
Platinum-catalyzed selective N-allylation of 2,3-disubstituted indoles with allylic acetates in water†
Bai-Jing
Peng
 ab,
Wen-Ting
Hsueh
a,
Ferenc
Fülöp
b and
Shyh-Chyun
Yang
*acd
ab,
Wen-Ting
Hsueh
a,
Ferenc
Fülöp
b and
Shyh-Chyun
Yang
*acd
aSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University, Kaohsiung 807, Taiwan
bInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary
cDepartment of Fragrance and Cosmetic Science, College of Pharmacy, Kaohsiung Medical University, Kaohsiung 807, Taiwan
dDepartment of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung 807, Taiwan. E-mail: scyang@kmu.edu.tw; Tel: +886-918-665-770
First published on 9th November 2018
Abstract
Due to their biological activity, indoles and substituted indoles have attracted considerable attention from both synthetic and medicinal scientists. Much effort has been directed toward the development of methods for the functionalization of the indole nucleus. The protocol uses a catalytic amount of catalyzed platinum as a promoting agent, producing N-allylated indoles in considerable yields. Moreover, water, with its large heat capacity, is one of the most abundant molecules on earth. The use of water as a solvent may bring about many environmental benefits. Herein, we have demonstrated that the platinum-catalyzed selective N-allylation of 2,3-disubstituted indoles proceeds in water. This method provides a simple, convenient, and efficient way to afford a high yield of N-allylated indoles.
Introduction
The asymmetric structure synthesis of N-allylic indoles is crucial for natural alkaloid synthesis and organic materials research.1 Particularly, indoles and indole-derived heterocycles consist of a 2,3-disubstituted construction in medicinal drugs.2 As a result, extensive functionalization efforts have been undertaken to develop catalytic asymmetric allylation of indole nuclei at the N1 or C3 sites.3 Nevertheless, the allylation attack at the N1 site on 2,3-disubstituted indoles to produce N-allylated indoles containing a new carbon–nitrogen bond is a difficult transformation because of the high nucleophilicity of the C3 site of the indole nucleus and the weak acidity of the N–H bond.4 For these reasons, efficient strategies for the synthesis of N-allylic 2,3-disubstituted indoles are still rare.4a Given the importance of N-allylation of 2,3-disubstituted indoles, we have been attracted toward this aspect of research.Transition metal catalysis has been and continues to be a predominant tool to form carbon–carbon, carbon–nitrogen, carbon–oxygen, and carbon–heteroatom bonds in organic synthesis.5 In particular, palladium-catalyzed Suzuki–Miyaura, Heck, and Negishi couplings are essential, as recorded by the 2010 Nobel Prize.6 The palladium-catalyzed allylation of nucleophiles has been proven to be an efficient, established, and highly chemo- and stereo-selective method.7 The catalytic cycles have been shown to proceed via the attack of nucleophiles on cationic η3-allylpalladium(II) complexes, an intermediate generated by the oxidative addition of allylic compounds, including carbamates, carbonates, esters, halides, phosphates, and related derivatives, to a Pd(0) complex.7c,8 Ruthenium, as well as palladium, have been used in the allylation reaction of 2,3-disubstituted indoles, but according to our knowledge, the use of platinum is rare.9 Platinum is a good transition metal, which is not often discussed.10 During our previous research on allylation with platinum as a catalyst agent, we established the application of processed platinum catalysis with desirable data.11
In recent years, our team has reported the Pt(II)-catalyzed allylation of 2,3-disubstituted indoles in benzene.12 However, benzene is a highly flammable and toxic liquid with a sweet smell, and increases the risk of cancer and other illnesses.13 In this scenario, considering both green chemistry and safety, it is crucial to find an alternative, environmentally-friendly solvent.14 Organic reactions in water have attracted much attention, because water is inexpensive, nonflammable, nontoxic, and has a large heat capacity.15 Water in its pure form is completely benign, so it would appear to be an attractive solvent.16 Therefore, the development of atom-economic reactions in water is one of the most important objectives of synthetic chemistry.11a,17 In this paper, we intend to disclose our study of the N-allylation of indoles with allylic acetates in the presence of a catalytic amount of platinum/phosphine complexes. The protocol allowed the N-selectivity of the reaction in water to be effectively controlled. This reaction system created a simple, convenient, and efficient way to afford a high yield of N-allylated indoles in water.
Results and discussion
The platinum-catalyzed allylation of 2,3-disubstituted indoles, such as 1,2,3,4-tetrahydrocarbazole, with allyl acetate was investigated under different scenarios (Scheme 1). When a mixture of 1,2,3,4-tetrahydrocarbazole (1a, 1 mmol) and allyl acetate (2a, 2 mmol) was refluxed in the presence of catalytic amounts of Pt(acac)2 (2.5 mmol%) and PPh3 (10 mmol%) in water for 2 h, N-allyl-1,2,3,4-tetrahydrocarbazole (3a) was formed in only 28% yield (entry 1 in Table 1). In initial studies, we examined the effectiveness of commercial sources for the ligands. Among the monodentate ligands inclusive of PPh3 (entry 1), (2-furyl)3P (entry 2), (2-CH3C6H4)3P (entry 3), (3-CH3C6H4)3P (entry 4), (4-CH3C6H4)3P (entry 5), (4-FC6H4)3P (entry 6), (4-ClC6H4)3P (entry 7), (4-CH3OC6H4)3P (entry 8), (n-butyl)3P (entry 9), [2,4,6-(CH3O)3C6H2]3P (entry 10), (2-pyridyl)Ph2P (entry 11), and [2,6-(CH3O)2C6H3]3P (entry 12) were used. In addition, bidentate ligands including BINAP (entry 13), dppf (entry 14), dppb (entry 15), dppm (entry 16), and dppe (entry 17) were evaluated in the reaction. The catalytic reactivity of the ligand (4-ClC6H4)3P was likely due to improved catalyst stability and produced N-allylation product 3a in a 95% yield (entry 7). It was obvious that the predominant product was N-allylated 1,2,3,4-tetrahydrocarbazole. In the absence of a ligand, the reaction did not occur (entry 18). The surrounding reaction conditions were also investigated. It was confirmed that when the temperature of the reaction was decreased, the yield also decreased (entry 19). Not only the temperature, but also reaction time affected the yield. As expected, as the reaction time became shorter, the yield of the predominate N-allylation derivative decreased too (entry 20). In the presence of various platinum catalysts, including Pt(acac)2 (entry 7), cis-PtCl2(PhCN)2 (entry 22), cis-PtCl2(PPh3)2 (entry 23), Pt(COD)Cl2 (entry 24), O[Si(CH3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2]2Pt (entry 25), PtCl2 (entry 26), PtI2 (entry 27), Pt(CN)2 (entry 28), Pt(CH2CH2)(PPh3)2 (entries 29 and 30), and Pt(PPh3)4 (entries 31 and 32), it was shown that the most effective platinum catalyst and the most regioselective for N-allylation is Pt(acac)2 (entry 7). However, using Pt(CH2CH2)(PPh3)2 or Pt(PPh3)4 with extra (4-ClC6H4)3P as a catalyst increased the yields of the products (entries 30 and 32). The reaction would not occur without any platinum species as a catalyst (entry 21). During the reaction, adding phosphine ligands could increase the activity of the platinum catalyst. A reduction in the ratio of Pt(acac)2 to (4-ClC6H4)3P, to a ratio of 1
CH2]2Pt (entry 25), PtCl2 (entry 26), PtI2 (entry 27), Pt(CN)2 (entry 28), Pt(CH2CH2)(PPh3)2 (entries 29 and 30), and Pt(PPh3)4 (entries 31 and 32), it was shown that the most effective platinum catalyst and the most regioselective for N-allylation is Pt(acac)2 (entry 7). However, using Pt(CH2CH2)(PPh3)2 or Pt(PPh3)4 with extra (4-ClC6H4)3P as a catalyst increased the yields of the products (entries 30 and 32). The reaction would not occur without any platinum species as a catalyst (entry 21). During the reaction, adding phosphine ligands could increase the activity of the platinum catalyst. A reduction in the ratio of Pt(acac)2 to (4-ClC6H4)3P, to a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (entry 33), decreased the yield of the reaction. Decreasing the relative amounts of allyl acetate disfavored the formation of product 3a (entry 34). It was known that several factors, such as the solvent and the nature of the nucleophile, could alter the product pattern in metal-catalyzed allylation. Six solvents were investigated (entries 7 and 35–39). To our surprise, when the reaction used an organic solvent like benzene, toluene, or dichloromethane, the C-allylation product yield was more superior to that of the N-allylation products (entries 35–37). In particular, the C-allylation product was the prominent product for reactions with dichloromethane as the solvent. Apart from this, the appropriate solvent for the reaction is water. Remarkably, no C-allylation products were detected in water.
:2 (entry 33), decreased the yield of the reaction. Decreasing the relative amounts of allyl acetate disfavored the formation of product 3a (entry 34). It was known that several factors, such as the solvent and the nature of the nucleophile, could alter the product pattern in metal-catalyzed allylation. Six solvents were investigated (entries 7 and 35–39). To our surprise, when the reaction used an organic solvent like benzene, toluene, or dichloromethane, the C-allylation product yield was more superior to that of the N-allylation products (entries 35–37). In particular, the C-allylation product was the prominent product for reactions with dichloromethane as the solvent. Apart from this, the appropriate solvent for the reaction is water. Remarkably, no C-allylation products were detected in water.
 | ||
| Scheme 1 Allylation of 1,2,3,4-tetrahydrocarbazole (1a) with allyl acetate (2a). | ||
| Entry | Ligand | Platinum catalyst | Solvent | Yield (%) (3a + 4a) | Yieldb (%) of 3a | Yieldb (%) of 4a |
|---|---|---|---|---|---|---|
| a Reaction conditions: 1a (1 mmol), 2a (2 mmol), Pt catalyst (0.025 mmol), and ligand (0.1 mmol) in solvent (5 mL) were refluxed for 2 h. b Isolated yield. c (±)-2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene. d 1,1-Bis(diphenylphosphino)ferrocene. e 1,4-Bis(diphenylphosphino)butane. f 1,1-Bis(diphenylphosphino)methane. g 1,2-Bis(diphenylphosphino)ethane. h Stirred at 80 °C. i Refluxed for 1 h. j (4-ClC6H4)3P (0.05 mmol). k 2a (1.2 mmol). | ||||||
| 1 | PPh3 | Pt(acac)2 | H2O | 28 | 28 | 0 |
| 2 | (2-Furyl)3P | Pt(acac)2 | H2O | 2 | 2 | 0 |
| 3 | (2-CH3C6H4)3P | Pt(acac)2 | H2O | 3 | 3 | 0 |
| 4 | (3-CH3C6H4)3P | Pt(acac)2 | H2O | 17 | 17 | 0 |
| 5 | (4-CH3C6H4)3P | Pt(acac)2 | H2O | 9 | 0 | 9 |
| 6 | (4-FC6H4)3P | Pt(acac)2 | H2O | 18 | 14 | 4 |
| 7 | (4-ClC6H4)3P | Pt(acac)2 | H2O | 95 | 95 | 0 |
| 8 | (4-CH3OC6H4)3P | Pt(acac)2 | H2O | 8 | 0 | 8 |
| 9 | (n-Butyl)3P | Pt(acac)2 | H2O | 12 | 12 | 0 |
| 10 | [2,4,6-(CH3O)3C6H2]3P | Pt(acac)2 | H2O | 8 | 0 | 8 |
| 11 | (2-Pyridyl)Ph2P | Pt(acac)2 | H2O | 4 | 4 | 0 |
| 12 | [2,6-(CH3O)2C6H3]3P | Pt(acac)2 | H2O | 5 | 5 | 0 |
| 13c | BINAP | Pt(acac)2 | H2O | 19 | 19 | 0 |
| 14d | dppf | Pt(acac)2 | H2O | 7 | 7 | 0 |
| 15e | dppb | Pt(acac)2 | H2O | 5 | 5 | 0 |
| 16f | dppm | Pt(acac)2 | H2O | 12 | 6 | 7 |
| 17g | dppe | Pt(acac)2 | H2O | 29 | 29 | 0 |
| 18 | — | Pt(acac)2 | H2O | 0 | 0 | 0 |
| 19h | (4-ClC6H4)3P | Pt(acac)2 | H2O | 67 | 67 | 0 |
| 20i | (4-ClC6H4)3P | Pt(acac)2 | H2O | 70 | 70 | 0 |
| 21 | (4-ClC6H4)3P | — | H2O | 0 | 0 | 0 |
| 22 | (4-ClC6H4)3P | cis-PtCl2(PhCN)2 | H2O | 93 | 86 | 7 |
| 23 | (4-ClC6H4)3P | cis-PtCl2(PPh3)2 | H2O | 82 | 72 | 10 |
| 24 | (4-ClC6H4)3P | Pt(COD)Cl2 | H2O | 12 | 12 | 0 |
| 25 | (4-ClC6H4)3P | O[Si(CH3)2CCH2]2Pt |
H2O | 89 | 68 | 21 |
| 26 | (4-ClC6H4)3P | PtCl2 | H2O | 13 | 13 | 0 |
| 27 | (4-ClC6H4)3P | PtI2 | H2O | 12 | 9 | 3 |
| 28 | (4-ClC6H4)3P | Pt(CN)2 | H2O | 9 | 0 | 9 |
| 29 | — | Pt(CH2CH2)(PPh3)2 |
H2O | 11 | 8 | 3 |
| 30 | (4-ClC6H4)3P | Pt(CH2CH2)(PPh3)2 |
H2O | 34 | 23 | 11 |
| 31 | — | Pt(PPh3)4 | H2O | 3 | 3 | 0 |
| 32 | (4-ClC6H4)3P | Pt(PPh3)4 | H2O | 11 | 11 | 0 |
| 33j | (4-ClC6H4)3P | Pt(acac)2 | H2O | 50 | 50 | 0 |
| 34k | (4-ClC6H4)3P | Pt(acac)2 | H2O | 60 | 60 | 0 |
| 35 | (4-ClC6H4)3P | Pt(acac)2 | Benzene | 34 | 10 | 24 |
| 36 | (4-ClC6H4)3P | Pt(acac)2 | Toluene | 28 | 9 | 19 |
| 37 | (4-ClC6H4)3P | Pt(acac)2 | CH2Cl2 | 34 | 2 | 31 |
| 38 | (4-ClC6H4)3P | Pt(acac)2 | CH3OH | 17 | 17 | 0 |
| 39 | (4-ClC6H4)3P | Pt(acac)2 | C2H5OH | 26 | 26 | 0 |
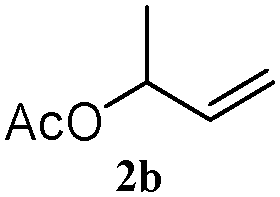
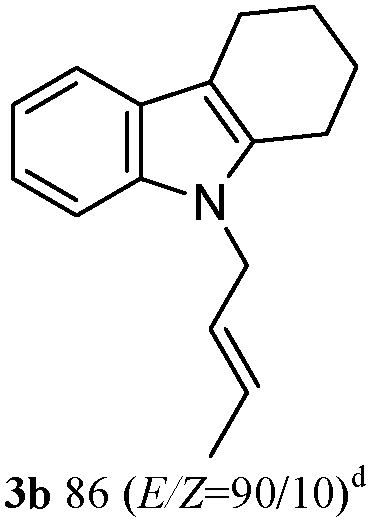
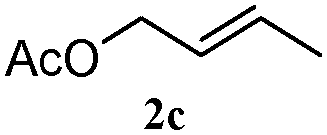
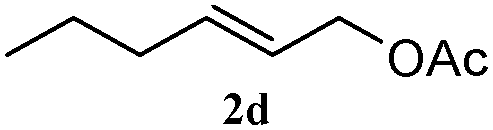
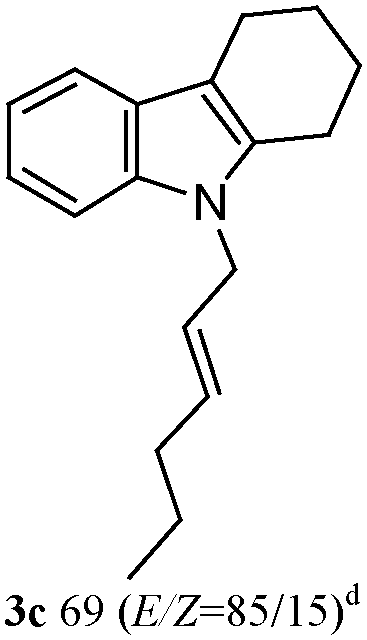
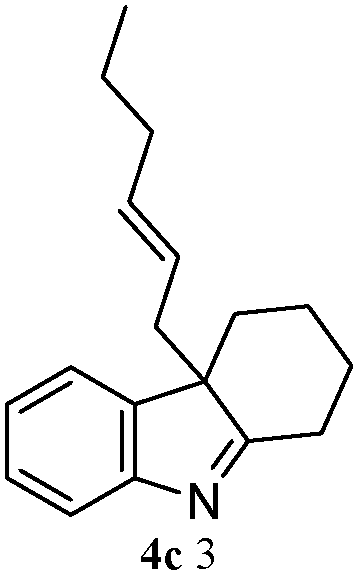
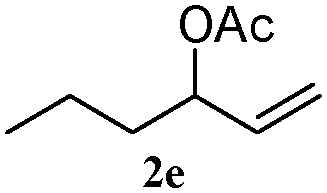
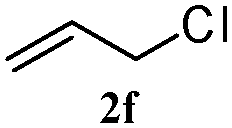
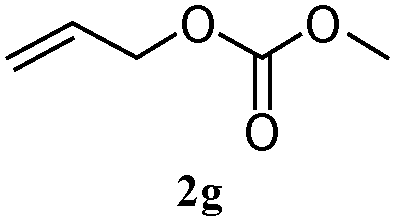
The outcomes of the reaction conditions examined above were found to be applicable to a wide variety of allylic compounds. The results for the allylation of a number of allylic compounds (2b–g) with 1,2,3,4-tetrahydrocarbazole (1a) using Pt(acac)2 and (4-ClC6H4)3P are compiled in Table 2. The allylation of 3-buten-2-yl acetate (2b) gave N-allylated tetrahydrocarbazole 3b in a yield of 86% (entry 1). The N-allylated tetrahydrocarbazole E/Z ratio of 3b was determined by GC. Obviously, the E alkene product was generated from the more thermodynamically-stable syn complex. It was in a 73% overall yield that the corresponding reaction with crotyl acetate (2c) afforded N-allylated and C-allylated tetrahydrocarbazole (entry 2). These N-allylated products might all originate from the same π-allylic intermediate, which could be attacked at the C-1 position. The formation of the regioisomeric product was not observed. It is probable that the C-3 position products, which were involved in the internal N-allylation, were not generated in this scenario. Moreover, the reaction was considered to proceed via π-allylplatinum intermediates. The loss of stereochemistry in the starting acetate 2b was due to the more rapid σ ↔ η3 ↔ σ interconversion of the intermediates compared to the rate of allylation. trans-2-Hexen-1-yl acetate (2d), which reacted with 1a, gave 3c and 4c in 69% and 3% yields, respectively (entry 3). In the reaction of hex-1-en-3-yl acetate (2e), the corresponding N-allylated products were formed in 81% overall yields (entry 4). Unfortunately, the reaction of allyl chloride (2e), which is not an appropriate reagent for allylation, only produced 3a in a yield of 12% (entry 5). Increasing the amount of the reagents Pt(acac)2 and (4-ClC6H4)3P to twice the original dose could improve yields of 3a up to 57% (entry 6). The catalyst could not affect the yield, but this concept was built on the use of a sufficient amount of catalyst. In our catalyst system, we established N-allylation with trace amounts of platinum. Last but not least, with allyl carbonate (2g), the reaction afforded 3a and 4a in a 97% overall yield (entry 7). 3a was still the dominant product and was obtained in a yield of 92% in the allylation of 2g.
| Entry | 2 | Yieldb (%) | |
|---|---|---|---|
| a Reaction conditions: 1a (1 mmol), 2a (2 mmol), Pt(acac)2 (0.025 mmol), and (4-ClC6H4)3P (0.1 mmol) in solvent (5 mL) were refluxed for 2 h. b Isolated yield. c Pt(acac)2 (0.05 mmol) and (4-ClC6H4)3P (0.2 mmol). d Determined by GC. | |||
| 1 |
|
|

|
| 2 |
|
3b 71 (E/Z = 89/11)d | 4b 2 |
| 3 |
|
|
|
| 4 |
|
3c 81 (E/Z = 85/15)d | 4c 0 |
| 5 |
|
3a 12 | 4a 0 |
| 6c | 2f | 3a 57 | 4a 0 |
| 7 |
|
3a 92 | 4a 5 |
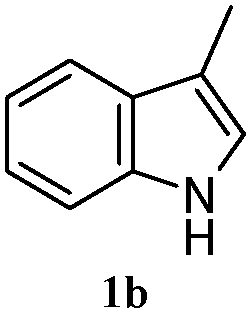
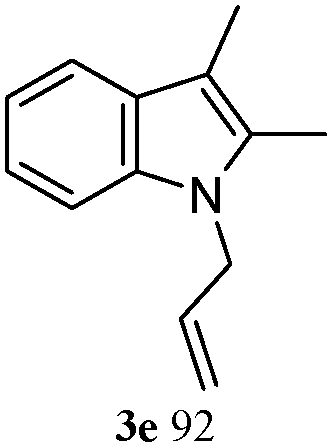
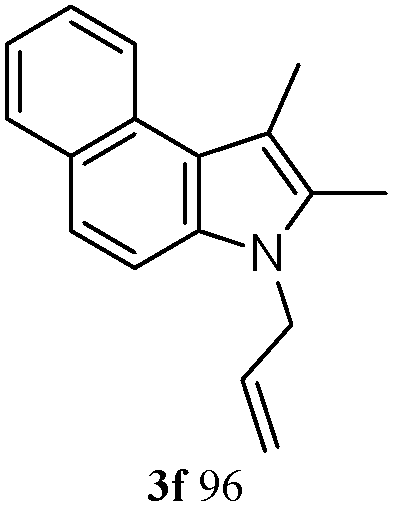
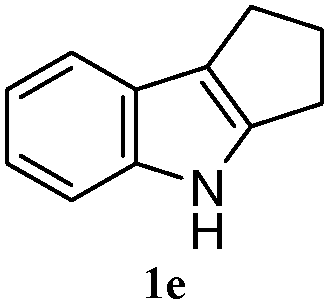
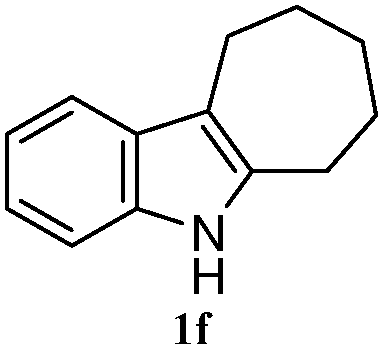
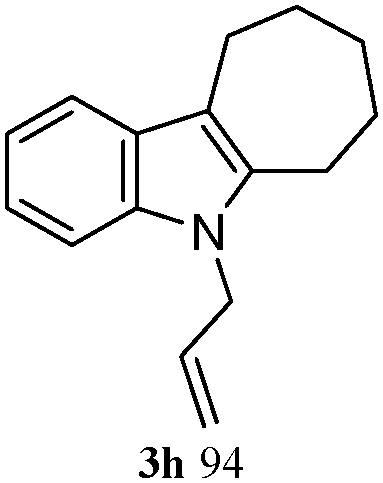
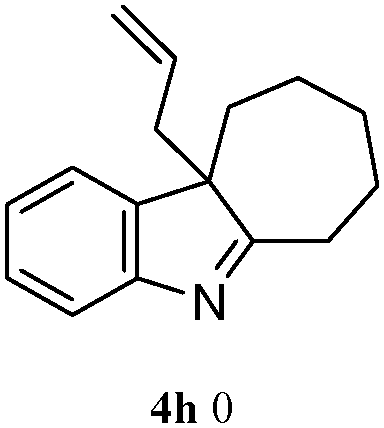
The good efficiency of the allylation reactions described above prompted us to extend the reaction to corresponding indole derivatives (Table 3). The results summarized in Table 3 showed that the allylation of allyl acetaete (2a) with indoles, using Pt(acac)2 and (4-ClC6H4)3P, gave generally good yields of a variety of allylic indoles (entries 1–5). First, the allylation of the simpler 3-methylindole 1b was investigated. The overall yield was 73% (entry 1). 2,3-Dimethyl indole (1c) was a desirable substrate and generated N-allylated tetrahydrocarbazole in a 92% yield (entry 2). 1,2-Dimethyl-3H-benzoindole (1d), which consisted of the substituted benzene ring of tetrahydrocarbazole, was well tolerated and gave 96% yields of the corresponding N-allylated products (entry 3). The cyclopentane- and cycloheptane-fused indoles 1e and 1f participated in the reaction and gave high yields of the corresponding N-allylated compounds (entries 4 and 5).
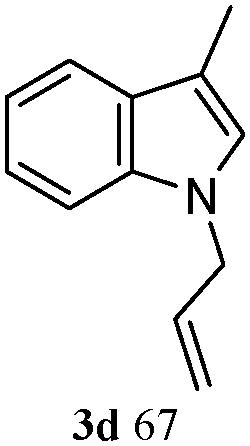
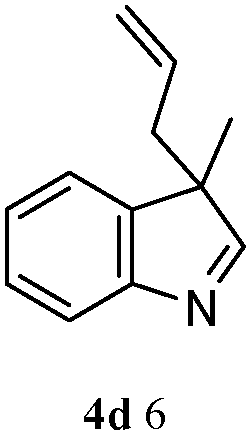


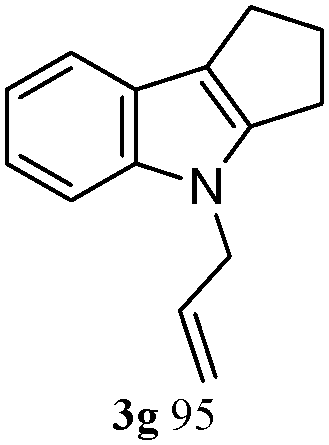
A possible mechanism for the formation of N-allyl-2,3-disubstituted indoles from 1 and 2 is illustrated in Scheme 2, in which the substituent on allylic acetate is omitted. The circulation indicates that a Pt(II)-assisted mechanism is the feasible mechanism for C–N formation and activation. The entire pathway consists of three steps: allyl acetate 2 with Pt(0)Ln, allylation of the 2,3-disubstituted indole, and elimination from the π-allylplatinum intermediate. As detailed, 2 reacts with Pt(0) and the phosphine ligand species, which generate in situ, to produce π-allylplatinum intermediate 5. Subsequently, the reaction of 5 with 2,3-disubstituted indole 1 gives the π-allylplatinum intermediate with indole 6. Finally, the C–N bond formation is followed by the elimination of the π-allylplatinum intermediate with indole 6. Then, the whole system gives N-allyl-2,3-disubstituted indole 3.
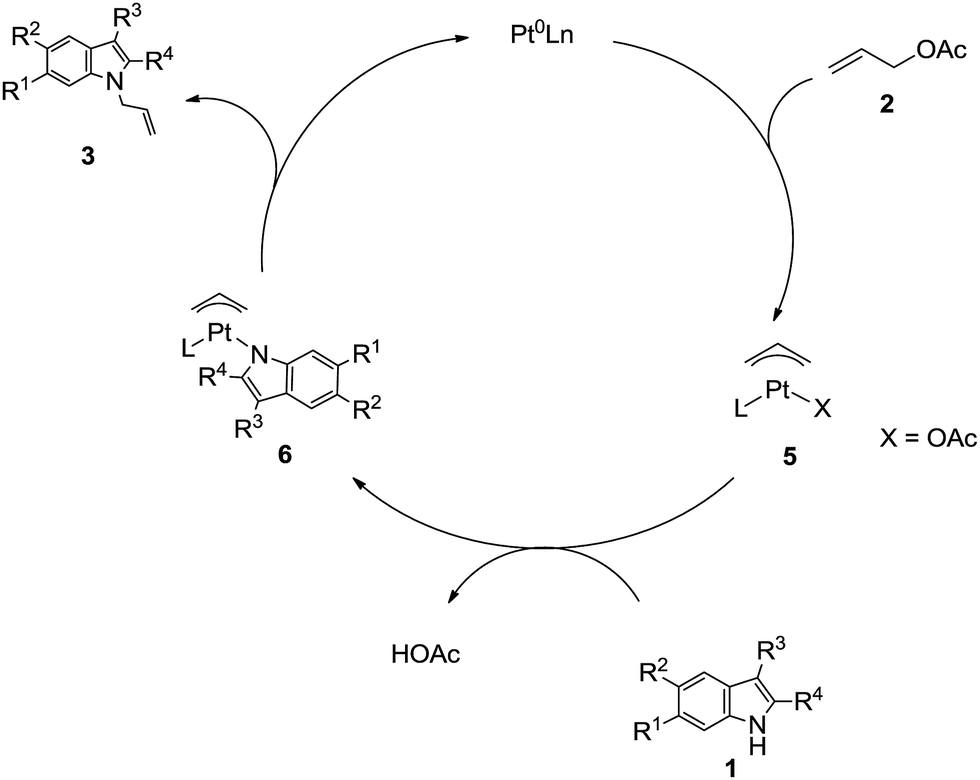 | ||
| Scheme 2 Proposed mechanism. | ||
Conclusions
In conclusion, we have shown that the platinum(0)-catalyzed allylation of allylic acetates using heterocycle fused indoles is a simple, convenient, and efficient way to achieve C–N bond formation. The reaction did not occur in the absence of a platinum catalyst and phosphine ligands. It is obvious that the best solvent is water in the reaction. The allylation of allylic acetates worked well with carbazoles, giving generally good yields of the corresponding allylic carbazoles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the National Science Council of the Republic of China (NSC101-2113-M-037-008-MY3).Notes and references
- (a) H. Yan, H. Wang, X. Li, X. Xin, C. Wang and B. Wan, Angew. Chem., Int. Ed., 2015, 54, 10613–10617 CrossRef CAS; (b) M. C. Tong, X. Chen, J. Li, R. Huang, H. Tao and C. J. Wang, Angew. Chem., Int. Ed., 2014, 53, 4680–4684 CrossRef CAS; (c) W. Gul and M. T. Hamann, Life Sci., 2005, 78, 442–453 CrossRef CAS.
- (a) N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159–184 CrossRef CAS; (b) E. V. Villemson, E. M. Budynina, O. A. Ivanova, D. A. Skvortsov, I. V. Trushkov and M. Y. Melnikov, RSC Adv., 2016, 6, 62014–62018 RSC; (c) G. W. Gribble, Indole ring synthesis: From natural products to drug discovery, John Wiley & Sons, 2016 Search PubMed.
- (a) J. Cheng, J. Sun, J. Yan, S. Yang, P. Zheng, Z. Jin and Y. R. Chi, J. Org. Chem., 2017, 82, 13342–13347 CrossRef CAS; (b) M. J. James, R. E. Clubley, K. Y. Palate, T. J. Procter, A. C. Wyton, P. O’Brien, R. J. Taylor and W. P. Unsworth, Org. Lett., 2015, 17, 4372–4375 CrossRef CAS; (c) L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 134, 14563–14572 CrossRef CAS; (d) M. Kimura, M. Futamata, R. Mukai and Y. Tamaru, J. Am. Chem. Soc., 2005, 127, 4592–4593 CrossRef CAS.
- (a) K. Xu, T. Gilles and B. Breit, Nat. Commun., 2015, 6, 7616 CrossRef; (b) N. Otero, M. Mandado and R. A. Mosquera, J. Phys. Chem. A, 2007, 111, 5557–5562 CrossRef CAS; (c) S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088–9095 CrossRef CAS PubMed; (d) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS; (e) S. Nunomoto, Y. Kawakami, Y. Yamashita, H. Takeuchi and S. Eguchi, J. Chem. Soc., Perkin Trans. 1, 1990, 111–114 RSC.
- (a) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS; (b) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS; (c) S. H. Leenders, R. Gramage-Doria, B. De Bruin and J. N. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC; (d) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS; (e) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS; (f) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef; (g) J. Tsuji, Transition metal reagents and catalysts: innovations in organic synthesis, John Wiley & Sons, 2002 CrossRef; (h) E. Negishi, A. O. King and N. Okukado, J. Org. Chem., 1977, 42, 1821–1823 CrossRef CAS; (i) K. B. Sharpless and R. Michaelson, J. Am. Chem. Soc., 1973, 95, 6136–6137 CrossRef CAS.
- (a) H. Li, C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS; (b) C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS.
- (a) S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278–15284 CrossRef CAS; (b) W. Wang, F. Huan, Y. Sun, J. Fang, X.-Y. Liu, Q.-Y. Chen and Y. Guo, J. Fluorine Chem., 2015, 171, 46–55 CrossRef CAS; (c) K. Ohmatsu, M. Ito and T. Ooi, Chem. Commun., 2014, 50, 4554–4557 RSC; (d) E. W. Abel, F. G. A. Stone and G. Wilkinson, Comprehensive Organometallic Chemistry II: Heteronuclear metal–metal bonds, Elsevier, 1995 RSC; (e) K. Sonogashira, Comprehensive organic synthesis, Pergamon Press, New York, 1991, vol. 3, p. 521 Search PubMed.
- (a) J. Tsuji, Palladium reagents and catalysts, Wiley & sons, 1995 Search PubMed; (b) J. Tsuji, Synthesis, 1990, 739–749 CrossRef CAS; (c) B. M. Trost, Angew. Chem., Int. Ed., 1989, 28, 1173–1192 CrossRef; (d) W. Oppolzer, Angew. Chem., Int. Ed., 1989, 28, 38–52 CrossRef; (e) J. D. Senra, A. C. Silva, R. V. Santos, L. F. B. Malta and A. B. Simas, J. Chem., 2017, 2017 Search PubMed; (f) A. Kondoh, Y. Kamata and M. Terada, Org. Lett., 2017, 19, 1682–1685 CrossRef CAS PubMed; (g) J. M. Bauer, W. Frey and R. Peters, Chem. – Eur. J., 2016, 22, 5767–5777 CrossRef CAS; (h) P. Yin, M. Y. Wong, J. Tham and T.-P. Loh, Org. Chem. Front., 2014, 1, 1266–1269 RSC; (i) M. Kawatsura, S. Terasaki, M. Minakawa, T. Hirakawa, K. Ikeda and T. Itoh, Org. Lett., 2014, 16, 2442–2445 CrossRef CAS; (j) S. Rajesh, B. Banerji and J. Iqbal, J. Org. Chem., 2002, 67, 7852–7857 CrossRef CAS PubMed.
- (a) K. Takamoto, S. Ohno, N. Hyogo, H. Fujioka and M. Arisawa, J. Org. Chem., 2017, 82, 8733–8742 CrossRef CAS; (b) S. Ohno, K. Takamoto, H. Fujioka and M. Arisawa, Org. Lett., 2017, 19, 2422–2425 CrossRef CAS; (c) Y. Masuya, M. Tobisu and N. Chatani, Org. Lett., 2016, 18, 4312–4315 CrossRef CAS; (d) R. Larock, E. Yum and M. Refvik, J. Org. Chem., 1998, 63, 7652–7662 CrossRef CAS.
- (a) K. Q. Huynh, C. A. Seizert, T. J. Ozumerzifon, P. A. Allegretti and E. M. Ferreira, Org. Lett., 2017, 19, 294–297 CrossRef CAS PubMed; (b) Y. Tsuji, R. Takeuchi, H. Ogawa and Y. Watanabe, Chem. Lett., 1986, 293–294 CrossRef CAS.
- (a) K.-H. Gan, C.-J. Jhong, Y.-J. Shue and S.-C. Yang, Tetrahedron, 2008, 64, 9625–9629 CrossRef CAS; (b) S.-C. Yang, W.-H. Feng and K.-H. Gan, Tetrahedron, 2006, 62, 3752–3760 CrossRef CAS; (c) S.-C. Yang, Y.-C. Tsai and Y.-J. Shue, Organometallics, 2001, 20, 5326–5330 CrossRef CAS.
- B.-J. Peng, W.-T. Wu and S.-C. Yang, Molecules, 2017, 22, 2097 CrossRef PubMed.
- (a) J. E. French, D. M. Gatti, D. L. Morgan, G. E. Kissling, K. R. Shockley, G. A. Knudsen, K. G. Shepard, H. C. Price, D. King and K. L. Witt, Environ. Health Perspect., 2015, 123, 237 CrossRef CAS PubMed; (b) Y.-M. Chang, J.-M. Tseng, C.-M. Shu and K.-H. Hu, Korean J. Chem. Eng., 2005, 22, 803–812 CrossRef CAS; (c) A. Yardley-Jones, D. Anderson and D. Parke, Occup. Environ. Med., 1991, 48, 437–444 CrossRef CAS; (d) B. D. Goldstein, J. Toxicol. Environ. Health. Suppl., 1977, 2, 69–105 CAS; (e) L. A. Cox and P. F. Ricci, Risk Anal., 1992, 12, 401–410 CrossRef.
- (a) F. Kerton and R. Marriott, Alternative solvents for green chemistry, 2013 Search PubMed; (b) J. H. Clark, Green Chem., 1999, 1, 1–8 RSC.
- (a) E. Levin, E. Ivry, C. E. Diesendruck and N. G. Lemcoff, Chem. Rev., 2015, 115, 4607–4692 CrossRef CAS; (b) U. M. Lindstrom, Organic reactions in water: principles, strategies and applications, John Wiley & Sons, 2008 Search PubMed.
- H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC.
- (a) A. Shaabani and S. E. Hooshmand, Ultrason. Sonochem., 2018, 40, 84–90 CrossRef CAS; (b) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy and D. A. Perry, Green Chem., 2008, 10, 31–36 RSC; (c) S.-C. Yang, Y.-C. Hsu and K.-H. Gan, Tetrahedron, 2006, 62, 3949–3958 CrossRef CAS; (d) B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj05051a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |