
Scalable synthesis of a foam-like FeS2 nanostructure by a solution combustion–sulfurization process for high-capacity sodium-ion batteries†
Rudan
Hu
a,
Hongan
Zhao
a,
Jianli
Zhang
a,
Qinghua
Liang
b,
Yining
Wang
a,
Bailing
Guo
a,
Raksha
Dangol
b,
Yun
Zheng
b,
Qingyu
Yan
 *b and
Junwu
Zhu
*a
*b and
Junwu
Zhu
*a
aKey Laboratory for Soft Chemistry and Functional Materials (Nanjing University of Science and Technology), Ministry of Education, Nanjing 210094, China. E-mail: zhujw@njust.edu.cn
bSchool of Material Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore. E-mail: alexyan@ntu.edu.sg
First published on 16th November 2018
Abstract
Pyrite-type FeS2 is regarded as a promising anode material for sodium ion batteries. The synthesis of FeS2 in large quantities accompanied by an improved cycling stability, as well as retaining high theoretical capacity, is highly desirable for its commercialization. Herein, we present a scalable and simple strategy to prepare a foam-like FeS2 (F-FeS2) nanostructure by combining solution combustion synthesis and solid-state sulfurization. The obtained F-FeS2 product is highly uniform and built from interconnected FeS2 nanoparticles (∼50 nm). The interconnected feature, small particle sizes and porous structure endow the product with high electrical conductivity, good ion diffusion kinetics, and high inhibition capacity of volume expansion. As a result, high capacity (823 mA h g−1 at 0.1 A g−1, very close to the theoretical capacity of FeS2, 894 mA h g−1), good rate capability (581 mA h g−1 at 5.0 A g−1) and cyclability (754 mA h g−1 at 0.2 A g−1 with 97% retention after 80 cycles) can be achieved. The sodium storage mechanism has been proved to be a combination of intercalation and conversion reactions based on in situ XRD. Furthermore, high pseudocapacitive contribution (i.e. ∼87.5% at 5.0 mV s−1) accounts for the outstanding electrochemical performance of F-FeS2 at high rates.
Introduction
Over the past decades, lithium-ion batteries (LIBs) have dominated the commercial market of energy storage.1–7 However, the limited resources and high price of lithium hinder their wide application.8–10 Hence, sodium-ion batteries (SIBs) become one of the most promising alternatives to LIBs considering the similar working mechanism and abundant resources.11–13 As the size of the Na+ ion (1.02 Å) is larger than that of the Li+ ion (0.69 Å), it is crucial to develop viable Na-host materials for commercializing the SIB technology.Pyrite-type FeS2 has been widely regarded as one of the promising anode materials for SIBs owing to its high theoretical capacity (894 mA h g−1), abundance, cost-effectiveness and eco-friendly nature.14–16 However, FeS2-based electrodes suffer from volume expansion up to 280% during the cycling process.17 Much effort has been devoted to optimize the electrochemical performance of this material. For example, some researchers tuned the cut-off voltage to avoid large volume change,18–20 and others composited FeS2 with carbon to buffer the expansion.21–23 Although improved cyclability has been achieved through reported methods, their low yield and partial capacity sacrifice have posed a great challenge towards commercialization.24,25 Thus, exploring a facile and cost effective strategy for the large-scale production of FeS2 while retaining high theoretical capacity is highly desirable.
Herein, we report a scalable and simple strategy to prepare a FeS2 nanostructure by combining solution combustion synthesis and solid-state reaction. The first step released large amounts of gaseous products and heat, yielding a porous Fe2O3 precursor by a solution combustion procedure. Then, the precursor was sulfurized to synthesize a foam-like FeS2 (F-FeS2) nanostructure. The uniform F-FeS2 exhibits high capacity, good rate capability and cyclability. It is found that the unique foam-like structure of F-FeS2 accounts for its good electrical conductivity, ion diffusion kinetics, and tolerance ability of volume expansion. Furthermore, in situ X-ray diffraction (XRD) has been conducted to study the sodium storage mechanism. This facile strategy could open opportunities for preparing other foam-like metal sulfide materials in considerable amount and with impressive performance in electrochemical fields.
Experimental
Materials
Iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, ≥98.5%, Sinopharm Chemical Reagent), glycine (C2H5NO2, 99.5–100.5%, Macklin), sublimed sulfur (S, ≥99.5%, Sinopharm Chemical Reagent), sodium hydroxide (NaOH, ≥97%, Macklin), and iron(III) chloride hexahydrate (FeCl3·6H2O, ≥99%, Macklin) were directly used as received without any purification.Synthesis of the porous Fe2O3 precursor
The Fe2O3 precursor was prepared by a simple solution combustion synthesis.26 15 mmol Fe(NO3)3·9H2O was first dissolved in 50 mL water. Then 15 mmol C2H5NO2 was added to the as-prepared solution under stirring at 80 °C. Subsequently, the mixture solution was continuously stirred at the same temperature for 2 h. The resultant solution was transferred into a preheated evaporating dish. After removing the water, the mixture became a homogeneous colloid and self-ignited rapidly. With a great deal of smoke spurting out, the foam-like Fe2O3 precursor was prepared. This synthesis process (Video S1†) and the digital photo of the foam-like Fe2O3 precursor (Fig. S1†) can be found in the ESI.†Synthesis of foam-like FeS2 (F-FeS2)
In a typical synthesis, 20 mg of the as-obtained Fe2O3 precursor and 25 mg of sublimed sulfur were ground together gently and loaded into a quartz tube evacuated with a mechanical pump and sealed by using a Partulab MRVS-1002 system. The sealed tubes were placed in a muffle furnace and annealed at 450 °C for 3 h. The heating rate was 10 °C min−1 before the temperature reached 400 °C. After that, the heating rate was changed to 5 °C min−1. The macroporous FeS2 was obtained after cooling to ambient temperature. The sulfurized quantity of the Fe2O3 precursor can be easily extended to be more than 500 mg in one synthesis step, by using quartz tubes with a larger length and diameter.Synthesis of routine FeS2
For comparison, the routine FeS2 was also prepared via a conventional precipitation method. Firstly, 150 mmol NaOH was dissolved in 100 mL water. Subsequently, 50 mmol FeCl3 was dissolved in 50 mL water and added dropwise into the above solution under stirring. The obtained precipitate was washed three times with water and then annealed at 600 °C under air for 5 h to form Fe2O3. The as-prepared Fe2O3 was sulfurized with the same method mentioned above.Materials characterization
The phase of the as-obtained products was confirmed by X-ray diffraction (XRD) on a Bruker D8 Advance diffractometer (40 kV, 40 mA, Cu Kα radiation, λ = 0.15418 nm), and the measurement settings were 0.02° and 0.25 seconds per step. X-ray photoelectron spectra (XPS) were recorded using a Thermo ESCALAB 250Xi system with monochromatic Al Kα radiation as the excitation source. Scanning electron microscopy (SEM, JEOL JSM-7001F) and transmission electron microscopy (TEM, Tecnai G2 F30 S-TWIN) were used to characterize the morphology. Energy dispersive X-ray spectroscopy (EDX) elemental mapping and selected area electron diffraction (SAED) were conducted on a Tecnai G2 F30 S-TWIN system. Nitrogen adsorption–desorption isotherms were recorded at 77 K on a Micromeritics TriStar II surface area and porosity instrument. Inductively coupled plasma (ICP) determined elemental constituents were measured on a PerkinElmer RFIC system.Electrochemical measurements
The electrode slurry was made up of 70 wt% active material, 10 wt% acetylene black, 10 wt% SWCNTs and 10 wt% polyvinylidene fluoride (PVDF) binder homogeneously mixed in 1-methyl-2-pyrrolidinone (NMP) solvent. After stirring for 12 h, the obtained slurry was pasted onto copper foil, and heated under air at 80 °C for 30 min, and then transferred into a vacuum oven for another 4 h of drying at 120 °C to remove NMP. The electrode mass loading is ∼1 mg cm−2. Electrochemical measurements were carried out via CR2032 coin-type half cells using Na metal as the counter/reference electrode, Whatman glass microfiber filter GF/D membrane as the separator, and the solution of 1 M NaClO4 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol% with 5 vol% fluoroethylene carbonate (FEC)) as the electrolyte (∼0.4 mL per cell). All coin-type cells were assembled in an argon-filled glove box (H2O ≤ 0.1 ppm, O2 ≤ 0.1 ppm). Cyclic voltammetry (CV) tests were conducted at various scan rates (0.1–5.0 mV s−1) in a potential window of 0.01–3 V. The electrochemical impedance spectra (EIS) were recorded over a frequency range of 0.01 to 105 Hz with an amplitude of 5 mV. Both CV and EIS tests were conducted using a Biologic VMP3 system. Galvanostatic charge–discharge (GCD) tests were carried out on a LANHE battery tester from 0.01–3 V.
:1 vol% with 5 vol% fluoroethylene carbonate (FEC)) as the electrolyte (∼0.4 mL per cell). All coin-type cells were assembled in an argon-filled glove box (H2O ≤ 0.1 ppm, O2 ≤ 0.1 ppm). Cyclic voltammetry (CV) tests were conducted at various scan rates (0.1–5.0 mV s−1) in a potential window of 0.01–3 V. The electrochemical impedance spectra (EIS) were recorded over a frequency range of 0.01 to 105 Hz with an amplitude of 5 mV. Both CV and EIS tests were conducted using a Biologic VMP3 system. Galvanostatic charge–discharge (GCD) tests were carried out on a LANHE battery tester from 0.01–3 V.
Results and discussion
The foam-like FeS2 was synthesized as described in the synthesis section. For comparison, the routine FeS2 prepared by a conventional precipitation method was also employed. Fig. 1a shows the XRD patterns of the obtained samples, and the related Rietveld refined patterns are shown in Fig. 1b and c. Both the patterns can be well indexed to pure pyrite FeS2 (JCPDS no. 24-0076, space group 205). Besides, the lattice parameters evaluated by Rietveld refinement are a = b = c = 5.406 Å for F-FeS2 and a = b = c = 5.407 Å for the routine FeS2, which fit well with the JCPDS data (card no. 24-0076, a = b = c = 5.418 Å). The chemical compositions were further determined by XPS (Fig. 1d–g). For F-FeS2, two peaks located at 720.8 and 708.0 eV can be assigned to Fe 2p1/2 and Fe 2p3/2, respectively (Fig. 1d), which are accompanied by the associated satellite peaks.27 Two peaks around 164.5 and 163.3 eV can be attributed to the S 2p1/2 and S 2p3/2 species, respectively (Fig. 1e). The XPS spectra of FeS2 show similar results to those of F-FeS2. The Fe 2p1/2 and Fe 2p3/2 peaks are positioned at 720.7 and 707.9 eV, and the S 2p1/2 and S 2p3/2 ones are located at 164.4 and 163.2 eV, respectively (Fig. 1f and g).28–30 The results from both XRD and XPS confirm that F-FeS2 and FeS2 have no difference in phase and composition.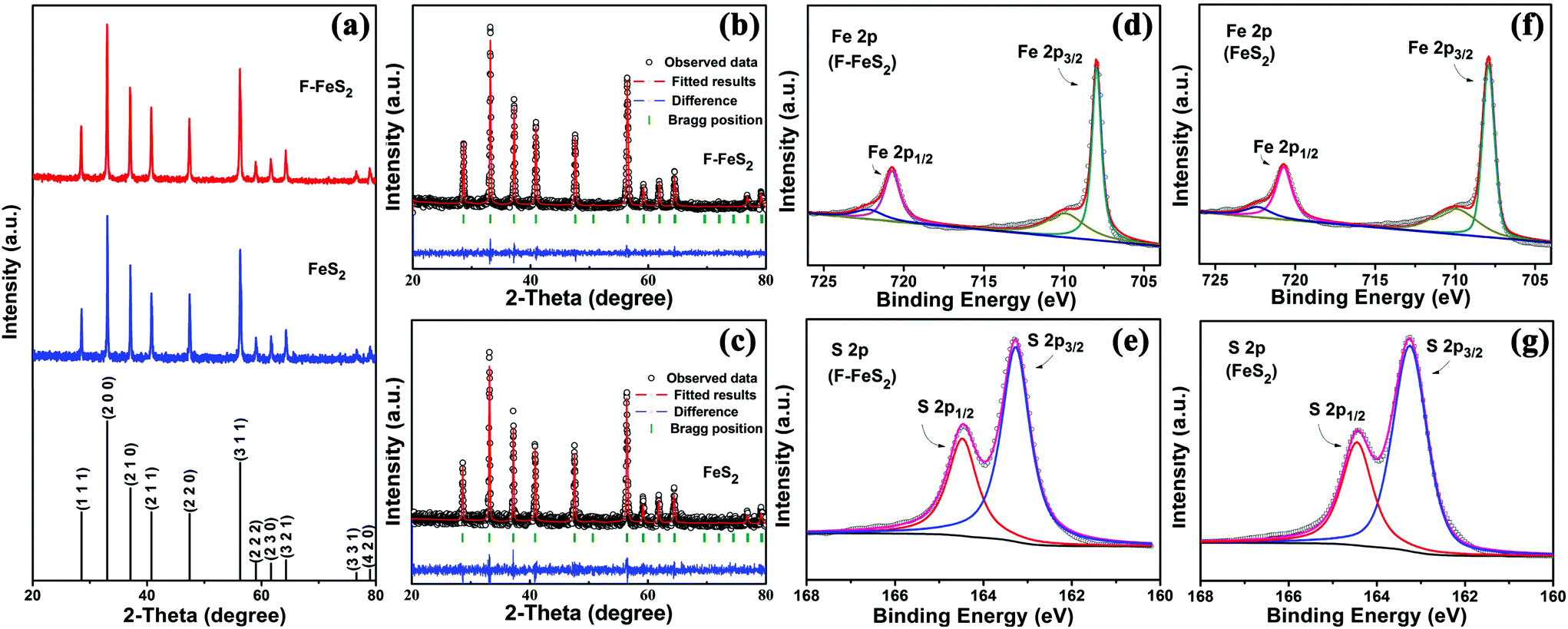 | ||
| Fig. 1 The phase and composition characterization of the as-prepared FeS2. (a) XRD patterns of F-FeS2 and FeS2. Rietveld refined patterns of (b) F-FeS2 and (c) FeS2. XPS spectra of Fe 2p and S 2p of (d–e) F-FeS2 and (f–g) FeS2. | ||
The morphology of F-FeS2 was studied by SEM and TEM, and the surface area was determined using the BET method. The as-prepared F-FeS2 has foam-like interconnected 3D porous morphology, as shown in Fig. 2a and Fig. S2 (ESI†). Such a porous structure is mainly formed due to the release of a large amount of gaseous products during liquid combustion synthesis.31,32 Interestingly, the skeleton of the foam structure is composed of FeS2 nanoparticles with diameters ranging from 25 to 120 nm (Fig. 2b), and most particles stick closely to each other, building up a continuous frame. The EDX elemental mapping results (Fig. 2c) show uniform distribution of Fe and S, confirming that the porous Fe2O3 was totally sulfurized into FeS2. The SAED pattern shown in Fig. 2d indicates the polycrystalline nature of F-FeS2. Moreover, the as-observed diffraction rings can be well indexed to the (111), (200), (210), (211), (220), (311) planes, respectively, agreeing well with the XRD result. In contrast, FeS2 shows the morphology of aggregated particles with an average size of 230 nm (Fig. 2e). Meanwhile, the BET result in Fig. 2f indicates that F-FeS2 offers a larger specific surface area (9.8 m2 g−1) than FeS2 (5.3 m2 g−1). Obviously, the larger specific surface area of F-FeS2 is attributed to the porous structure and the smaller particle sizes.
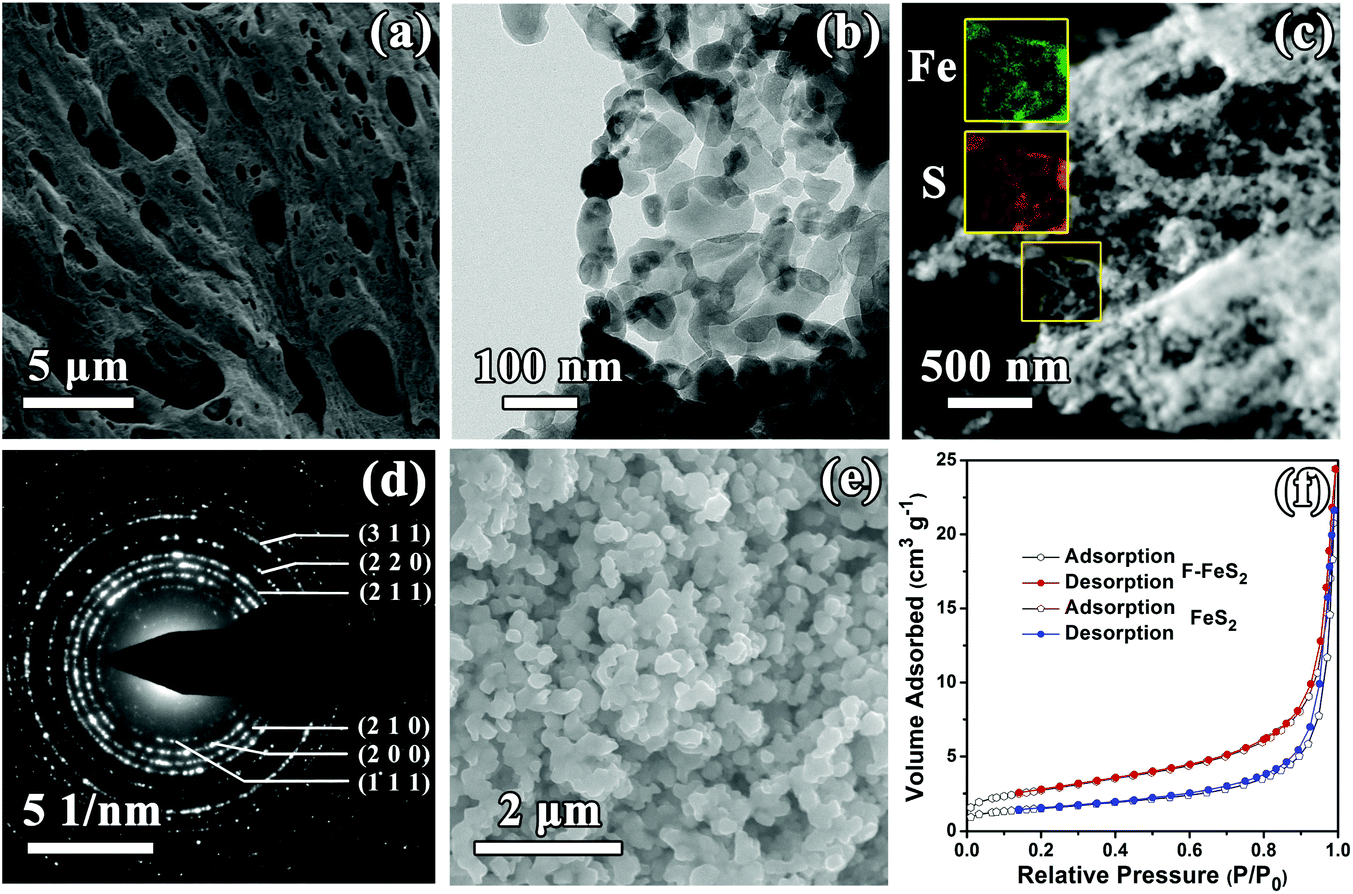 | ||
| Fig. 2 (a) SEM and (b) TEM images of F-FeS2. (c) EDX elemental mapping of Fe (green) and S (brown) in F-FeS2. (d) SAED image of F-FeS2. (e) SEM image of FeS2. (f) Nitrogen adsorption–desorption isotherms of F-FeS2 and FeS2. | ||
A half-cell configuration was assembled to evaluate the sodium storage behavior of F-FeS2. Fig. 3a shows the initial five CV curves of the F-FeS2 electrode recorded between 0.01 and 3 V at a scan rate of 0.1 mV s−1. As can be seen from the cathodic scan, two groups of peaks can be observed. A sharp cathodic peak located at 1.07 V is observed in the first cycle, which disappears in the subsequent cycles. This indicates an irreversible reaction process. It is substituted with a new reduction peak at 2.12 V. Another group of peaks, belonging to the discharging process, can be detected below 0.76 V in all cycles. The associated in situ XRD patterns in Fig. 3b show that the intensity of the F-FeS2 peaks gradually decreases during the discharging process and no peak shift can be observed. No other crystalline compound is detected until the Na2S was formed during the discharging process. These observations indicate that the phase of F-FeS2 has changed around 1 V and the iron sulphides converted into Na2S below 0.7 V. For the charging process, two groups of peaks, associated with Na+ extraction, are located around 1.32 and 2.54 V as seen in Fig. 3a. Each group contains at least two obvious peaks, indicating the corresponding multi-stepped reactions.33 Along the same process, no other diffraction peak change occurs except for the decomposition of Na2S from the lowest potential. This decomposition accounts for the first group of reaction peaks in the charging process, whereas another group of peaks appear to correspond to a different desodiation process. Previous studies have reported that the formation of NaxFeS2 can be observed during the first cycle discharging process, as well as a charging/discharging potential above 0.8 V in all subsequent scans.14,18 Besides, the lattice fringe of NaFeS2 can be observed by ex situ HRTEM for the samples charged/discharged to the mentioned potentials (Fig. S3, ESI†) in our study. Moreover, our CV plots present a similar profile as in the previous report, which was associated with an intercalation reaction.34 It is reasonable to believe that the intercalation reaction occurs in this electrochemical process. Hence, we propose the following reaction mechanism:
| FeS2 + xNa+ + xe− → NaxFeS2 (x < 2) | (1) |
Nax−yFeS2 + yNa+ + ye− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) NaxFeS2 (y < x < 2) NaxFeS2 (y < x < 2) | (2) |
| NaxFeS2 + (4 − x)Na+ + (4 − x)e− 2Na2S + Fe | (3) |
 | ||
| Fig. 3 (a) Initial five CV cycles of F-FeS2 for SIBs at 0.1 mV s−1. (b) In situ XRD patterns of F-FeS2 for SIBs from OC voltage discharging to 0.01 V, then recharging to 3 V at a rate of 0.2 A g−1; the black symbols represent the diffraction peaks of Be and BeO, the red arrow represents the initial peaks of F-FeS2 and the star mark represents the strongest Na2S peaks. (c) Initial five GCD curves of F-FeS2 for SIBs at 0.1 A g−1. | ||
Eqn (1) is associated with a sharp peak located at 1.07 V in the first cycle, and it is substituted by eqn (2) in the following cycles. The cathodic peak at 2.12 V and the anodic peaks around 2.54 V originate from this reaction. The irreversible phase change of FeS2 matches well with the previous report.21Eqn (3) describes the reduction below 0.76 V and the oxidation around 1.32 V in all cycles. In particular, we observed the peak shift of Na2S near 23.6° as seen from the magnified in situ XRD patterns in Fig. S4 (ESI†). The Na2S diffraction peak associated with 0.01 V matches exactly with that of pure Na2S. It is interesting to find that these peaks tend to appear at lower angles before discharging to 0.01 V, and keep shifting to larger angles after it. This may be attributed to the interphase generated in the conversion process, whose interplanar spacing shrinks from 3.79 to 3.74 Å during the process. In addition, the typical diffraction peaks of Be and BeO can also be seen in Fig. 3b, which are attributed to the Be window used in the in situ XRD test.
Fig. 3c presents the corresponding initial five GCD profiles of the F-FeS2 anode at 0.1 A g−1. The first cycle delivers a discharge specific capacity of 1341 mA h g−1 and a charge specific capacity of 840 mA h g−1. The initial capacity loss is due to the irreversible phase transformation reaction and the formation of a solid–electrolyte interface (SEI) layer. The SEI generating reaction also accounts for the capacity decrease between the first scan curve and the subsequent curves. This matches well with the intensity decay in the reduction peaks below 0.76 V and the oxidation peaks around 1.32 V shown in Fig. 3a. The discharge plateaus are observed around 2.1 V and 0.5 V, while the charge plateaus locate around 1.3 V and 2.5 V. This result also matches well with the redox peaks in the CV curves. Both CV curves and GCD profiles overlap well from the second cycle onward, demonstrating the good reversibility of the reaction.
The rate capabilities and cycling stabilities of the F-FeS2 and FeS2 electrodes are presented in Fig. 4a and b. The F-FeS2 anode exhibits specific capacities of 823, 771, 721, 693, 662, 630, 608, 582, and 861 mA h g−1 at current densities of 0.1, 0.2, 0.5, 1.0, 2.0, 3.0, 4.0, 5.0, and 0.1 A g−1, respectively. These values are much higher than those of FeS2 (Fig. 4a). Besides, the rate performance of F-FeS2 demonstrates the highest specific capacities among previous reports (Fig. S5, ESI†). For cyclability, F-FeS2 shows a specific capacity of 755 mA h g−1 at 0.2 A g−1 after 80 cycles, with 97% capacity retention. Meanwhile, the 20th, 50th, and 80th GCD curves during cycling can be found in Fig. S6 (ESI†), and the similar profiles reveal the stability of our F-FeS2. The coulombic efficiency below 99% after tens of cycles might be attributed to the side reactions between the ester-based electrolyte and sulfur anionic groups.6,18,25,35 In comparison, FeS2 only delivers 194 mA h g−1 at 0.2 A g−1 after 80 cycles, with 33% capacity retention. The slight increase of the discharge capacity of F-FeS2 during the initial fifty cycles can be ascribed to the activation process, which has been observed in other transition metal sulfides.36–38 In contrast, the specific capacity of FeS2 decreases rapidly in the first few cycles. Considering the same phase and composition of F-FeS2 and FeS2, it is reasonable to believe that the foam-like structure enables the improved electrochemical performance of F-FeS2. In addition, we also investigated the sodium storage performance of SWCNTs, and it displays a 5th-cycle specific capacity of 91.8 mA h g−1 at 0.1 A g−1 (Fig. S7, ESI†). Taking the mass percentage (10% of the loading mass) into account, the capacity contribution from the SWCNT additive was negligible.
 | ||
| Fig. 4 (a) Rate capabilities of F-FeS2 and FeS2 for SIBs from 0.1–5.0 A g−1. (b) Cycling stabilities of F-FeS2 and FeS2 at 0.2 A g−1. (c) EIS of F-FeS2 and FeS2 half cells. | ||
To reveal the reaction kinetics for such good Na-storage performance of F-FeS2, both samples were subjected to EIS. Both samples show similar Nyquist plots obtained at open circuit voltage. A depressed semicircle at high frequency followed by a diffusion drift at low frequency can be clearly observed (Fig. 4c). Obviously, F-FeS2 has a smaller depressed semicircle diameter than FeS2 at high frequency. This suggests that the F-FeS2 electrode has better charge transfer performance than FeS2, which might be attributed to the interconnected feature of F-FeS2. The low frequency line is associated with the ion diffusion of electrodes.15 The line slope of FeS2 is smaller than that of F-FeS2, indicating the faster transport of ions in the F-FeS2 electrode cells. The porous structure and relatively small particle sizes offer a larger surface area. This allows more contact sites with the electrolyte, thus shortening the ion transporting distance.15,20 The EIS spectra have also been fitted to an equivalent circuit, and the calculated resistances and Warburg coefficients are agreeable with the analysis above (Fig. S8, ESI†). All of these account for the high discharge capacity at each scan rate. Besides, the pores of the foam structure of FeS2 offer more space for volume expansion during the sodium storing process. This guarantees the structural stability of F-FeS2, resulting in a high capacity F-FeS2 electrode with good cyclability.
To better understand the good high-rate performance of F-FeS2, the reaction kinetics was further analyzed by distinguishing the surface-controlled pseudocapacitive capacity and diffusion-controlled capacity. Fig. 5a shows the CV curves of F-FeS2 for various scan rates ranging from 0.1–5 mV s−1. It can be found that the peaks of all the CV curves show a similar shape, although broadened peaks can be observed at increased scan rates. The preserved peaks even at high scan rates indicate small polarization during the reaction process.39 The contribution of pseudocapacitive effects is qualitatively revealed by the b value from the equation log(i) = blog(v) + log(a) where both a and b are constants, and i and v represent the current and scan rate.40–42 The series of peak currents marked in Fig. 5a were chosen to determine the b value by fitting lines based on this equation. The calculated b value of F-FeS2 is 0.93. As the value of b is very close to 1, it suggests that there is a large contribution from the pseudocapacitive storage in the F-FeS2 electrode.43–45 To quantify the pseudocapacitive contribution, the equation i = k1v + k2v0.5 was used according to previous reports, where i and v represent the current and scan rate, respectively, and k is a constant related to a fixed potential.46–48 By fitting the value of k1 at different voltage stages, we calculated a series of pseudocapacitive contributions to the current. As a result, the direct view of pseudocapacitive storage contribution (red part) for F-FeS2 at 1.0 mV s−1 is shown in Fig. 5c, and it is determined to be ∼82.5%. This indicates the surface pseudocapacitance-dominated process in the F-FeS2 electrode. The overall pseudocapacitive contributions at different scan rates were obtained using the same method, as shown in Fig. 5d. As the scan rates increase, the pseudocapacitive contribution also increases gradually. A maximum value of ∼87.5% can be obtained at 5.0 mV s−1. It suggests that the pseudocapacitive behavior dominates the whole reaction process, especially at high scan rates. It is well known that the surface-controlled pseudocapacitive process is much faster than the diffusion-controlled process.19 Such high pseudocapacitive contribution may be ascribed to the unique foam-like structure with relatively large surface area and small particle sizes, resulting in good rate capability of the F-FeS2 electrode.
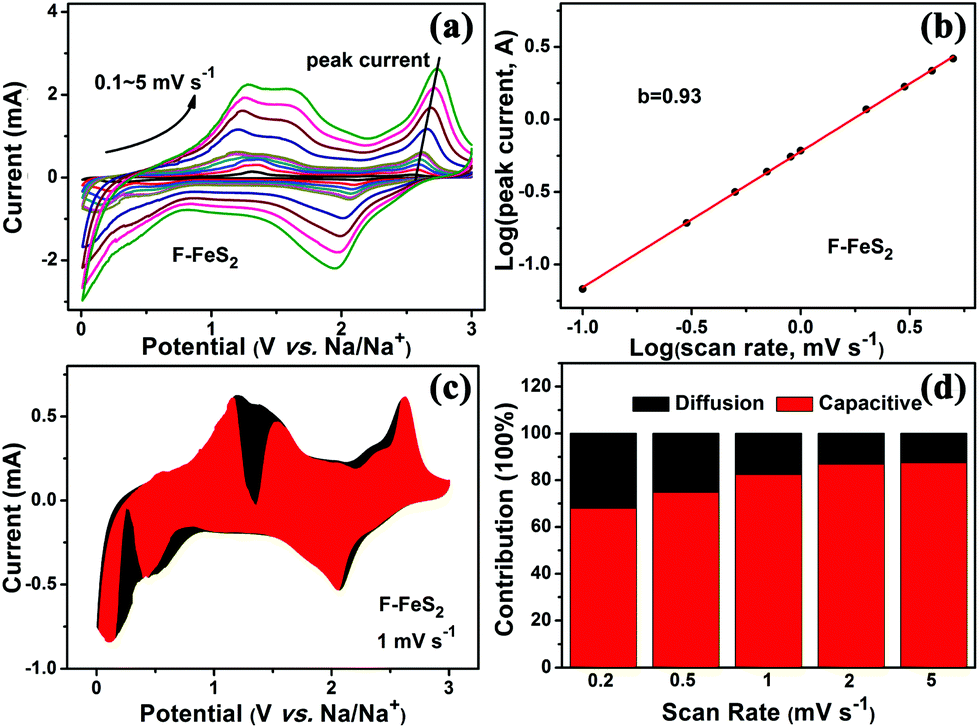 | ||
| Fig. 5 (a) CV curves of F-FeS2 for scan rates from 0.1 to 5 mV s−1. (b) Functional relationship of peak current (i) vs. scan rate (v). (c) Capacity contribution of F-FeS2 at 1 mV s−1. (d) Capacitive contribution comparison at different scan rates. | ||
Besides, the good electrochemical performance of F-FeS2, which benefits from the foam-like structure, can also be explained from the structural mechanistic aspect. As shown in Fig. S9 (ESI†), Na, S and Fe elements distribute evenly throughout the 3rd-cycle discharged material, indicating a thorough chemical conversion process. This means that our porous foam-like structure consisting of small FeS2 nanoparticles is suitable for kinetically reversible reactions.34 In addition, FeS2 tends to react with alkali metals via a radial movement of a reaction front triggered by the contact of two materials.49,50 Thus, the sufficient contact area and ion transport pathway offered by the porous structure are beneficial for sodium storage. Besides, Fe ions, based on their short diffusion length,51 tend to nucleate uniformly alongside Na2S in the resultant particle during the conversion reaction process.34,49,52 As a result, the conversion products only showed a slight increase in the diameters of the original particles, retaining the initial shape. Meanwhile, the neighboring pores in our porous foam-like structure can accommodate the associated volume expansion, maintaining the initial contact of particles. Hence, the interconnected foam-like structure can be preserved, even undergoing volume expansion after the discharge/charge process (Fig. S10, ESI†).
Conclusions
In summary, the facile and scalable synthesis of foam-like F-FeS2 with high purity is successfully realized through the combination of solution combustion synthesis and solid-state sulfurization. It is observed that the interconnected nanoparticles make up the skeleton of porous F-FeS2. For the sodium storage in F-FeS2, CV analysis and in situ XRD techniques were used to prove that the reversible reaction is based on the combination of insertion and conversion reaction mechanisms. Benefiting from the unique structure, the as-prepared F-FeS2 manifests impressive sodium storage properties with high specific capacity (823 mA h g−1 at 0.1 A g−1), good rate capability (581 mA h g−1 at 5 A g−1) and cyclability (754 mA h g−1 at 0.2 A g−1 with 97% retention after 80 cycles). Additionally, the resistance analysis explains its high capacity, and the proven pseudocapacitance contribution clarifies its good rate capability. This work holds great promise for commercializing high capacity FeS2-based SIBs, offering scientific insights into the rational design of structures to enhance their electrochemical performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This investigation was supported by the Natural Science Foundation of China (No. 51472122, 51772152), the Fundamental Research Funds for the Central Universities (No. 30915011201, NJ20160030), the “333 project” of Jiangsu and the PAPD of Jiangsu.References
- Y. Fang, Y. Lv, F. Gong, A. A. Elzatahry, G. Zheng and D. Zhao, Adv. Mater., 2016, 28, 9385–9390 CrossRef CAS PubMed.
- S. Xia, J. Ni, S. V. Savilov and L. Li, Nano Energy, 2018, 45, 407–412 CrossRef CAS.
- Y. Zhang, Y. Wang, L. Wang, C.-M. Lo, Y. Zhao, Y. Jiao, G. Zheng and H. Peng, J. Mater. Chem. A, 2016, 4, 9002–9008 RSC.
- X. Li, Y. Ma, G. Cao and Y. Qu, J. Mater. Chem. A, 2016, 4, 12487–12496 RSC.
- R. Dangol, Z. Dai, A. Chaturvedi, Y. Zheng, Y. Zhang, K. N. Dinh, B. Li, Y. Zong and Q. Yan, Nanoscale, 2018, 10, 4890–4896 RSC.
- W. Yao, J. Chen, L. Zhan, Y. Wang and S. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 39371–39379 CrossRef CAS PubMed.
- C. Wang, S. Wang, Y.-B. He, L. Tang, C. Han, C. Yang, M. Wagemaker, B. Li, Q.-H. Yang, J.-K. Kim and F. Kang, Chem. Mater., 2015, 27, 5647–5656 CrossRef CAS.
- J. Ni and L. Li, Adv. Funct. Mater., 2018, 28, 1704880 CrossRef.
- T. Wu, C. Zhang, H. Hou, P. Ge, G. Zou, W. Xu, S. Li, Z. Huang, T. Guo, M. Jing and X. Ji, Adv. Funct. Mater., 2018, 28, 1705744 CrossRef.
- D. Wu, G. Zhang, D. Lu, L. Ma, Z. Xu, X. Xi, R. Liu, P. Liu and Y. Su, J. Mater. Chem. A, 2018, 6, 13613–13618 RSC.
- X. Zhu, D. Liu, D. Zheng, G. Wang, X. Huang, J. Harris, D. Qu and D. Qu, J. Mater. Chem. A, 2018, 6, 13294–13301 RSC.
- Z. Xu, K. Yao, Z. Li, L. Fu, H. Fu, J. Li, L. Cao and J. Huang, J. Mater. Chem. A, 2018, 6, 10535–10542 RSC.
- P. F. Wang, H. Xin, T. T. Zuo, Q. Li, X. Yang, Y. X. Yin, X. Gao, X. Yu and Y. G. Guo, Angew. Chem., Int. Ed., 2018, 57, 8178–8183 CrossRef CAS PubMed.
- W. Chen, S. Qi, L. Guan, C. Liu, S. Cui, C. Shen and L. Mi, J. Mater. Chem. A, 2017, 5, 5332–5341 RSC.
- B. H. Hou, Y. Y. Wang, J. Z. Guo, Q. L. Ning, X. T. Xi, W. L. Pang, A. M. Cao, X. Wang, J. P. Zhang and X. L. Wu, Nanoscale, 2018, 10, 9218–9225 RSC.
- D. Li, Y. Sun, S. Chen, J. Yao, Y. Zhang, Y. Xia and D. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 17175–17182 CrossRef CAS PubMed.
- M. Walter, T. Zund and M. V. Kovalenko, Nanoscale, 2015, 7, 9158–9163 RSC.
- Z. Hu, Z. Zhu, F. Cheng, K. Zhang, J. Wang, C. Chen and J. Chen, Energy Environ. Sci., 2015, 8, 1309–1316 RSC.
- W. Zhao, C. Guo and C. M. Li, J. Mater. Chem. A, 2017, 5, 19195–19202 RSC.
- Y. Chen, X. Hu, B. Evanko, X. Sun, X. Li, T. Hou, S. Cai, C. Zheng, W. Hu and G. D. Stucky, Nano Energy, 2018, 46, 117–127 CrossRef CAS.
- Z. Liu, T. Lu, T. Song, X.-Y. Yu, X. W. Lou and U. Paik, Energy Environ. Sci., 2017, 10, 1576–1580 RSC.
- M. J. Choi, J. Kim, J. K. Yoo, S. Yim, J. Jeon and Y. S. Jung, Small, 2018, 14, 1702816 CrossRef PubMed.
- F. Bu, P. Xiao, J. Chen, M. F. Aly Aboud, I. Shakir and Y. Xu, J. Mater. Chem. A, 2018, 6, 6414–6421 RSC.
- L. Li, S. Peng, N. Bucher, H.-Y. Chen, N. Shen, A. Nagasubramanian, E. Eldho, S. Hartung, S. Ramakrishna and M. Srinivasan, Nano Energy, 2017, 37, 81–89 CrossRef CAS.
- K. Zhang, M. Park, L. Zhou, G. H. Lee, J. Shin, Z. Hu, S. L. Chou, J. Chen and Y. M. Kang, Angew. Chem., Int. Ed., 2016, 55, 12822–12826 CrossRef CAS PubMed.
- K. Deshpande, A. Mukasyan and A. Varma, Chem. Mater., 2004, 16, 4896–4904 CrossRef CAS.
- J. Zhang, C. Du, Z. Dai, W. Chen, Y. Zheng, B. Li, Y. Zong, X. Wang, J. Zhu and Q. Yan, ACS Nano, 2017, 11, 10599–10607 CrossRef CAS PubMed.
- X. Wen, X. Wei, L. Yang and P. K. Shen, J. Mater. Chem. A, 2015, 3, 2090–2096 RSC.
- Y. Liang, P. Bai, J. Zhou, T. Wang, B. Luo and S. Zheng, CrystEngComm, 2016, 18, 6262–6271 RSC.
- W. Ma, X. Liu, X. Lei, Z. Yuan and Y. Ding, Chem. Eng. J., 2018, 334, 725–731 CrossRef CAS.
- W. Wen and J.-M. Wu, RSC Adv., 2014, 4, 58090–58100 RSC.
- F. T. Li, Q. Wang, J. Ran, Y. J. Hao, X. J. Wang, D. Zhao and S. Z. Qiao, Nanoscale, 2015, 7, 1116–1126 RSC.
- Z. Shadike, Y.-N. Zhou, F. Ding, L. Sang, K.-W. Nam, X.-Q. Yang and Z.-W. Fu, J. Power Sources, 2014, 260, 72–76 CrossRef CAS.
- A. Douglas, R. Carter, L. Oakes, K. Share, A. P. Cohn and C. L. Pint, ACS Nano, 2015, 9, 11156–11165 CrossRef CAS PubMed.
- Y. Zhou, J. Tian, H. Xu, J. Yang and Y. Qian, Energy Storage Mater., 2017, 6, 149–156 CrossRef.
- B. H. Hou, Y. Y. Wang, J. Z. Guo, Y. Zhang, Q. L. Ning, Y. Yang, W. H. Li, J. P. Zhang, X. L. Wang and X. L. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 3581–3589 CrossRef CAS PubMed.
- D. Li, D. Yang, X. Yang, Y. Wang, Z. Guo, Y. Xia, S. Sun and S. Guo, Angew. Chem., Int. Ed., 2016, 55, 15925–15928 CrossRef CAS PubMed.
- J. Li, D. Yan, X. Zhang, S. Hou, T. Lu, Y. Yao and L. Pan, J. Mater. Chem. A, 2017, 5, 20428–20438 RSC.
- Z. Lu, N. Wang, Y. Zhang, P. Xue, M. Guo, B. Tang, Z. Bai and S. Dou, Electrochim. Acta, 2018, 260, 755–761 CrossRef CAS.
- W. Ren, H. Zhang, C. Guan and C. Cheng, Adv. Funct. Mater., 2017, 27, 1702116 CrossRef.
- R. Sun, S. Liu, Q. Wei, J. Sheng, S. Zhu, Q. An and L. Mai, Small, 2017, 13, 1701744 CrossRef PubMed.
- P. Ge, C. Zhang, H. Hou, B. Wu, L. Zhou, S. Li, T. Wu, J. Hu, L. Mai and X. Ji, Nano Energy, 2018, 48, 617–629 CrossRef CAS.
- G. Zou, H. Hou, C. W. Foster, C. E. Banks, T. Guo, Y. Jiang, Y. Zhang and X. Ji, Adv. Sci., 2018, 5, 1800241 CrossRef PubMed.
- Y. Long, J. Yang, X. Gao, X. Xu, W. Fan, J. Yang, S. Hou and Y. Qian, ACS Appl. Mater. Interfaces, 2018, 10, 10945–10954 CrossRef CAS PubMed.
- F. Niu, J. Yang, N. Wang, D. Zhang, W. Fan, J. Yang and Y. Qian, Adv. Funct. Mater., 2017, 27, 1700522 CrossRef.
- C. Zhang, S.-H. Park, S. E. O'Brien, A. Seral-Ascaso, M. Liang, D. Hanlon, D. Krishnan, A. Crossley, N. McEvoy, J. N. Coleman and V. Nicolosi, Nano Energy, 2017, 39, 151–161 CrossRef CAS.
- D. Yu, Q. Pang, Y. Gao, Y. Wei, C. Wang, G. Chen and F. Du, Energy Storage Mater., 2018, 11, 1–7 CrossRef.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- M. T. McDowell, Z. Lu, K. J. Koski, J. H. Yu, G. Zheng and Y. Cui, Nano Lett., 2015, 15, 1264–1271 CrossRef CAS PubMed.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- J. H. Chen and W. W. Harvey, Metall. Trans. B, 1975, 6, 331 CrossRef.
- F. Wang, H. C. Yu, M. H. Chen, L. Wu, N. Pereira, K. Thornton, A. Van der Ven, Y. Zhu, G. G. Amatucci and J. Graetz, Nat. Commun., 2012, 3, 1201 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr06675b |
| This journal is © The Royal Society of Chemistry 2019 |