
Rapid microwave-assisted distillation–precipitation polymerization for the synthesis of magnetic molecular imprinted polymers coupled to HPTLC determination of perphenazine in human urine†
Mehdi
Safdarian
 and
Zahra
Ramezani
*
and
Zahra
Ramezani
*
Nanotechnology Research Center, Medicinal Chemistry Department, Faculty of Pharmacy, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran. E-mail: zramezani@ajums.ac.ir; zahramezani@gmail.com; Fax: +98 6133738380; Tel: +98 6133738379
First published on 19th November 2018
Abstract
Microwave-assisted distillation–precipitation polymerization (MWDPP) for the synthesis of magnetic molecularly imprinted polymers (MMIPs) under atmospheric pressure is reported. The suggested synthesis methodology is fast and was performed in two steps. In the first step, methacrylic acid (MAA)-coated Fe3O4 (Fe3O4@MAA) was prepared under a nitrogen atmosphere for about 30 min at 60 °C using a maximum microwave power of 400 W. In the second step, the Taguchi orthogonal array was used to design experiments for optimization of the MMIP's synthesis at 82 °C (500 W), having considered sorption capacity for the template as the response. In Taguchi design, three factors including the amount of Fe3O4@MAA, the template to monomer ratio, and the MAA to cross linker ratio at three levels were considered. Under optimal conditions, MMIPs were synthesized in 60 minutes. Different microscopic, spectroscopic, and thermal methods were used to evaluate the MMIPs. The results illustrated the formation of mono-dispersed, highly-selective super paramagnetic MMIPs with a mean size of 13 nm, an adsorption capacity of 86.84 mg g−1, and an imprinting factor of 5.39 with fast adsorption and desorption kinetics for perphenazine (PPZ) as the template. Finally, the as-prepared MMIPs were used for pre-concentration and cleanup of PPZ in urine, without any treatment, coupled to high performance thin layer chromatography equipped with a photodiode array detector (HPTLC-PDA). Under optimized conditions, the calibration curve was linear in the range of 2.5–50 ng spot−1 (R2 = 0.9938) with the detection limit of 0.56 ng per spot−1. Parameters affecting the analytical performance of HPTLC were also optimized. A correction factor was introduced and compared with the internal standard to improve the precision and reliability of the HPTLC results. Spiked urine samples were analyzed after preconcentration. The HPTLC-PDA results were in good agreement with the corresponding high performance liquid chromatography coupled with photodiode array detection (HPLC-PDA) as a standard method.
1. Introduction
Molecularly imprinted polymers (MIPs) are artificial antibodies or enzymes with a long lifetime and selectivity, and, as a result, have a vast variety of applications in chemistry, biology, and medical sciences.1,2 Because of their molecular recognition ability, MIPs are used in separation, cleanup, and pre-concentration of a specific analyte in complicated matrices such as environmental, clinical, and biological samples, turning into one of the most important fields of scientific research.3–10 For fast and easy separation of particles from solutions, magnetic MIPs (MMIPs) were developed.9–11 In addition, MMIPs, compared to bulk MIPs, have a higher surface area to volume ratio, a more accessible imprinted cavity, and a higher capacity. MMIPs are synthesized by polymerization around a magnetic core such as Fe3O4. To initiate polymerization, the Fe3O4 surface is modified by reagents such as silica compounds,12 polyethylene glycol (PEG), oleic acid,13 and a small molecule such as methacrylic acid (MAA).14,15 MMIPs prepared by each modifier show different magnetic properties. Magnetism, in addition to the concentration of the magnetic core, is size-dependent. Then, the thickness of the modified layer around the Fe3O4 determines the magnetism and, consequently, the final size of the MMIPs. Surface modifiers such as MAA reduce the size of MMIPs and increase their magnetism. MMIP synthesis is not an easy undertaking, and there are different synthetic protocols such as grafting methods, emulsion, suspension, and precipitation polymerizations1,10 introduced to obtain MMIPs with a low template leak, a high mass transfer and binding capacity, and an excellent site accessibility at a short time with a reduced amount of reagents. Among different protocols, precipitation polymerization (pp) has attracted researchers' attention because it is a simple technique and produces monodispersed, spherical micro and nanoparticles in good yield. In 2004,16 distillation PP (DPP) was introduced to produce narrow dispersity polymer microspheres. In DPP, more than 60% of the solvent was distilled during precipitation–polymerization. To tune the particle size in DPP, accurate control over the reaction condition is required. Conventional heating and UV light are ordinarily applied for temperature control in DPP. Different polymers including MMIPs were prepared by a DPP method. The polymeric particles were produced in at least 15 h.17–20 Polymerization via conventional heating is time-consuming and produces large MMIP particles. Microwave heating has been introduced as an alternative choice in MMIP synthesis. High penetration of microwave accelerates polymerization. In addition to high speed, microwave synthesis has control over particle morphology and size.13,21,22 To date, no reports on microwave-assisted distillation–precipitation polymerization (MWDPP) of MMIP synthesis are available.In the synthesis of MMIPs, many parameters can influence the morphology, size, and properties of the particles.10 In this respect, the optimization of the effective parameters is of vital importance. In most studies, optimization involves the variation of one parameter while keeping the others constant. This method, besides being time-consuming, cannot estimate the variables’ interactions. However, a statistical design of experimental methods provides an easier and more efficient approach to optimize several operational variables.23 The Taguchi's optimization technique is a powerful optimization discipline that allows optimization with a minimum number of experiments. The Taguchi experimental design reduces the cost, improves the quality, and provides robust design solutions. Another advantage of the Taguchi method over the other optimization methods is that numerous factors can be simultaneously optimized. Still, more quantitative information can be extracted from fewer experimental trials.23,24
A variety of analytical techniques have been used for the determination of the target analyte after separation and pre-concentration. The selection of an instrumental method is based on the selectivity of the sample treatment and the extent of analyte pre-concentration. Since MMIPs act selectively toward structurally-related compounds, chromatographic methods have been extensively introduced in their quantitation and detection. Depending on the analyte volatility, HPLC or GC is suggested. HPTLC, an automated form of thin layer chromatography, enables simultaneous processing of sample and standard under similar conditions and gives acceptable analytical precision and accuracy.25 Additionally, extreme flexibility for the selection of stationary phase, mobile phase, developing technique, and detection with or without pre or post-development derivatization are some of the advantages of HPTLC. It is a powerful technique in fingerprint determination of herbal plants and is extensively used in pharmacognosy.26,27–33 Because of the high sample throughput and very low amount of mobile phase compared to HPLC, HPTLC quantitative analysis is growing fast, and the number of papers has increased recently.34–44 However, a serious limitation is the plate-to-plate variation that affects the accuracy and precision when calibration standards and samples are spotted in the different plates. This problem is formally solved by adding a suitable internal standard (IS) to both calibration and sample solutions prior to analysis. The IS is a compound with close structural relationship to the analyte. It may be a synthetic modification or isotopic version of the analyte. The following points must be considered in IS selection: it must have well-resolved peaks for both analyte and IS, be preferably eluted after the analyte, be sufficiently stable during the sample preparation and chromatographic analysis processes, be available in a pure form, and be compatible with the detector response. The principal disadvantage to the internal standard calibration is that the IS must be from compounds that are not found in the samples to be analyzed.
The objective of the present research is to evaluate the effect of microwave irradiations in DPP synthesis of MMIPs. Perphenazine (PPZ) was selected as a model template to demonstrate the effect of microwaves on MMIP morphology, size, binding capacity, imprinting factor, and time of synthesis compared to the conventional heating reported previously.14 The specific feature of the present synthesis procedure is that MAA-modified Fe3O4 (MAA@Fe3O4) has also been prepared through microwave heating. Finally, the as-prepared particles were used in pre-concentration and separation of PPZ in human urine samples without any pretreatment or dilution using HPTLC. PPZ is a typical antipsychotic drug and its quantification by HPTLC has not yet been reported. An area correction factor was introduced to compensate the plate-to-plate variation in HPTLC instead of using the IS calibration method.
2. Materials and methods
2.1 Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O), iron(II) chloride tetrahydrate (FeCl2·4H2O), potassium dihydrogen phosphate (KH2PO4), ethanol (EtOH), butanol (But-OH), propanol (Pro-OH), ammonia, hydrochloric acid, glacial acetic acid (HOAc), tetrabutylammonium bromide (TBABr), hexane (n-Hex), ethyl acetate (Et-Ac) and cetyltrimethylammonium bromide (CTAB) were purchased from Merck (Darmstadt, Germany) and used without additional purification. Ethylene glycol dimethacrylate (EGDMA), methacrylic acid (MAA) and HPLC-grade acetonitrile (ACN) were also obtained from Merck (Darmstadt, Germany). Perphenazine (PPZ), trifluoperazine (TFP), thioridazine hydrochloride (TRZ), amitriptyline hydrochloride (AML), chlorpromazine hydrochloride (CPZ), hydroxyzine (HXZ), promazine (PRM), and 2,2-azobisisobutyronitrile (AIBN) were bought from Sigma-Aldrich (St. Louis, MO, Germany). HPLC-grade methanol (MeOH) was supplied by Fanavaran Petrochemical Company (Tehran, Iran). Stock solutions (1000 mg L−1) of all standards were prepared in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HOAc (9:1) and stored at 4 °C, protected from light. Working standards were prepared by serial dilution of these stock solutions.
:HOAc (9:1) and stored at 4 °C, protected from light. Working standards were prepared by serial dilution of these stock solutions.
2.2 Apparatus
HRTEM was acquired by Philips CM30 (Philips, Eindhoven, The Netherlands) operating at 150 kV. FESEM was obtained by Mira3-XMU FESEM (Tescan, Czech Republic). All FTIR spectra were recorded by KBr pellets using a vertex 70 FTIR spectrometer (Bruker, Germany). TGA analyses were performed simultaneously on thermal analyzers (STA503, BÄHR-Thermoanalyse GmbH, Germany). An Operon freeze dryer (Korea) was used to lyophilize the as-prepared MMIP suspensions. UV/vis measurements were made on a Jasco 7850 spectrophotometer (Japan). An HPTLC equipped with Linomat 5 sample application, a CAMAG horizontal developing chamber, a TLC Scanner equipped with photodiode array detector (PDA), and an image documentation system were used throughout this study. The instrument was controlled via Win CATs-version 1.3.3. Table S1 (ESI†) lists the instrumental details and the parameters set for the PPZ analysis. HPLC was performed on a Waters 600 instrument comprising two reciprocating pumps, an AF in-line degasser, a high-pressure manual Rheodyne injection valve (20 μL injection loop), and a Waters 2998 photodiode array detector (PDA). Data acquisition and processing were performed using the Empower 2 software.2.3 Synthesis of MMIPs
:Fe3+ (1:2) in 0.5 M HCl was added drop-wise to the alkaline solution at 60 °C. Then 1 mL MAA was added to the particles, the supernatant was sampled at different time intervals, its absorbance was determined at 290 nm (maximum absorption wavelength of MAA), and the solution was returned to the reaction vessel. When the absorbance reached zero, another portion of 1 mL MAA was added. Successive addition, collection, and measurements were performed three times to reach the plateau (Fig. S1A(a–c), ESI†).
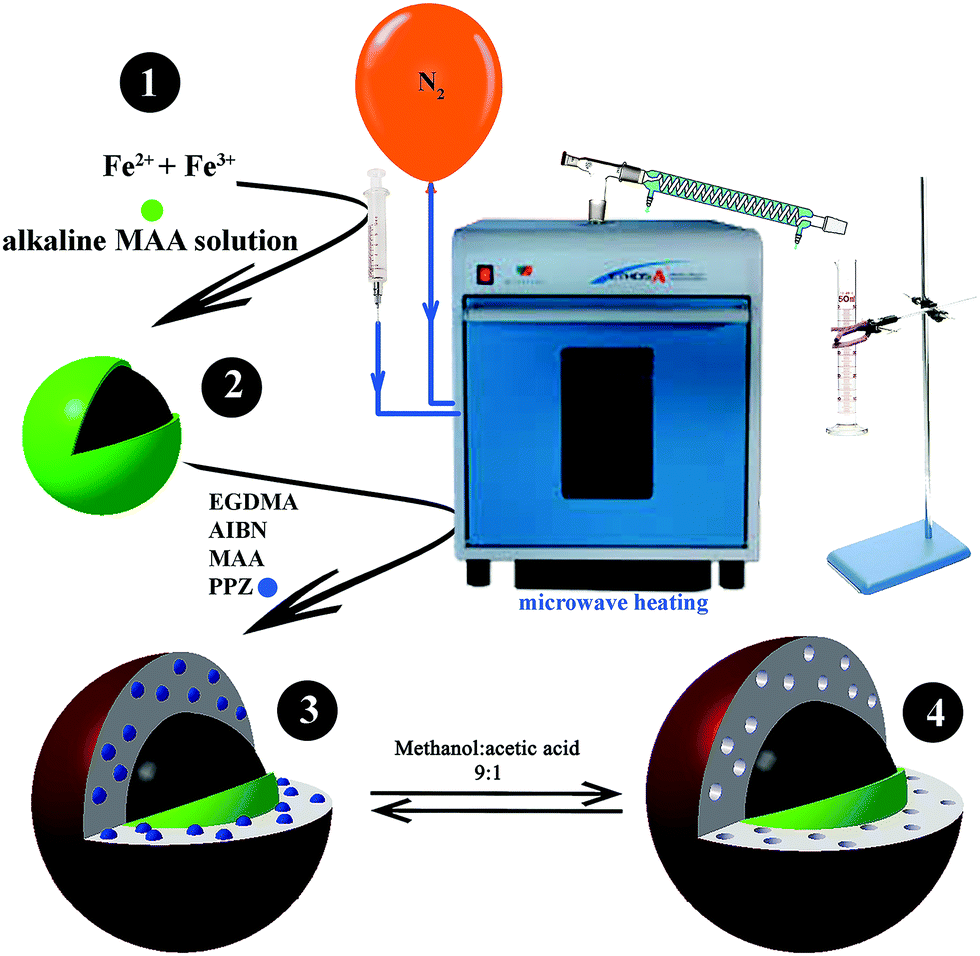 | ||
| Fig. 1 Setup of the microwave system for PPZ MMIP synthesis. | ||
2.3.3.1 MMIP synthesis under optimum conditions. To begin the synthesis, 0.1 mmol PPZ and 85 μL MAA were mixed with 25 mL ACN and stirred for 30 min to form a template–monomer complex (pre-polymerization solution).29 Then 1.7 mL cross linker (EGDMA) and 10 mL 20% (w/v) Fe3O4@MAA suspension were added. The whole solution was transferred to a three-necked rounded-bottom flask and placed in a Milestone MicroSYNTH microwave oven under a nitrogen atmosphere. After 1 min irradiation at 400 W (Fig. S1C; ESI†), the temperature had increased to 65 °C, and 0.05 g AIBN in 5 mL ACN was rapidly added through the injection port indicated in Fig. 1 (total solution volume was 50 mL). The mixture remained at 65 °C for 6 min, and was later increased to 80 °C. After 20 min, the temperature was increased to 84 °C to start distillation, and about 20 mL ACN was distilled in 2 min and gathered in a graduated cylinder outside the microwave (Fig. 1). The system was cooled down, and MMIP nanoparticles were separated by a 1.4 Tesla magnet. The particles were washed five times with re-dispersion in 50 mL MeOH
:HOAc (9:1, v/v) and the collection was made by the magnet to remove the template molecules (PPZ) as completely as possible. The extent of template removal was determined by detecting PPZ in supernatants with both the UV-vis spectrophotometer and the HPLC at 253 nm. The MMIPs were washed with deionized water until the eluents became neutral. Finally, it was re-dispersed in deionized water to make a 50 mg mL−1 MMIP suspension and stored at 4 °C for future use.
Magnetic non-imprinted polymers (MNIPs) were also prepared via the same procedure in the absence of the template molecule.
2.4 Chromatographic analysis of PPZ
cm × 10cm glass HPTLC plates coated with silica gel 60 F254 (Merck, Darmstadt, Germany). Chromatographic steps including sample spotting, visualization, and scanning the plates were performed automatically. 10 μL of samples or standards were spotted on the plate using an ATS4 CAMAG automatic sampler. Then, the chromatographic plates were developed in a horizontal Teflon chamber. The plates were scanned at 256 nm, and a PDA detector recorded the spot spectra in the range of 200 to 400 nm. Ten standard solutions of PPZ in the range of 2.5–50 ng spot−1 (25–5000 ng mL−1) were used for calibration.
2.5 Procedure of PPZ extraction
At first, 100 μL suspension of the prepared MMIPs (containing 5 mg of dried particles) was washed by 2 mL deionized water and decanted under a magnetic field, 5 mL of the sample or standard PPZ solutions at pH 6 was added, and the resulting mixture was stirred for 1 min. The solution was decanted by a magnet, and the isolated particles were washed with 2 mL deionized water to remove any possible interference. Finally, PPZ was eluted from the MMIP particles using two 200 μL portions of MeOH:HOAc (9:1, v/v) followed by sonication for 1 min. The solvents were mixed, evaporated at 60 °C in a water bath, and the dry residue was re-dissolved in 250 μL MeOH:HOAc (9:1). For PPZ quantitation, 10 μL and 20 μL of the solution were applied to HPTLC and HPLC, respectively. The whole procedure from synthesis to quantitation is illustrated in Scheme 1.
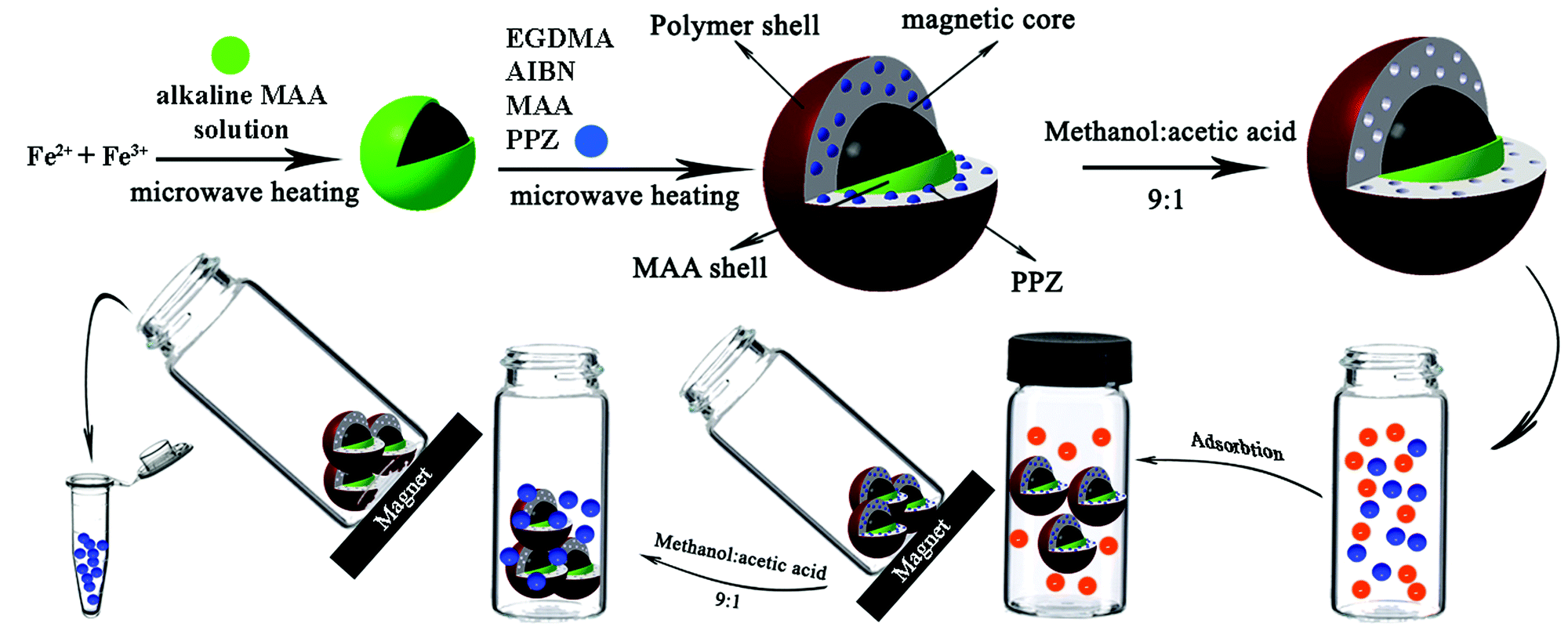 | ||
| Scheme 1 Schematic representation of the whole procedure, from synthesis to PPZ extraction. | ||
2.6 Urine sample preparation
Drug-free human urine samples were collected from healthy volunteers with informed consent and stored at −20 °C before use. The PPZ extraction procedure was conducted without any sample pretreatments and/or dilution. Calibration curves were also obtained in drug-free human urine. PPZ spiked urine samples were analyzed by HPTLC. The accuracy of the measurements was determined by comparing the results with those of HPLC as the reference method.143. Results and discussion
3.1 Microwave-assisted synthesis of MMIPs
Taguchi orthogonal array (Table S2, ESI†) was used to optimize the reagents ratio in the microwave synthesis of MMIPs (Fig. S1C, ESI†) since the Taguchi method emphasizes that the mean performance characteristic value be close to the target value, thus improving the MMIP quality. By analyzing the Taguchi results using Minitab16 statistical software considering the sorbent capacity for PPZ as a response, and the rule “the higher is the best”, the optimum amount of the reagents was obtained. The output graphs indicate (Fig. S2, ESI†) 10 mL of 20% w/v Fe3O4@MAA, 0.1 mmol PPZ, 85 μL MAA in 25 mL ACN, 1.7 mL EGDM, 50 mg AIBN in 5 mL ACN, and 8.3 mL ACN are the optimum values for preparing high capacity nano MMIPs. Finally, MMIPs and magnetic nonimprinted polymers (MNIPS) synthesized under the optimum conditions were kept refrigerated for future use and were stable for nine months. The stability over nine months was not checked.
 | ||
| Fig. 2 (A) HR-TEM images of Fe3O4@MAA, (B) HR-TEM, (C) FESEM, and (D) EDCX of the MMIPs. | ||
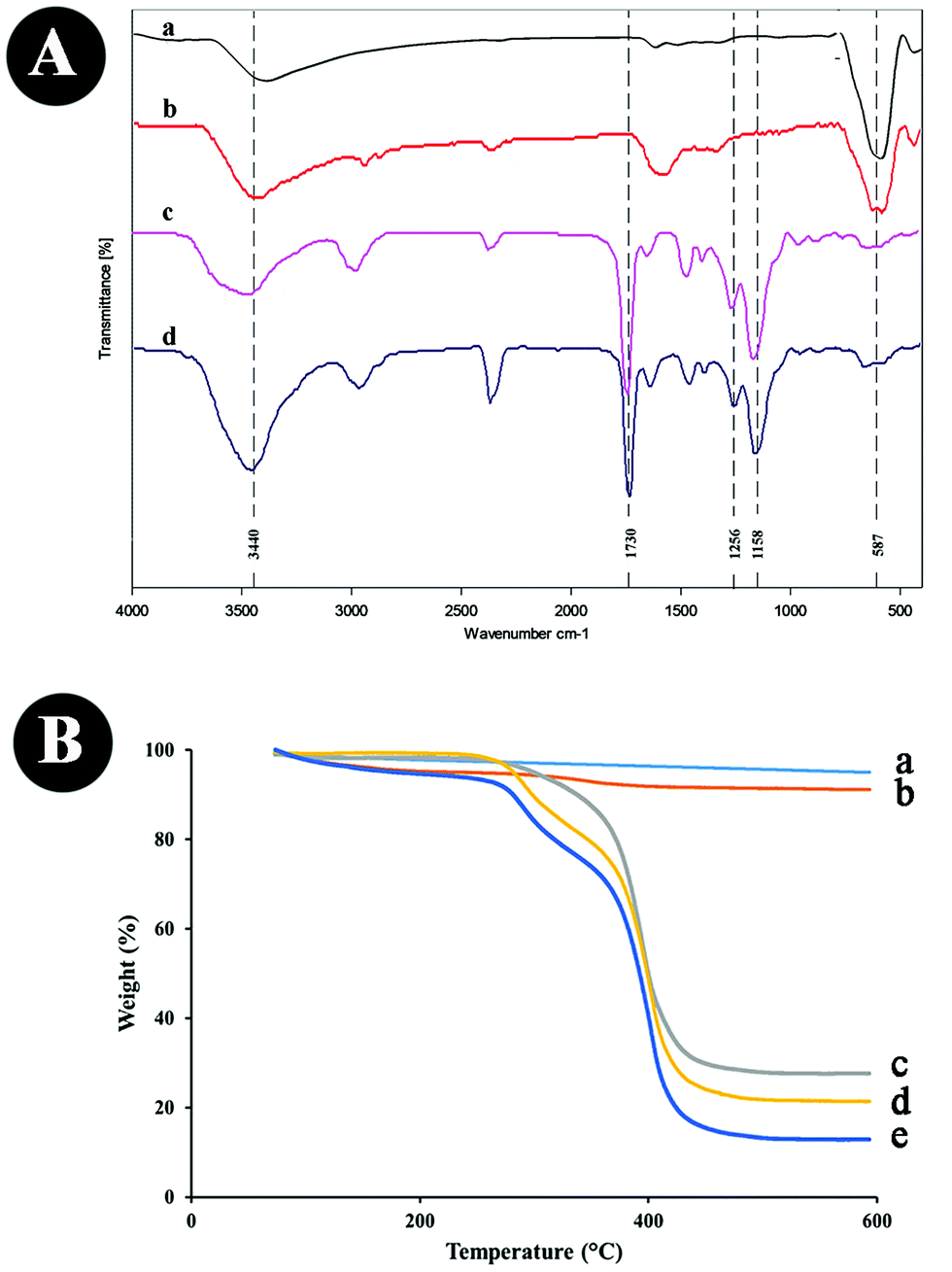 | ||
| Fig. 3 (A) FTIR of (a) Fe3O4, (b) Fe3O4@MAA, (c) MMIPs, (d) MNIPs; and (B) TGA analysis of (a) Fe3O4, (b) Fe3O4@MAA, (c) MNIPs, (d) MMIPs, (e) PPZ + MMIPs. | ||
The HRTEM image of the MMIPs in Fig. 2B indicates that a thin layer (below 10 nm) of the imprinted polymers (marked with yellow lines) has been formed around the magnetite core and the mean size of the as-prepared particles is 13 nm. The FESEM image in Fig. 2C shows uniform and porous MMIP nanoparticles, and the EDX analysis in Fig. 2D further confirms MMIP production by the presence of carbon, oxygen and Fe peaks. There is a slight shift in Fe–O peak (Fig. 3A(c and d)) for the as-prepared MMIPs and MNIPs compared to Fe3O4@MAA. C–O asymmetric (1158 cm−1) and symmetric stretching (1256 cm−1), and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at 1730 cm−1 in both MMIPs and MNIPs (Fig. 3A(c and d)) indicates the formation of the polymers. The thermal gravimetric analysis further proves the formation of MMIPs. The absence of template molecules increases the magnetite core content of the as-prepared particles. As a result, the TGA thermogram of the MNIPs (Fig. 3B(c)) shows a lower weight loss compared to those of MMIPs (Fig. 3B(d)). A 50 mL solution of 50 μg mL−1 PPZ was stirred with 50 mg MMIPs. Then, the particles were separated, dried, and their thermogram was recorded (Fig. 3B(d)). The difference in weight loss (8.84%) in Fig. 4B(d and e) is due to the presence of the adsorbed PPZ. The MMIP's capacity can be estimated from this weight loss, which is 88.4 mg g−1. It is close to the experimental value of 86.48 mg g−1, with about 2% error. This MMIP capacity for PPZ is as much as about 73.68% higher than those prepared in our previous MMIP synthesis using traditional heating.14
O band at 1730 cm−1 in both MMIPs and MNIPs (Fig. 3A(c and d)) indicates the formation of the polymers. The thermal gravimetric analysis further proves the formation of MMIPs. The absence of template molecules increases the magnetite core content of the as-prepared particles. As a result, the TGA thermogram of the MNIPs (Fig. 3B(c)) shows a lower weight loss compared to those of MMIPs (Fig. 3B(d)). A 50 mL solution of 50 μg mL−1 PPZ was stirred with 50 mg MMIPs. Then, the particles were separated, dried, and their thermogram was recorded (Fig. 3B(d)). The difference in weight loss (8.84%) in Fig. 4B(d and e) is due to the presence of the adsorbed PPZ. The MMIP's capacity can be estimated from this weight loss, which is 88.4 mg g−1. It is close to the experimental value of 86.48 mg g−1, with about 2% error. This MMIP capacity for PPZ is as much as about 73.68% higher than those prepared in our previous MMIP synthesis using traditional heating.14
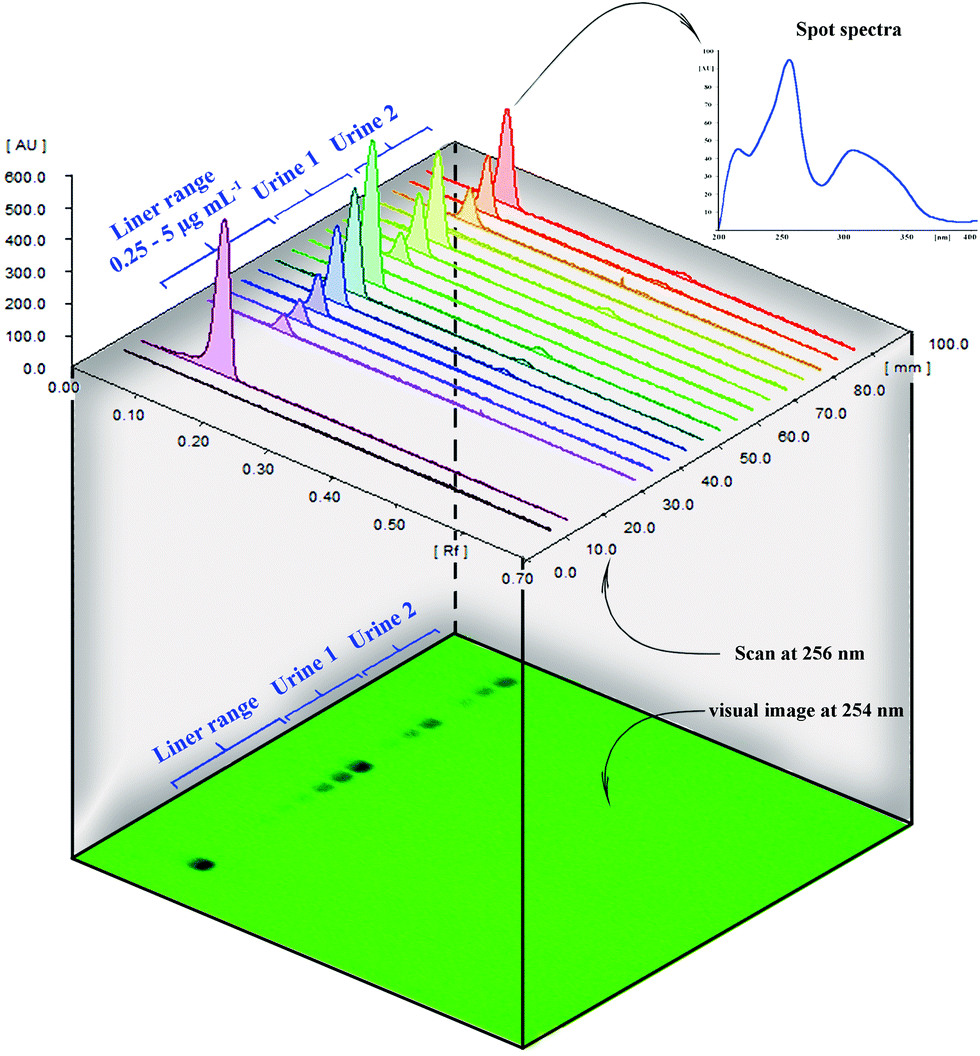 | ||
| Fig. 4 HPTLC 3D chromatogram and their plate photo at 254 nm as well as PPZ spectra at Rf = 0.09 recorded by photodiode array detector. Calibration and urine spots are also identified in the photo. | ||
3.2 HPTLC evaluations
:Et-Ac:10 mmol TBABr in EtOH:But-OH:NH3. Further evaluation of the resolution for phenothiazine mixtures (PPZ, TFP, CPZ, AMI, HXZ) suggested n-Hex:Et-Ac:10 mmol TBABr in EtOH:But-OH:NH3 (5:2:0.75:1:0.1) as the best solvent mixture in HPTLC analysis of PPZ.
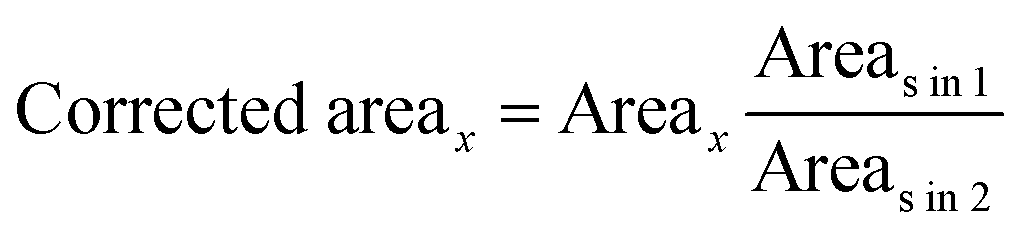 | (1) |
in1 and Areasin2 are the areas of the standard with a fixed concentration spotted on plates 1 and 2, respectively. Corrected Area is used to determine the unknown concentration from the calibration curve. Table 1 compares quantitation for three different amounts of PPZ by external calibration, internal standard, and the proposed area correction method. As the results imply, the area correction method has the same, or sometimes better, results than those obtained by the internal standard.
| Concentration (ng spot−1) | Found concentration (ng spot−1) | ||
|---|---|---|---|
| External calibration | Area correction method | Internal standard method | |
| Data in parentheses are recoveries for the tested standards. | |||
| 10 | 8.43 (84.30) | 10.28 (102.80) | 10.08 (100.80) |
| 20 | 21.04 (105.20) | 20.12 (100.58) | 19.08 (95.40) |
| 40 | 42.51 (106.27) | 40.13 (100.32) | 38.77 (96.93) |
3.3 Parameters affecting pre-concentration and separation of PPZ by MMIP
The MMIP amount, the pH, the contact time, the kind and volume of washing, and the desorbing solvents were optimized. Fig S4A (ESI†) shows that pH has no considerable effect on the extraction in the range of 4 to 9. In acidic solutions, a competition between protons and the drug for the carboxyl site within the polymer cavity happens, and protonation of the carboxyl acid groups might reduce PPZ adsorption at lower pH values. As a result, extraction recovery was decreased at pH below four. At pH above 8, de-protonation of PPZ (pKa = 7.94) reduces the hydrogen bonding between PPZ and carboxylic groups within the polymer cavity and, consequently, decreases adsorption.47 The effect of contact time on PPZ adsorption in Fig. S4B (ESI†) illustrates fast adsorption. The maximum sorption capacity determined after desorption of PPZ is about 86.48 mg g−1. Reusability of the particles was also investigated by six successive adsorptions and the desorption of 1 μg mL−1 PPZ. The minimum PPZ recoveries were 92%. Super paramagnetic properties of the particles prevent their losses during filtration under a magnetic field. No significant changes in the particles’ performance were observed during storage for nine months.3.4 Method validation
Linearity, accuracy, precision, specificity, limit of quantification (LOQ), limit of detection (LOD), robustness, and system suitability were evaluated according to ICH guidelines (Table 2, Tables S3 and S4, ESI†). Both external and in-urine calibration curves were linear in the range 25–5000 ng mL−1 (2.5–50 ng spot−1). Repeatability of the measurements was calculated by the analysis of 10, 20, and 40 ng spot−1 PPZ in triplicate on the same day with the same plate. Intermediate precision was determined for the three mentioned concentrations on four different days. The results in Table 2 demonstrate the excellent reproducibility and repeatability.| Parameter | HPLC | HPTLC | ||
|---|---|---|---|---|
| External calibration | In urine calibration | External calibration | In urine calibration | |
| a Concentration of PPZ added. b Concentration found in ng spot−1. | ||||
| Liner range | 20–5000 (ng mL−1) | 20–5000 (ng mL−1) | 2.5–50 (ng spot−1) | 2.5–50 (ng spot−1) |
| Slope ± SD | 2048508 ± 36442 |
2036006 ± 14787 |
6449 ± 44 | 6452 ± 140 |
| Intercept ± SD | 186434 ± 3481 |
159838 ± 3675 |
100 ± 9 | −151 ± 7 |
| R 2 ± SD | 0.9995 ± 0.02 | 0.9999 ± 0.006 | 0.9963 ± 0.001 | 0.9980 ± 0.0006 |
| LOD | 1.51 (ng mL−1) | 2.10 (ng mL−1) | 0.13 (ng spot−1) | 0.56 (ng spot−1) |
| LOQ | 5.30 (ng mL−1) | 7.03 (ng mL−1) | 1.03 (ng spot−1) | 1.80 (ng spot−1) |
| Mean recovery (n = 6) | 99.67 ± 1.67 | 100.8 ± 0.89 | 97.79 ± 1.70 | 96.44 ± 1.12 |
| RSD% (n = 6) | 1.89 | 1.41 | 1.74 | 1.16 |
| EF | 16.75 | — | 19.87 | — |
| PF | 20 | — | 20 | — |
| ME | — | 100 | — | 100.19 |
| PE | 99.67 | — | 97.97 | — |
| Repeatability | ||||
| 10a | — | 9.78 ± 0.312b (3.19) | ||
| 20a | — | 19.38 ± 0.370b (1.91) | ||
| 40a | — | 39.65 ± 0.828b (2.09) | ||
| Intermediate precision | ||||
| 10a | — | 10.10 ± 0.183b (1.81) | ||
| 20a | — | 19.75 ± 0.168b (0.84) | ||
| 40a | — | 39.47 ± 0.402b (1.20) | ||
The specificity of a method is proved by the accuracy of the analysis of PPZ in the matrix or a mixture. Sample preparation may be viewed as the main reason for the matrix effect (ME) and indicates the extent of the signal suppression or depression, depending on the value. ME value, 100% no effect, less than 100% signal suppression, and over 100% signal enhancement according to co-eluting compounds. MMIPs, due to their selectivity nature, are expected to clean up the samples and eliminate interferences as much as possible. Herein, the ME value was calculated by eqn (2)
| ME = (Smatrix/Ss) × 100 | (2) |
LOD and LOQ were calculated as three and ten times the signal to noise ratio, respectively. Enrichment factor (EF) was determined by dividing the slope of the method's calibration curve with the standard calibration curve before pre-concentration. As a result, EF, determined by the slope ratio, was 19.87. Pre-concentration factor (PF), calculated by Vf/Vi, where Vi and Vf are the volumes of the PPZ solution before and after pre-concentration, was 20. Process efficiency (PE) is the common term for expressing exactness in chromatographic separations and is calculated by eqn (3), where R stands for recovery percent.
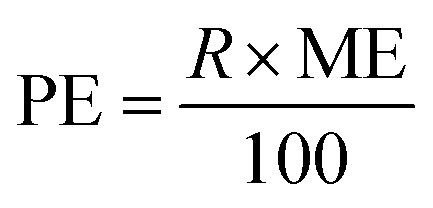 | (3) |
Data also show that good correlation between the HPTLC and HPLC method exists. HPTLC chromatograms, the plate photo, and the UV spectra at PPZ Rf are presented in Fig. 4. HPLC chromatograms are also provided in Fig. S5 (ESI†).
Deliberate small changes in some HPTLC parameters, listed in Table S4 (ESI†), did not lead to significant changes in the Rf of PPZ. Therefore, the system is robust.
In clinical analysis, urine is usually a good choice for pharmaceutical and metabolite analysis because it is available in large volumes. However, its complex matrix has a considerable impact on the direct analysis of a target compound, and a reliable pre-concentration and cleanup are required.8 Selective sorbents such as MIP reduce the urine matrix interferences. The reliability of the present pre-concentration procedure was evaluated by spiking human urine samples at three levels (0.5, 1, and 3 μg mL−1) and determining the spiked amount by both HPTLC and HPLC. The values in Table 3 illustrate the reliability of the HPTLC method and the pre-concentration ability of the as-prepared MMIPs in human urine directly with no need for any treatments or dilution. Urine chromatogram (Fig. 4) illustrates excellent cleanup.
| Samples | Added (μg mL−1) | HPLC | HPTLC | ||
|---|---|---|---|---|---|
| Found (μg mL−1) | Recovery (%) | Found (μg mL−1) | Recovery (%) | ||
| Human urine 1 | — | ND | — | ND | — |
| 0.5 | 0.497 (±0.029) | 99.41 | 0.480 (±0.021) | 96.05 | |
| 1 | 0.997 (±0.014) | 99.70 | 0.984 (±0.033) | 98.38 | |
| 3 | 3.034 (±0.025) | 101.14 | 2.890 (±0.066) | 96.20 | |
| Human urine 2 | — | ND | — | ND | — |
| 0.5 | 0.497 (±0.017) | 99.46 | 0.477 (±0.054) | 95.50 | |
| 1 | 0.998 (±0.019) | 99.83 | 0.971 (±0.067) | 97.10 | |
| 3 | 3.025 (±0.200) | 100.85 | 2.863 (±0.071) | 95.44 | |
3.5 Imprinting properties of MMIPs
The polymers’ selectivity evaluations were performed by determining the adsorption capacity of nanoparticles incubated in the mixture of PPZ and the other six selected compounds under optimized conditions. The adsorption capacity calculated by the residual concentrations in the supernatant identified no considerable difference between MMIPs and MNIPs for PPZ adsorption (Fig. 5, adsorption column). Nonspecific adsorption of the phenothiazine on the highly porous nano MMIPs is responsible for this high sorption capacity. To show the effect of nonspecific adsorption, the desorbed amount (as described in Section 2.7) was determined (Fig. 5, desorption). Then, a considerable difference was observed in the adsorption capacity of PPZ for MMIPs compared to MNIPs and the other tested compounds (Fig. 5).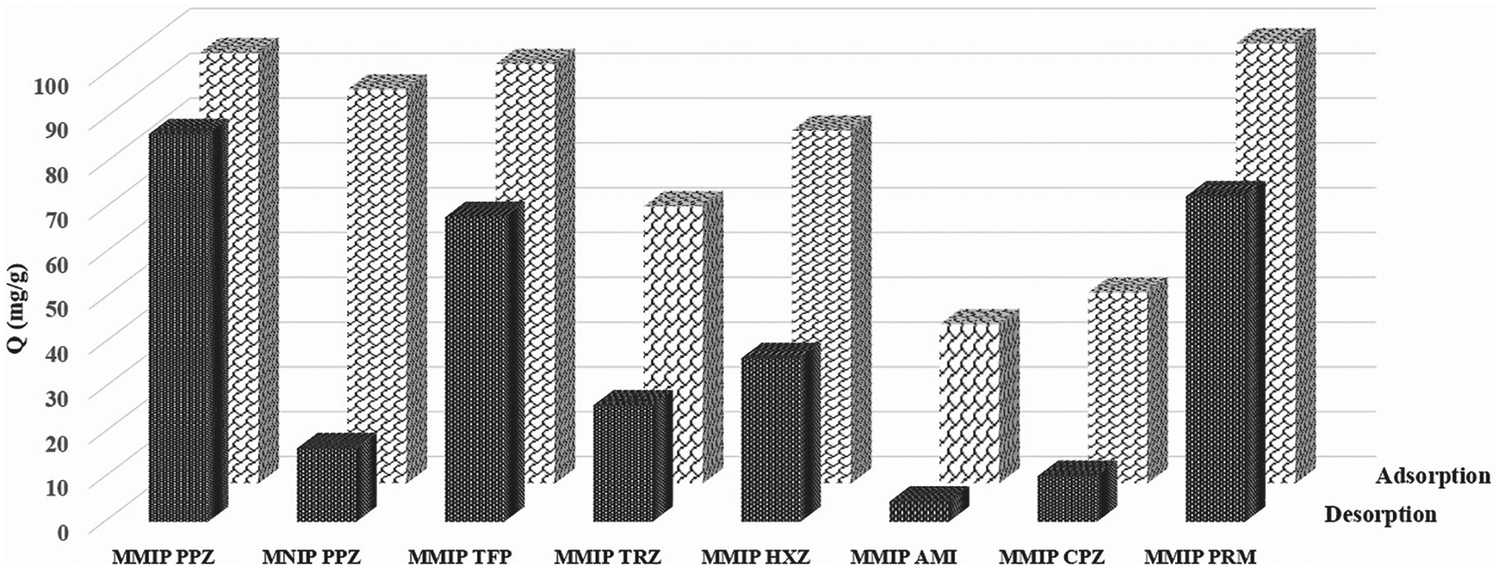 | ||
| Fig. 5 Selectivity of PPZ MMIPs and MNIPs prepared by the present method based on (a) equilibrium concentration after adsorption; and (b) desorbed amount of the compounds by MeOH:HOAc (9:1, v/v) as an eluting solvent. | ||
Imprinting factor (IF) defined as the ratio of the capacity factor (φ) between MMIPs and MNIPs (eqn (4)) is an easy way to highlight the recognition properties of imprinted materials. Calculated IF values for all six compounds are presented in Table S5 (ESI†). MMIPs prepared by microwave irradiation have an IF of 5.39 for PPZ which is much higher than those prepared by conventional heating (2.41).14 TFP, PRM, and HXZ have an IF of 3.69, 3.44, and 2.15, respectively. The high imprinting factor for PPZ, compared to other compounds, illustrates the high selectivity of the MMIPs for the template. The selectivity of the as-prepared MMIPs for TFP and PRM is also considerable, which can be explained by their similar chemical structure. The tendency of MNIPs toward adsorption of the phenothiazine is due to their nanometer sizes and the presence of the carboxyl group on the polymer, which was confirmed by FTIR.
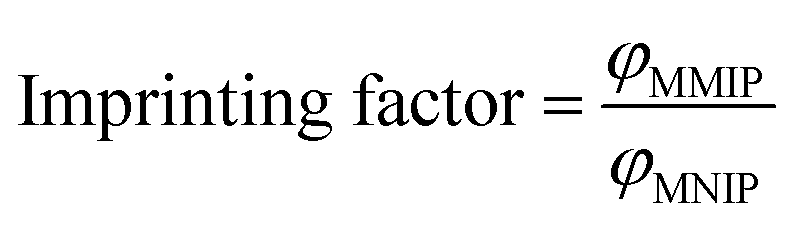 | (4) |
3.6 Comparisons with some reported methods
A specific feature of the present synthesis protocol of MMIPs over other reported microwave-assisted procedures is that all of the synthetic steps are conducted in the microwave under precise control of temperature. Moreover, it is the first method of microwave-assisted DPP synthesis of MMIPs. Besides, the number of synthesis steps, Fe3O4@MAA preparation time, polymerization time, and extraction time using the as-prepared particles are lower than those reported in the literature (Table S6, ESI†). The mean diameter of the present MMIPs is 13 nm, which is much smaller than all the other microwave-assisted procedures as well as the conventional method (Table S6, ESI†). In a recent study, Kazemi et al.48 applied precipitation polymerization for the synthesis of letrozole magnetic imprinted polymer in three steps using conventional heating. They modified the Fe3O4 surface by MAA in 18 h and prepared MMIPs with the mean size of 100 nm in 24 h.In the current study, PPZ was used as a template to evaluate the effect of the heating method in distillation–precipitation polymerization for the same compound. The capacity of the as-prepared MMIPs was 86.84 mg g−1, about 73.68% higher than that obtained by our previous report (50 mg g−1). The selectivity (IF 5.39 compared to 2.41) is also higher, and the size of the particles is smaller than that in the previous report.14
In Table S7 (ESI†), some recent sample preparation techniques coupled with different instrumental methods are listed for comparison. The majority of the reported protocols dilute the biological samples before PPZ assay because of the matrix interferences. The selective nature of MMIPs reduces the matrix interferences and no dilution is required. PPZ extraction time for the present sorbent is much lower than that in other reported techniques. Liquid–liquid extraction (LLE), dispersive liquid–liquid extraction (DLLME), and ionic liquid (IL-SE-UA-ME) coupled with HPLC or capillary electrophoresis (CE) have a better limit of detection and quantitation. These techniques, however, use organic solvents or sophisticated instruments, are not cost-effective and time-consuming. They also have a lower PPZ recovery compared to the present HPTLC system.
4. Conclusions
This is the first time, to the best of our knowledge that the synthesis of MMIPs by distillation–precipitation polymerizations and Fe3O4@MAA is thoroughly carried out in a microwave. The introduced protocol is fast, and the produced MMIPs are uniform and nano in size. Moreover, they have super-paramagnetic properties and a high imprinting factor. If MMIPs are used in sample preparation, an excellent clean-up of untreated human urine samples will be possible. Normal phase HPTLC was introduced both as a reliable PPZ quantitation technique in human urine and an alternative for HPLC. It has several advantages including lower organic solvent consumption, high sample throughput, and better resolution of co-eluting compounds compared with HPLC. The precision of measurements in HPTLC analysis, which is usually challenging, was improved by introducing an area correction factor as an available, easy-to-apply, and reliable alternative to IS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to gratefully acknowledge the research deputy of Ahvaz Jundishapur University of Medical Sciences and Iran National Science Foundation (INSF) who supported this work under grant number N112 and grant number 95849362, respectively. This paper is extracted from Mehdi Safdarian's PhD dissertation.References
- K. Haupt, Anal. Chem., 2003, 376A–383A CAS.
- B. Tse Sum Bui and K. Haupt, Anal. Bioanal. Chem., 2010, 398, 2481–2492 CrossRef.
- V. Pérez-Fernández, L. Mainero Rocca, P. Tomai, S. Fanali and A. Gentili, Anal. Chim. Acta, 2017, 983, 9–41 CrossRef.
- P. Muhammad, J. Liu, R. Xing, Y. Wen, Y. Wang and Z. Liu, Anal. Chim. Acta, 2017, 995, 34–42 CrossRef CAS.
- D. He, X. Zhang, B. Gao, L. Wang, Q. Zhao, H. Chen, H. Wang and C. Zhao, Food Control, 2014, 36, 36–41 CrossRef CAS.
- E. Turiel and A. Martín-Esteban, Anal. Chim. Acta, 2010, 668, 87–99 CrossRef CAS PubMed.
- L. Chen, X. Zhang, Y. Xu, X. Du, X. Sun, L. Sun, H. Wang, Q. Zhao, A. Yu, H. Zhang and L. Ding, Anal. Chim. Acta, 2010, 662, 31–38 CrossRef CAS.
- S. Ansari and M. Karimi, Talanta, 2017, 167, 470–485 CrossRef CAS PubMed.
- S. Ansari and M. Karimi, TrAC, Trends Anal. Chem., 2017, 89, 146–162 CrossRef CAS.
- S. Ansari, TrAC, Trends Anal. Chem., 2017, 90, 89–106 CrossRef CAS.
- K. Aguilar-Arteaga, J. A. Rodriguez and E. Barrado, Anal. Chim. Acta, 2010, 674, 157–165 CrossRef CAS.
- J.-M. Liu, S.-Y. Wei, H.-L. Liu, G.-Z. Fang and S. Wang, Polymers, 2017, 9, 546 CrossRef.
- Y. Zhang, R. Liu, Y. Hu and G. Li, Anal. Chem., 2009, 81, 967–976 CrossRef CAS PubMed.
- M. Safdarian, Z. Ramezani and A. A. Ghadiri, J. Chromatogr. A, 2016, 1455, 28–36 CrossRef CAS.
- M. Safdarian and Z. Ramezani, Colloids Surf., A, 2018, 541, 97–107 CrossRef CAS.
- F. Bai, X. Yang and W. Huang, Macromolecules, 2004, 37, 9746–9752 CrossRef CAS.
- X. Wang, L. Wang, X. He, Y. Zhang and L. Chen, Talanta, 2009, 78, 327–332 CrossRef CAS.
- M. Zhang, X. Zhang, X. He, L. Chen and Y. Zhang, Nanoscale, 2012, 4, 3141–3147 RSC.
- J. Cao, X. Zhang, X. He, L. Chen and Y. Zhang, J. Mater. Chem. B, 2013, 1, 3625–3632 RSC.
- X. Zhang, X. He, L. Chen and Y. Zhang, J. Mater. Chem. B, 2014, 2, 3254–3262 RSC.
- Z. Zhang, W. Tan, Y. Hu, G. Li and S. Zan, Analyst, 2012, 137, 968–977 RSC.
- S. Shi and J.-Y. Hwan, J. Miner. Mater. Charact. Eng., 2003, 2, 101–110 Search PubMed.
- G. Taguchi, S. Chowdhury and Y. Wu, Taguchi's Quality Engineering Handbook, John Wiley & Sons, Inc., Hoboken, New Jersey, USA/Canada, 2005 Search PubMed.
- E. Zeinali, Design of experiments with Taguchi method using Qualitek software, Petrochemical Research and Technology company, Tehran, Iran, 2008 Search PubMed.
- J. T. Oberlerchner, C. Fuchs, H. Grausgruber, A. Potthast and S. Böhmdorfer, J. Chromatogr. A, 2018, 1533, 193–198 CrossRef.
- Á. M. Móricz, P. G. Ott, I. Yüce, A. Darcsi, S. Béni and G. E. Morlock, J. Chromatogr. A, 2018, 1533, 213–220 CrossRef.
- S. Krüger, L. Winheim and G. E. Morlock, Food Chem., 2018, 239, 1182–1191 CrossRef.
- S. K. Kadam, V. V. Chandanshive, N. R. Rane, S. M. Patil, A. R. Gholave, R. V. Khandare, A. R. Bhosale, B. H. Jeon and S. P. Govindwar, Environ. Res., 2018, 160, 1–11 CrossRef CAS.
- V. Hynstova, D. Sterbova, B. Klejdus, J. Hedbavny, D. Huska and V. Adam, J. Pharm. Biomed. Anal., 2018, 148, 108–118 CrossRef CAS.
- E. Guzelmeric, P. Ristivojević, J. Trifković, T. Dastan, O. Yilmaz, O. Cengiz and E. Yesilada, LWT--Food Sci. Technol., 2018, 87, 23–32 CrossRef CAS.
- X. Zhou, E. Wen and C. Liu, J. Planar Chromatogr.--Mod. TLC, 2017, 30, 205–210 CrossRef CAS.
- N. Smyrska-Wieleba, K. K. Wojtanowski and T. Mroczek, Phytochem. Lett., 2017, 20, 339–349 CrossRef CAS.
- N. A. Sheikh and T. R. Desai, Int. J. Green Pharm., 2017, 11, S285–S291 Search PubMed.
- C. Oellig, J. Schunck and W. Schwack, J. Chromatogr. A, 2018, 1533, 208–212 CrossRef CAS PubMed.
- R. Ł. Gwarda and T. H. Dzido, J. Chromatogr. A, 2018, 1534, 179–187 CrossRef CAS PubMed.
- L. Stütz, S. C. Weiss, W. Schulz, W. Schwack and R. Winzenbacher, J. Chromatogr. A, 2017, 1524, 273–282 CrossRef.
- C. Oellig, J. Chromatogr. A, 2017, 1507, 124–131 CrossRef CAS.
- L. Morschheuser, K. Mink, R. Horst, C. Kallinich and S. Rohn, J. Chromatogr. A, 2017, 1526, 157–166 CrossRef CAS.
- A. Liu, Z. Shen, Y. Tian, R. Shi, Y. Liu, Z. Zhao and M. Xian, J. Chromatogr. A, 2017, 1526, 151–156 CrossRef CAS.
- E. Kranjc, A. Albreht, I. Vovk and V. Glavnik, J. Chromatogr. A, 2017, 1526, 137–150 CrossRef CAS PubMed.
- S. Agatonovic-Kustrin and D. W. Morton, J. Chromatogr. A, 2017, 1530, 197–203 CrossRef CAS PubMed.
- S. Agatonovic-Kustrin, D. W. Morton and P. Ristivojević, J. Chromatogr. A, 2016, 1468, 228–235 CrossRef CAS PubMed.
- J. Biller, L. Morschheuser, M. Riedner and S. Rohn, J. Chromatogr. A, 2015, 1415, 146–154 CrossRef CAS PubMed.
- Z. Zhang, W. Tan, Y. Hu and G. Li, J. Chromatogr. A, 2011, 1218, 4275–4283 CrossRef CAS PubMed.
- M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128–141 RSC.
- L. Zhu, C. Li, J. Wang, H. Zhang, J. Zhang, Y. Shen, C. Li, C. Wang and A. Xie, Appl. Surf. Sci., 2012, 258, 6326–6330 CrossRef CAS.
- Z. Ramezani, M. Shekarriz, A. A. Behfar and S. Kiamarzi, J. Braz. Chem. Soc., 2017, 28, 2172–2179 CAS.
- S. Kazemi, A. A. Sarabi and M. Abdouss, Korean J. Chem. Eng., 2016, 33, 3289–3297 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj05062g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |