdS QDs. This could be explained by increased polydispersity and increased total number of defects (with broader energy range of defect states) for the CdS QDs grown when additional AS is provided.55,58 The PL QY of CdS QDs was estimated to be 4.9% (Fig. S8,† Rhodamine B [QY = 31% in water] was used as a reference). Fig. 5c shows the dispersion of CdS QDs in hexane solution under room light (left) and UV light (right). The red-shift of absorbance and PL reflects the increasing size of CdS QDs with increasing quantity of S2−employed. Again, we propose that S2− can remove ligands from the CdS QDs, allowing unreacted Cd2+ to adsorb on the surface, react, and produce larger QDs. Further increasing the amount of AS from 1 ml to 4 ml produced minimal further red-shift, indicating minimal further growth of CdS QDs after consuming all of the Cd precursor (Fig. S9†).
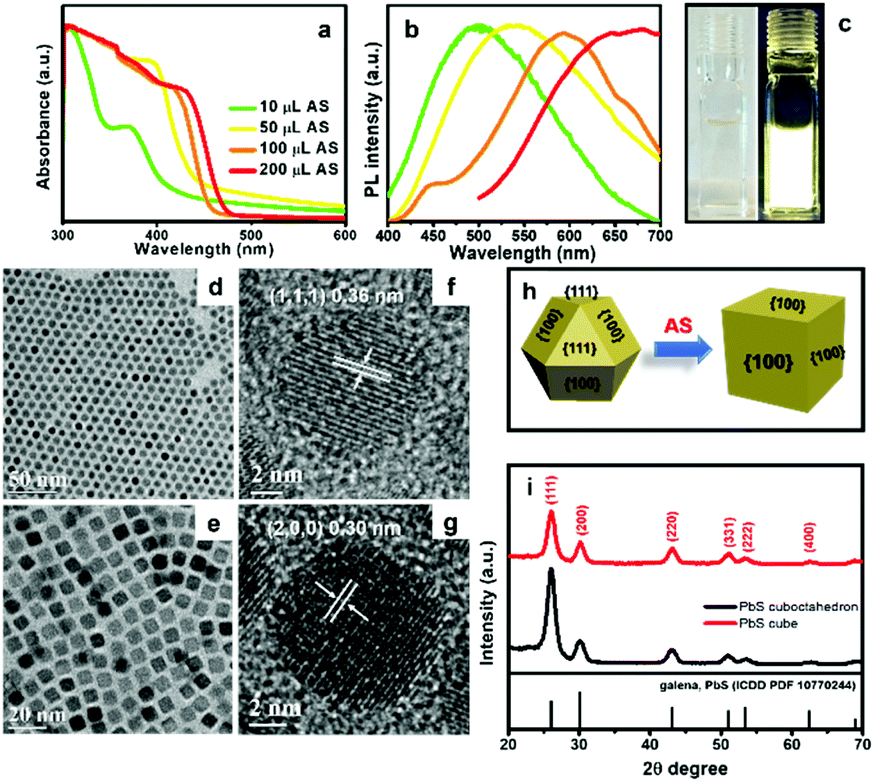 | ||
| Fig. 5 (a) Normalized UV-vis absorbance spectra of CdS QDs synthesized with increasing amounts of AS. (b) Normalized PL spectra of CdS QDs from (a). (c) Dispersion of CdS QDs in hexane under room light (left) and 365 nm UV light (right). (d), (f) TEM and HRTEM images of cuboctahedral PbS NCs. (e), (g) TEM and HRTEM images of PbS nanocubes obtained by reacting AS solution with cuboctahedral PbS NCs. (h) A schematic illustration of shape evolution of PbS NCs from cuboctahedral to cubic shape. (i) XRD patterns of PbS NCs before and after AS treatment. | ||
PbS NCs have great potential for use in thermoelectric, photocatalytic and photovoltaic devices.59,60 There have been some reports focused on shape evolution of PbS NCs by changing the molar ratio between Pb2+ and S2− precursors, reaction time, ligands, and temperature in the colloidal system.61,62 However, post-synthetic modification of PbS NCs using AS has not been studied. Here we introduce AS solution to cuboctahedral PbS NCs and observe their size and shape evolution. AS solution is a highly reactive sulphur precursor with potential to trigger shape evolution at room temperature. Cuboctahedral PbS NCs were synthesized according to Cademartiri's method.63 The TEM image in Fig. 5d reveals that the monodisperse PbS NCs have a diameter of 8.2 ± 0.6 nm. The HRTEM image in Fig. 5f shows the NCs are single crystalline, with an interplanar spacing of 0.36 nm, matching the (111) planes of galena PbS. We added 0.5 mL of AS solution into a toluene dispersion of these PbS NCs, at a concentration of ∼0.5 mmol Pb2+ at room temperature. Interestingly, the cuboctahedral PbS NCs were converted into PbS nanocubes (NCbs), with side length of 7.7 ± 0.4 nm. Consistent with prior reports of PbS shape evolution at higher temperatures, the shape evolution observed here can be generally described as follows.37,61,64 The surface energy of the (111) facets of cuboctahedral PbS NCs is much higher than the (100) facets and the atom occupancy of (111) can be considered as alternating Pb and S layers. Prior to AS addition, OAm can effectively stabilize the (111) facets. However, the (111) facets finally disappear in the presence of AS because of the high reactivity of AS and its ability to displace OAm from Pb-terminated (111) facets. A schematic illustration of this process is displayed in Fig. 5h. XRD analysis also provides evidence of shape evolution. Both cuboctahedral and cubic PbS NCs are in the galena phase. However, for the nanocubes, the peak corresponding to (111) planes is depressed compared to the peak corresponding to (200) planes, indicating (111) facets of PbS NCbs are diminished after the shape evolution.
We also observed that further addition of AS solution to the PbS NPls prepared at room temperature did not produce larger NPls. As shown in Fig. S10,† the PbS NPls slightly aggregate due to partial ligand removal upon exposure to more AS. Although the shape of the SnS NCs are not well-defined, the general size of SnS NCs also remained nearly the same upon introducing excess S2− (>2.5 mL, Fig. S11†). The limited additional growth of PbS and SnS NCs can be explained by nearly full conversion of all of the cation precursors when growing these NCs.
Conclusions
In summary, we have developed a room-temperature method for synthesis of a variety of monodispersed binary, ternary and quaternary metal sulphide nanocrystals. The crystallization and growth processes were rapid (complete in <60 seconds). This could make the present synthetic approach more convenient, scalable, and economical compared to conventional high-temperature routes. The rapid crystallization at room temperature is attributed to high reactivity of AS, which was used in excess. An additional colloidal annealing process was applied to improve the crystallinity of ternary and quaternary NCs. Moreover, we show that the concentration of metal cation and S2− can affect the size and morphology of MS NCs. The ability to tune the properties of these MS NCs in a convenient, low-temperature method can further expand the potential for practical application of this diverse family of semiconductor nanocrystals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the New York State Center of Excellence in Materials Informatics.References
- Y. Liu, M. Liu, D. Yin, L. Qiao, Z. Fu and M. T. Swihart, ACS Nano, 2018, 7803–7811 CrossRef CAS PubMed.
- Y. Liu, M. Liu and M. T. Swihart, J. Phys. Chem. C, 2017, 121, 13435–13447 CrossRef CAS.
- Q. A. Akkerman, A. Genovese, C. George, M. Prato, I. Moreels, A. Casu, S. Marras, A. Curcio, A. Scarpellini, T. Pellegrino, L. Manna and V. Lesnyak, ACS Nano, 2015, 9, 521–531 CrossRef CAS PubMed.
- D. Deng, Y. Chen, J. Cao, J. Tian, Z. Qian, S. Achilefu and Y. Gu, Chem. Mater., 2012, 24, 3029–3037 CrossRef CAS.
- Y. Ben-Shahar, F. Scotognella, I. Kriegel, L. Moretti, G. Cerullo, E. Rabani and U. Banin, Nat. Commun., 2016, 7, 10413 CrossRef CAS PubMed.
- L. Amirav and A. P. Alivisatos, J. Am. Chem. Soc., 2013, 135, 13049–13053 CrossRef CAS PubMed.
- X. J. Wu, X. Huang, X. Qi, H. Li, B. Li and H. Zhang, Angew. Chem., Int. Ed., 2014, 53, 8929–8933 CrossRef CAS PubMed.
- D. Wang, J. K. Baral, H. Zhao, B. A. Gonfa, V.-V. Truong, M. A. El Khakani, R. Izquierdo and D. Ma, Adv. Funct. Mater., 2011, 21, 4010–4018 CrossRef CAS.
- X. Chen, J. Yang, T. Wu, L. Li, W. Luo, W. Jiang and L. Wang, Nanoscale, 2018, 10, 15130–15163 RSC.
- S. Wang, A. Riedinger, H. Li, C. Fu, H. Liu, L. Li, T. Liu, L. Tan, M. J. Barthel, G. Pugliese, F. De Donato, M. Scotto D'Abbusco, X. Meng, L. Manna, H. Meng and T. Pellegrino, ACS Nano, 2015, 9, 1788–1800 CrossRef CAS PubMed.
- Y. Wang, X. Liu, G. Deng, J. Sun, H. Yuan, Q. Li, Q. Wang and J. Lu, Nanoscale, 2018, 10, 2866–2875 RSC.
- Z. Wang, P. Huang, O. Jacobson, Z. Wang, Y. Liu, L. Lin, J. Lin, N. Lu, H. Zhang, R. Tian, G. Niu, G. Liu and X. Chen, ACS Nano, 2016, 10, 3453–3460 CrossRef CAS PubMed.
- G. Ku, M. Zhou, S. Song, Q. Huang, J. Hazle and C. Li, ACS Nano, 2012, 6, 7489–7496 CrossRef CAS PubMed.
- C. B. Williamson, D. R. Nevers, T. Hanrath and R. D. Robinson, J. Am. Chem. Soc., 2015, 137, 15843–15851 CrossRef CAS PubMed.
- S. Lee, S. Baek, J. P. Park, J. H. Park, D. Y. Hwang, S. K. Kwak and S.-W. Kim, Chem. Mater., 2016, 28, 3337–3344 CrossRef CAS.
- Y. Du, B. Xu, T. Fu, M. Cai, F. Li, Y. Zhang and Q. Wang, J. Am. Chem. Soc., 2010, 132, 1470–1471 CrossRef CAS PubMed.
- W. K. Koh, S. R. Saudari, A. T. Fafarman, C. R. Kagan and C. B. Murray, Nano Lett., 2011, 11, 4764–4767 CrossRef CAS PubMed.
- X. Liu, X. Wang, B. Zhou, W.-C. Law, A. N. Cartwright and M. T. Swihart, Adv. Funct. Mater., 2013, 23, 1256–1264 CrossRef CAS.
- M. Kruszynska, H. Borchert, A. Bachmatiuk, M. H. Rummeli, B. Buchner, J. Parisi and J. Kolny-Olesiak, ACS Nano, 2012, 6, 5889–5896 CrossRef CAS PubMed.
- M. Liu, X. Xue, C. Ghosh, X. Liu, Y. Liu, E. P. Furlani, M. T. Swihart and P. N. Prasad, Chem. Mater., 2015, 27, 2584–2590 CrossRef CAS.
- Q. Tian, G. Wang, W. Zhao, Y. Chen, Y. Yang, L. Huang and D. Pan, Chem. Mater., 2014, 26, 3098–3103 CrossRef CAS.
- H. Zhang, B. R. Hyun, F. W. Wise and R. D. Robinson, Nano Lett., 2012, 12, 5856–5860 CrossRef CAS PubMed.
- S. H. Choi, K. An, E. G. Kim, J. H. Yu, J. H. Kim and T. Hyeon, Adv. Funct. Mater., 2009, 19, 1645–1649 CrossRef CAS.
- Y. Liu, D. Yin and M. T. Swihart, Chem. Mater., 2018, 30, 1399–1407 CrossRef CAS.
- L. Cademartiri, J. Bertolotti, R. Sapienza, D. S. Wiersma, G. von Freymann and G. A. Ozin, J. Phys. Chem. B, 2006, 110, 671–673 CrossRef CAS PubMed.
- Y. Zou, X. Su and J. Jiang, J. Am. Chem. Soc., 2013, 135, 18377–18384 CrossRef CAS PubMed.
- X. Liu, X. Wang and M. T. Swihart, Chem. Mater., 2013, 25, 4402–4408 CrossRef CAS.
- X. Liu, X. Wang and M. T. Swihart, Chem. Mater., 2015, 27, 1342–1348 CrossRef CAS.
- J. W. Thomson, K. Nagashima, P. M. Macdonald and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5036–5041 CrossRef CAS PubMed.
- Y. Liu, M. Liu, D. Yin, W. Wei, P. N. Prasad and M. T. Swihart, Chem. Mater., 2017, 29, 3555–3562 CrossRef CAS.
- L. De Trizio, H. Li, A. Casu, A. Genovese, A. Sathya, G. C. Messina and L. Manna, J. Am. Chem. Soc., 2014, 136, 16277–16284 CrossRef CAS PubMed.
- S. N. Park, S. J. Sung, J. H. Sim, K. J. Yang, D. K. Hwang, J. Kim, G. Y. Kim, W. Jo, D. H. Kim and J. K. Kang, Nanoscale, 2015, 7, 11182–11189 RSC.
- Y. Kim, K. Woo, I. Kim, Y. S. Cho, S. Jeong and J. Moon, Nanoscale, 2013, 5, 10183–10188 RSC.
- B. I. Park, Y. Hwang, S. Y. Lee, J. S. Lee, J. K. Park, J. Jeong, J. Y. Kim, B. Kim, S. H. Cho and D. K. Lee, Nanoscale, 2014, 6, 11703–11711 RSC.
- J. Wang, X. Xin and Z. Lin, Nanoscale, 2011, 3, 3040–3048 RSC.
- W. Chen, K. Chen, Q. Peng and Y. Li, Small, 2009, 5, 681–684 CrossRef CAS PubMed.
- K. S. Cho, D. V. Talapin, W. Gaschler and C. B. Murray, J. Am. Chem. Soc., 2005, 127, 7140–7147 CrossRef CAS PubMed.
- G. Zhou, M. Lu, Z. Xiu, S. Wang, H. Zhang, Y. Zhou and S. Wang, J. Phys. Chem. B, 2006, 110, 6543–6548 CrossRef CAS PubMed.
- P. J. Morrison, R. A. Loomis and W. E. Buhro, Chem. Mater., 2014, 26, 5012–5019 CrossRef CAS.
- J. Breternitz, R. Gunder, H. Hempel, S. Binet, I. Ahmet and S. Schorr, Inorg. Chem., 2017, 56, 11455–11457 CrossRef CAS PubMed.
- D. Yin, C. Dun, X. Gao, Y. Liu, X. Zhang, D. L. Carroll and M. T. Swihart, Small, 2018, 14, 1801949 CrossRef PubMed.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS PubMed.
- K. T. Nguyen and Y. Zhao, Nanoscale, 2014, 6, 6245–6266 RSC.
- M. Z. Fahmi and J. Y. Chang, Nanoscale, 2013, 5, 1517–1528 RSC.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- Y. Liu, M. Liu and M. T. Swihart, J. Am. Chem. Soc., 2017, 139, 18598–18606 CrossRef CAS PubMed.
- Y. Liu, M. Liu and M. T. Swihart, Chem. Mater., 2017, 29, 4783–4791 CrossRef CAS.
- G. Xu, S. Zeng, B. Zhang, M. T. Swihart, K. T. Yong and P. N. Prasad, Chem. Rev., 2016, 116, 12234–12327 CrossRef CAS PubMed.
- C. Li, Y. Zhang, M. Wang, Y. Zhang, G. Chen, L. Li, D. Wu and Q. Wang, Biomaterials, 2014, 35, 393–400 CrossRef CAS PubMed.
- H. Chen, B. Li, M. Zhang, K. Sun, Y. Wang, K. Peng, M. Ao, Y. Guo and Y. Gu, Nanoscale, 2014, 6, 12580–12590 RSC.
- J. I. Wong, N. Mishra, G. Xing, M. Li, S. Chakrabortty, T. C. Sum, Y. Shi, Y. Chan and H. Y. Yang, ACS Nano, 2014, 8, 2873–2879 CrossRef CAS PubMed.
- T. D. Nguyen, W. Y. Hamad and M. J. MacLachlan, Adv. Funct. Mater., 2014, 24, 777–783 CrossRef CAS.
- B. L. Cao, Y. Jiang, C. Wang, W. H. Wang, L. Z. Wang, M. Niu, W. J. Zhang, Y. Q. Li and S. T. Lee, Adv. Funct. Mater., 2007, 17, 1501–1506 CrossRef CAS.
- K. T. Yong, R. Hu, I. Roy, H. Ding, L. A. Vathy, E. J. Bergey, M. Mizuma, A. Maitra and P. N. Prasad, ACS Appl. Mater. Interfaces, 2009, 1, 710–719 CrossRef CAS PubMed.
- Y. Liu, D. Kim, O. P. Morris, D. Zhitomirsky and J. C. Grossman, ACS Nano, 2018, 12, 2838–2845 CrossRef CAS PubMed.
- W. Zhai, J. Lin, Q. Li, K. Zheng, Y. Huang, Y. Yao, X. He, L. Li, C. Yu, C. Liu, Y. Fang, Z. Liu and C. Tang, Chem. Mater., 2018, 30, 3714–3721 CrossRef CAS.
- G. Nagamine, H. B. Nunciaroni, H. McDaniel, A. L. Efros, C. H. de Brito Cruz and L. A. Padilha, Nano Lett., 2018, 18, 6353–6359 CrossRef CAS PubMed.
- H. Utzat, K. E. Shulenberger, O. B. Achorn, M. Nasilowski, T. S. Sinclair and M. G. Bawendi, Nano Lett., 2017, 17, 6838–6846 CrossRef CAS PubMed.
- T. Bielewicz, M. M. Ramin Moayed, V. Lebedeva, C. Strelow, A. Rieckmann and C. Klinke, Chem. Mater., 2015, 27, 8248–8254 CrossRef CAS.
- H. Fu and S. W. Tsang, Nanoscale, 2012, 4, 2187–2201 RSC.
- A. H. Khan, U. Thupakula, A. Dalui, S. Maji, A. Debangshi and S. Acharya, J. Phys. Chem. C, 2013, 117, 7934–7939 CrossRef CAS.
- M. C. Weidman, M. E. Beck, R. S. Hoffman, F. Prins and W. A. Tisdale, ACS Nano, 2014, 8, 6363–6371 CrossRef CAS PubMed.
- L. Cademartiri, J. Bertolotti, R. Sapienza, D. S. Wiersma, G. von Freymann and G. A. Ozin, J. Phys. Chem. B, 2006, 110, 671–673 CrossRef CAS PubMed.
- C. Schliehe, B. H. Juarez, M. Pelletier, S. Jander, D. Greshnykh, M. Nagel, A. Meyer, S. Foerster, A. Kornowski, C. Klinke and H. Weller, Science, 2010, 329, 550–553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr07483f |
| This journal is © The Royal Society of Chemistry 2019 |