
Fe2P4O12–carbon composite as a highly stable electrode material for electrochemical capacitors†
Roby
Soni
ab and
Sreekumar
Kurungot
 *ab
*ab
aPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Pune-411008, India. E-mail: k.sreekumar@ncl.res.in
bAcademy of Scientific and Innovative Research, New Delhi-110001, India
First published on 19th November 2018
Abstract
Supercapacitors are important energy storage devices for high power applications. Carbon materials, metal oxides and conducting polymers have been known to show capacitive charge storage but transition metal phosphates are not as well-known as electrode materials in supercapacitors. In this study, an iron cyclotetraphosphate (Fe2P4O12)–carbon composite has been synthesized and demonstrated as an electrode material for supercapacitors. The Fe2P4O12–carbon composite has been thoroughly characterized by X-ray photoelectron spectroscopy, Raman spectroscopy, X-ray diffraction, electron microscopy, etc. The composite shows predominant capacitive behaviour only in acidic medium; its charge storage properties are relatively poor in alkaline medium, indicating the important role of H+ in the charge storage mechanism. The composite shows a capacitance of 251 F g−1 at a current density of 1 A g−1. The composite is highly stable in 0.5 M H2SO4 and does not show any apparent capacitance loss after 9000 charge–discharge cycles recorded at a current density of 5 A g−1.
Introduction
Supercapacitors are playing an increasingly important role as energy storage devices in the realm of green and clean energy technologies owing to their features of high power density, long cycle life, environmental benignity and significant energy density.1 Various carbon morphologies such as graphene,2 carbon nanotubes,3 and carbon nanofibers,4 metal oxides such as manganese oxide (MnO2),5 ruthenium oxide (RuO2),6 and iron oxide (Fe2O3),7 conducting polymers such as polyethylene dioxythiophene,8 polyaniline,9 and polypyrrole,10 and a few metal phosphates such as manganese phosphates11 have been demonstrated as highly efficient materials for supercapacitors. Among the metal phosphates, manganese phosphate has been shown to exhibit capacitive behaviour11 when synthesized by the chemical precipitation method, but the capacitance performance was inferior when compared to well-known capacitive materials. Transition metal oxides have been widely reported as materials for energy storage in batteries and supercapacitors. Among the transition metals, iron is an earth-abundant metal that is environmentally benign, non-toxic, and inexpensive,12 and therefore it possesses high potential for commercial applications. Iron shows multiple valance states, Fe0, Fe2+, and Fe3+, and it also exhibits rich redox chemistry (Fe2+/Fe3+, Fe0/Fe2+, and Fe0/Fe3+).13 Due to the aforementioned properties, iron and its compounds have great potential for application in charge storage devices. Oxides of iron such as FeO,14 Fe2O3,15 and Fe3O416 and their composites have been demonstrated as capacitive materials in supercapacitors. For instance, a multi-walled carbon nanotube–Fe3O4 composite electrode was synthesized by Park and Kim; it exhibited a capacitance of 165 F g−1 in 1 M Na2SO4 electrolyte.17 Moreover, ordered Fe2O3 nanotubes prepared by the sacrificial template-accelerated hydrolysis treatment method showed a capacitance of 184 mF cm−2 with good rate capability.18 However, poor electronic conductivity and slow ion diffusion are the pertinent issues that hinder the application of iron oxides in supercapacitors. Till now, only oxides of iron have been used as electrode materials in supercapacitors; whereas although phosphates of iron have been used in lithium ion batteries as cathode materials, they have not been used in supercapacitors. Phosphates of iron possess certain desirable properties as they are less expensive, easy to synthesise and non-toxic; also, they possess a high theoretical capacity.19Among the phosphates of iron, iron cyclotetraphosphate or iron tetrametaphosphate (Fe2P4O12) belongs to the group of inorganic compounds of the cyclotetraphosphate phase of M2P4O12, where M is the transition metal ion.20 This material has been extensively studied for its magnetic properties and is used in magnetic recording.20 It also has prospective applications in ferroelectrics, lithium ion batteries, ferrofluids, waste water purification, etc. In addition, it is an environmentally friendly and non-toxic phosphate that is used as a food additive. From the electrochemical point of view, iron phosphate has a redox centre, Fe2+, which is expected to show the redox inter-conversion of Fe2+ to Fe3+. The redox centre Fe2+ present in Fe2P4O12 can show a rapid transition to the Fe3+ state on the application of a potential. Thus, the amount of charge involved in the redox reaction of Fe2P4O12 is expected to be potential-dependent, and this can be used for facilitating charge storage in supercapacitors.
In this work, intensive electrochemical tests are performed to understand the capacitive behaviour at different pH values and in different electrolytes. Also, full cell testing has been carried out in aqueous electrolyte using 0.5 M H2SO4 and in the solid-state configuration using PVA–H2SO4 gel electrolyte. The Fe2P4O12–carbon composite shows good capacitive behaviour compared with many other iron-based systems; it exhibits high capacitance, good rate capability, and very high stability.
Experimental section
Material
Anhydrous iron chloride (FeCl3), phytic acid, potassium hydroxide, and polyvinyl alcohol (PVA) were purchased from Sigma-Aldrich. Sulfuric acid (H2SO4) was purchased from Thomas Baker. De-ionized (DI) water was used in all the experiments.Characterization
SEM analysis was carried out on a Quanta and Nova SEM 450. XRD analysis was performed using a Rigaku SmartLab diffractometer with Cu Kα radiation (λ = 1.5406 Å) at a scan rate of 2° min−1. XPS spectra were recorded using a Thermo Fisher Scientific Instruments, U.K. instrument at 1.28 × 10−8 mbar pressure of the sample loading chamber with an Al K alfa X-ray source (monochromatic) with 6 mA beam current and 12 kV. The spot size of the X-ray beam on the sample is 400 μm. The surface area and pore size distribution were determined using ultra-pure N2 on a Quantachrome Quadrasorb automatic volumetric instrument. A Bio-logic VMP-3 PG Stat was employed for all the cyclic voltammetry, electrochemical impedance spectroscopy and galvanostatic charge/discharge measurements. Electrochemical data were processed with EC-Lab software V10.44.Synthesis of carbon–iron cyclotetraphosphate (C–Fe2P4O12) composite
In a typical synthesis of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-C–Fe2P4O12 composite, where the ratio represents the molar ratio of FeCl3 and phytic acid, 6.9 millimoles (1.1 g) of iron(III) chloride (FeCl3) and 14 millimoles (9.2 g) of phytic acid were mixed to obtain a solution. The above solution was then stirred for 15 minutes to produce a brown coloured turbid solution of iron phytate. The obtained brown solution was then dried in an oven for 12 h at 80 °C to obtain iron phytate powder. Further, the obtained iron phytate was pyrolyzed at 800 °C for 3 h to obtain the C–Fe2P4O12 composite. The obtained material was then treated with 0.5 M H2SO4 at 80 °C to remove un-reacted metal; it was then washed with deionised water, filtered and dried at 100 °C for 10 hours in an oven. Here, phytic acid acts as a source of carbon as well as phosphorus. For preparing the 1:4-C–Fe2P4O12 composite, the materials used were 6.9 millimoles of FeCl3 and 30 millimoles of phytic acid and subsequent steps of the synthesis procedure were repeated as in the previous case.
:2-C–Fe2P4O12 composite, where the ratio represents the molar ratio of FeCl3 and phytic acid, 6.9 millimoles (1.1 g) of iron(III) chloride (FeCl3) and 14 millimoles (9.2 g) of phytic acid were mixed to obtain a solution. The above solution was then stirred for 15 minutes to produce a brown coloured turbid solution of iron phytate. The obtained brown solution was then dried in an oven for 12 h at 80 °C to obtain iron phytate powder. Further, the obtained iron phytate was pyrolyzed at 800 °C for 3 h to obtain the C–Fe2P4O12 composite. The obtained material was then treated with 0.5 M H2SO4 at 80 °C to remove un-reacted metal; it was then washed with deionised water, filtered and dried at 100 °C for 10 hours in an oven. Here, phytic acid acts as a source of carbon as well as phosphorus. For preparing the 1:4-C–Fe2P4O12 composite, the materials used were 6.9 millimoles of FeCl3 and 30 millimoles of phytic acid and subsequent steps of the synthesis procedure were repeated as in the previous case.
Electrochemical testing of C–Fe2P4O12
For electrochemical characterization, the composite was drop coated on a carbon current collector, having an active area of 1 cm2. C–Fe2P4O12 was dispersed in N-methyl pyrrolidone (NMP) with a concentration of 1 mg/100 μl through sonication for 1 h. 100 μl of the slurry was drop coated on a carbon current collector and was subsequently dried in an oven at a temperature of 150 °C for 6 h. The electrode was then used for electrochemical testing using 0.5 M H2SO4 and 0.1 M KOH as electrolytes. The amount of active material used in the electrochemical characterization was 1 mg cm−2 in all cases.Results and discussion
Fig. 1 shows the schematic diagram of the process used for the preparation of carbon-supported iron cyclotetraphosphate nanoparticles (C–Fe2P4O12). Phytic acid in the synthesis serves as the source of both the phosphorus for the formation of Fe2P4O12 and the phosphorus-doped carbon that helps to enhance the electrical conductivity of the composite.21 First, the crystal structure and phase of the composite were examined by recording the X-ray diffraction (XRD) pattern, which is shown in Fig. 2a. The XRD pattern shows very sharp peaks, which indicate the highly crystalline nature of Fe2P4O12, with the crystal structure matching with ICDD file number 78-2285 that confirms the formation of the Fe2P4O12 phase of iron phosphate. The ICDD database indicates that the iron cyclotetraphosphate formed here crystallizes in the form of a monoclinic crystal, which has four atoms per unit cell.22 It consists of two distinct FeO6 with ring-shaped P4O124− anion tetrahedra. The most intense peak at the 2Θ value of 29.59° represents the (022) plane while the other major reflections at 14.28°, 20.82°, and 27.67° correspond to the (111), (112), and (310) planes of Fe2P4O12 (ICDD 78-2285).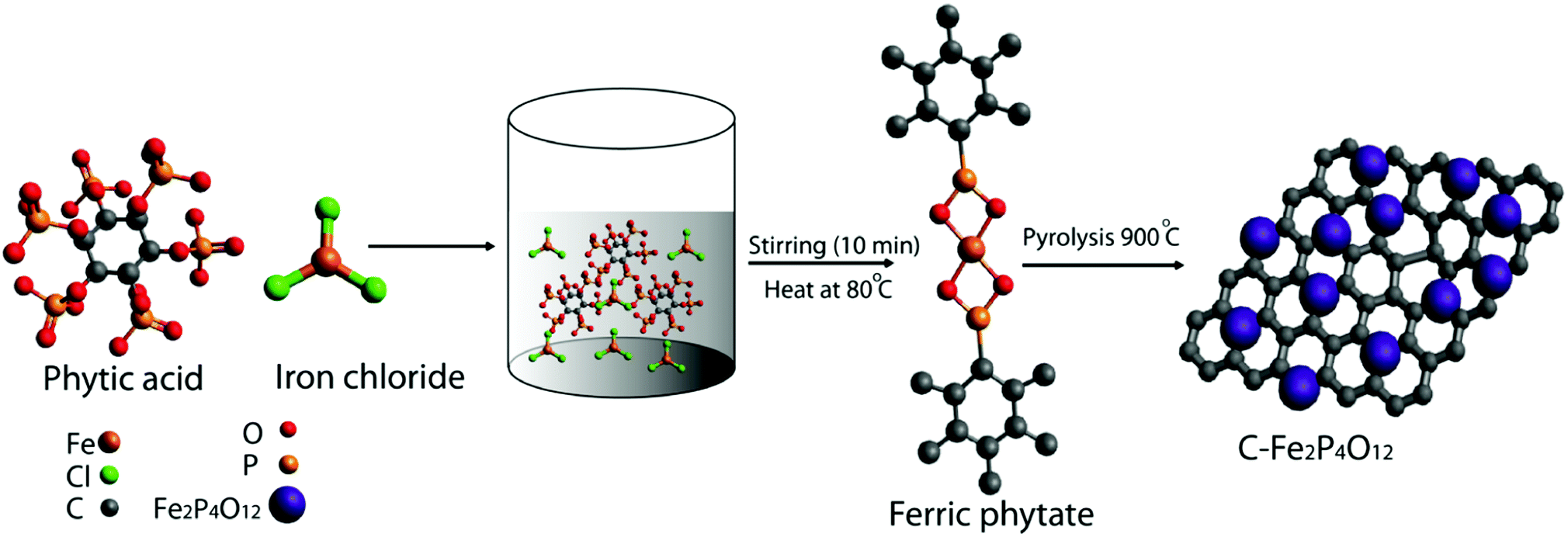 | ||
| Fig. 1 Schematic representation of the methodology developed to prepare carbon supported iron cyclotetraphosphate, Fe2P4O12 (C–Fe2P4O12). | ||
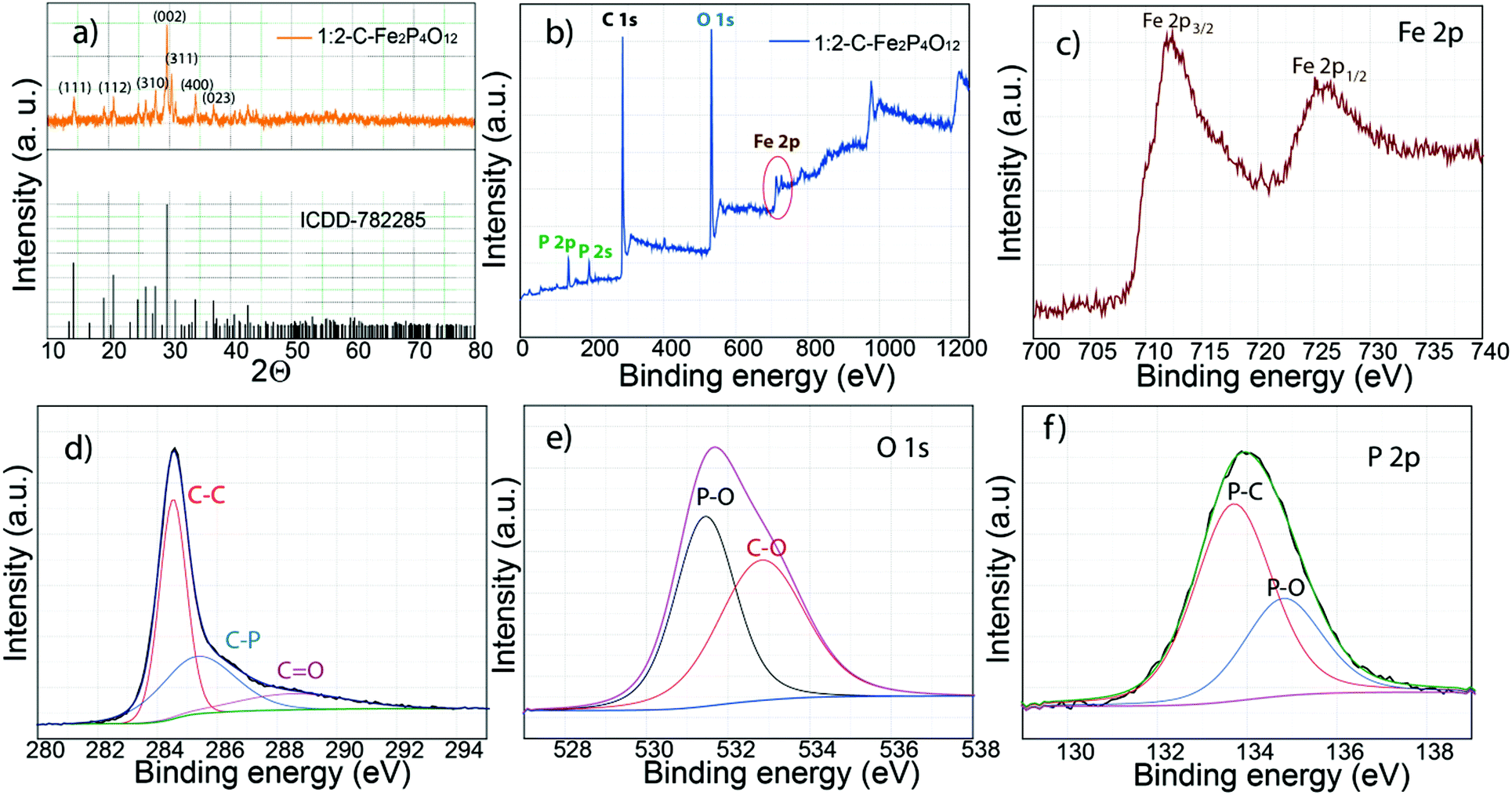 | ||
| Fig. 2 XRD and XPS characterization of 1:2-C–Fe2P4O12: (a) XRD pattern and the corresponding ICDD data, (b) XPS survey spectrum, (c) high resolution XPS Fe 2p spectrum where the 2p peak is split into two components due to spin–orbit coupling, (d) deconvoluted C 1s spectra, which are resolved into three components, (e) deconvoluted O 1s spectra, (f) resolved P 2p spectra. | ||
The composition and bonding characteristics of C–Fe2P4O12 were analysed through X-ray photoelectron spectroscopy (XPS). The survey spectrum given in Fig. 2b shows the typical XPS peaks of carbon (284.5 eV), iron (712.3 Fe 2p3/2 and 725.6 eV Fe 2p1/2), phosphorus (133.2 eV) and oxygen (531.2 eV).23 The XPS spectra of O, P, and C were deconvoluted to understand the composition and nature of bonding in the composite. The percentage of carbon measured from the survey spectra was 66.93% while that measured for iron, phosphorus and oxygen was 1.12, 7.04, and 24.91%, respectively, of the composite. Fig. 2c represents the XPS spectra of iron; it shows two distinct peaks at 712.3 eV for Fe 2p3/2 and 725.6 eV for Fe 2p1/2 caused by the spin–orbit coupling.24 The binding energies obtained for Fe 2p occur at higher values compared to the usual binding energies of iron in its oxides.24 This can be explained by the coordination of Fe with six oxygen atoms in the FeO6 moiety.20 The deconvoluted carbon 1s spectrum is shown in Fig. 2d; it could be resolved into three regions with binding energies of 284.5, 285.3 and 288.5 eV corresponding to the C–C bonds, C–P bonds, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively. The quantification of the peaks reveals 49.33% C–C bonds, 35.93% C–P bonds and 14.74% CO bonds comprising the total carbon present in the sample. The above results show that the carbon obtained is heavily doped with phosphorus, which is advantageous in terms of the capacitive performance of the composite.25
O bonds, respectively. The quantification of the peaks reveals 49.33% C–C bonds, 35.93% C–P bonds and 14.74% CO bonds comprising the total carbon present in the sample. The above results show that the carbon obtained is heavily doped with phosphorus, which is advantageous in terms of the capacitive performance of the composite.25
In addition, the carbon surface is highly oxidized due to the presence of oxygen functionalities, which could have a negative effect on the conductivity of the composite. The above results indicate that the low conductivity of the carbon derived from phytic acid is due to the oxidized surface and low graphitization, which result from the non-aromatic nature of phytic acid. Deconvolution of oxygen spectra can give information about the structure and the binding states of Fe2P4O12 with the underlying carbon. The oxygen 1s spectra given in Fig. 2e show two binding states, which can be attributed to the P–O and C–O bonds with corresponding binding energies of 531.4 and 532.8 eV, respectively.23 The P–O binding energy represents the P–O linkage present in the P4O124− anion, which has been discussed in the previous section. Furthermore, the deconvoluted P 2p spectra shown in Fig. 2f show two regions, which represent the P–C bond at a binding energy of 133.7 eV and the P–O bond at a binding energy of 134.8 eV. The binding energy difference between the two peaks is 1.1 eV, which is the same as previously reported values.26 In the XPS spectra, the Fe–P linkage is not observed, indicating that iron phosphide is not formed in the composite, which corroborates the XRD results. From the XPS results, it is highly likely that Fe2P4O12 particles interact with carbon through the C–P linkage, as the high percentage of C–P bond indicates.
Raman spectroscopy was carried out to detect the P–O vibration modes of Fe2P4O12 in the 1:2 composite and in Fe2P4O12 after the removal of carbon. Fig. 3a compares the Raman spectra of 1:2-C–Fe2P4O12 and Fe2P4O12. 1:2-C–Fe2P4O12 shows the ID peak that originates from the various defects present in the carbon honeycomb lattice at the vibrational frequency of 1325 cm−1, while the IG peak that signifies the degree of graphitization in carbon appears at the frequency of 1600 cm−1. For pristine carbon, the IG peak appears at 1580 cm−1; however, a shift of 20 cm−1 is observed in the present case, which can be attributed to the doping of phosphorus in the carbon lattice.27 The ID/IG ratio calculated from the intensities of the corresponding peaks is 1.3, which indicates a high degree of defects and low graphitization level that have also been observed in the results of the XPS analysis. In the composite, however, vibrational modes of Fe2P4O12 are not observed. A plausible reason for this may be the coverage of the Fe2P4O12 particles by the carbon that blocks the access of the laser to the former as Raman spectroscopy is only able to analyse the surface. However, after the removal of carbon, the vibrational modes corresponding to P–O become conspicuous, as can be seen in Fig. 3a. As the P–O bond in PO22− is stronger than the P–O–P bond, the P–O vibration frequency is expected to occur at higher frequencies. The frequencies at 1388, 1327, and 1264 cm−1 can be ascribed to the asymmetric stretching of the P–O bond while the frequencies at 1076 and 1111 cm−1 are ascribed to the symmetric stretching of the P–O bond.28 Similarly, the lower vibrational modes at 709 and 1010 cm−1 are due to the symmetric and asymmetric stretching, respectively, of the P–O–P bond.28
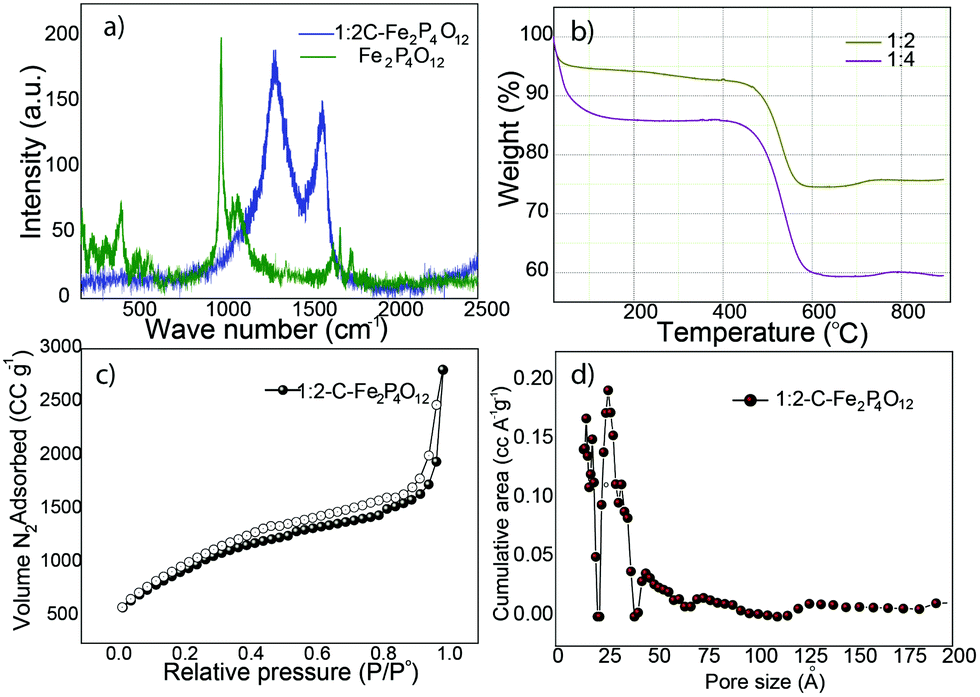 | ||
| Fig. 3 (a) Raman spectra of 1:2-C–Fe2P4O12 and Fe2P4O12 after the removal of carbon by calcination, (b) TGA plots that compare the weight loss profiles of the 1:2 and 1:4-C–Fe2P4O12 composites, (c) N2 gas adsorption isotherm for 1:2-C–Fe2P4O12 and (d) pore size distribution profile of 1:2-C–Fe2P4O12. | ||
The quantitative measurements of the carbon and Fe2P4O12 content in the different composites was carried out by thermogravimetric analysis (TGA) in an oxygen atmosphere at a heating rate of 10 °C min−1. The TGA plot shown in Fig. 3b compares the results obtained for the 1:2 and 1:4 composites. Both profiles show an initial weight loss up to the temperature of 100 °C, which can be ascribed to the loss of the moisture initially present in the sample. The weight remains unchanged till 450 °C, beyond which the weight decreases continually due to the removal of carbon from the system. The weight loss continues till 580 °C, after which it becomes constant, indicating the complete loss of carbon; at this stage, all that remains is Fe2P4O12. The percentage of Fe2P4O12 obtained from the TGA analysis is 82 for the 1:2 composite. The 1:4 composite, as observed from the TGA data, contains only 60% Fe2P4O12, indicating clearly that the increase in phytic acid content in the reaction mixture leads to increased carbon content in the composite.
Surface area and pore size distribution are important parameters to judge the suitability of any material for application in capacitors. The surface area of 1:2-C–Fe2P4O12 was determined through N2 gas adsorption measurements. The surface area measured for 1:2-C–Fe2P4O12 by the Barrett–Joyner–Halenda (BJH) method is 1043 m2 g−1, which is very high compared to many metal oxide–carbon composites29,30 and comparable or even higher than that of many other carbon morphologies.31,32 The adsorption isotherm shown in Fig. 3c indicates type IV behaviour and it displays the features of monolayer–multilayer adsorption. The adsorption isotherm features also show complete pore filling, which indicates the presence of a highly accessible surface for gas adsorption. The pore size distribution profile given in Fig. 3d shows most pores to be in the range of 1.3–2.6 nm, and so, it has both microporous and mesoporous structures with an average pore size of 1.32 nm. Such a combination of micro and mesoporosity is advantageous for charge storage applications. Large surface area and the micro–mesoporous size distribution lead to a high pore volume of 3.0 cc g−1. These surface properties are expected to have a positive effect on the charge storage properties of 1:2-C–Fe2P4O12.
Field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) were carried out to analyse the morphology of 1:2-C–Fe2P4O12 and Fe2P4O12. The FESEM image of C–Fe2P4O12 is shown in Fig. 4a and the SEM images are shown in Fig. S1 (ESI†); the image shows an interconnected fibrous network of carbon along with large and small pores that are uniformly distributed over the entire surface. Moreover, no distinct particles can be seen in the SEM image, which implies close integration of Fe2P4O12 and the carbon. Such a porous structure is expected to allow for the easy movement of electrolyte ions during the charge–discharge cycle. In order to examine the morphology of Fe2P4O12 alone, the composite was calcined at 600 °C in a muffle furnace for 4 h to remove the carbon from the composite. The SEM images of the Fe2P4O12 particles obtained after calcination are given in Fig. 4b and Fig. S2 (ESI†); spindle-shaped large particles of iron cyclotetraphosphate can be observed in the images. These large particles may have been formed during the calcination process; as the carbon is removed from the system, it is likely that Fe2P4O12 particles that were embedded in the carbon start to coalesce together, forming spindle-shaped entities. To obtain detailed information about the morphology and structure, TEM analysis of 1:2-C–Fe2P4O12 was carried out. The TEM image given in Fig. 4c for 1:2-C–Fe2P4O12 shows a uniform distribution of Fe2P4O12 particles, and aggregates were not observed even in the low-magnification, large-area images of the composite. Such an association is important for facile charge-transfer during the electrochemical process. A high resolution TEM image of 1:2-C–Fe2P4O12 is shown in Fig. 4d, in which a spherical particle of size 15–20 nm can be seen in close integration with carbon. The selected area electron diffraction (SAED) pattern of the composite is given in Fig. S3 (ESI†), which shows bright spots arranged in the circular pattern characteristic of highly crystalline materials.33 The SAED results corroborate well with the XRD pattern, both the depicting crystalline nature of Fe2P4O12.
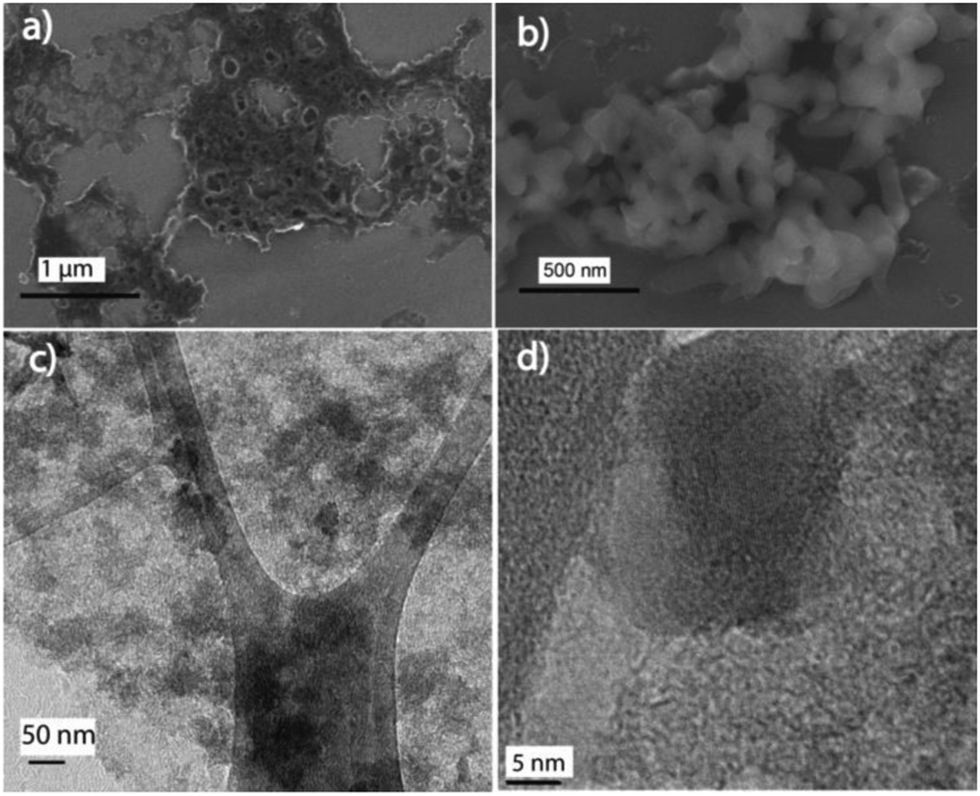 | ||
| Fig. 4 (a) FESEM image of 1:2-C–Fe2P4O12, (b) FESEM image of Fe2P4O12 particles obtained after removal of carbon, (c) TEM image of 1:2-C–Fe2P4O12 depicting the uniform distribution of Fe2P4O12 and (d) high resolution TEM image of 1:2-C–Fe2P4O12 showing an individual Fe2P4O12 particle embedded in carbon. | ||
Electrochemical characterization of the C–Fe2P4O12 composites, Fe2P4O12 and phytic acid-derived carbon was carried out through cyclic voltammetry, galvanostatic charge–discharge, and electrochemical impedance spectroscopy (EIS). The cyclic voltammograms shown in Fig. 5a describe the electrochemical behaviour of 1:2-C–Fe2P4O12, Fe2P4O12 and phytic acid-derived carbon. The CV of 1:2-C–Fe2P4O12 shows distinct redox peaks at 0 and 0.1 V, which can be assigned to the Fe2+/Fe3+ couple. During the redox reaction, Fe oscillates between the +3 and +2 oxidation states resulting in pseudocapacitive charge storage.34 The CV indicates that this material can be utilized as a negative electrode in asymmetric supercapacitors as it has more charge storage in the negative region of the potential window from −0.8 V to 0 V, providing a potential window of 0.8 V in the negative regime. However, the CV of Fe2P4O12 in which the carbon has been removed by the calcination of C–Fe2P4O12 in air at 600 °C covers much less area compared to 1:2-C–Fe2P4O12. The low capacitance of Fe2P4O12 results from its low conductivity, which hinders the charge transfer and current distribution to the current collector. Similarly, the aggregation of particles during the calcination decreases its active surface unlike 1:2-C–Fe2P4O12. The IR spectrum of the phytic acid-derived carbon, given in Fig. S4 (ESI†), shows the presence of carboxylic (1381 cm−1), –OH (2963 cm−1 and 3434 cm−1), and P–O bond stretching (1165 cm−1) functional groups along with the stretching modes of CC (1591 cm−1) and C–H (2923 cm−1).35,36 The CV of the phytic acid-derived carbon shows typical double-layer behaviour along with a feeble redox couple in the same potential range as 1:2-C–Fe2P4O12. The redox couple arises due to the phosphorus and functional groups (as indicated by the IR analysis) present in the phytic acid-derived carbon and induces pseudocapacitance in the carbon.37Fig. 5b shows the CV of 1:2-C–Fe2P4O12 recorded at different scan rates of 10, 20, 50, 100, 200, and 500 mV s−1, and it can be observed that with the increase in the scan rate, the area under the CV also increases accordingly, which is a characteristic feature of capacitive materials.
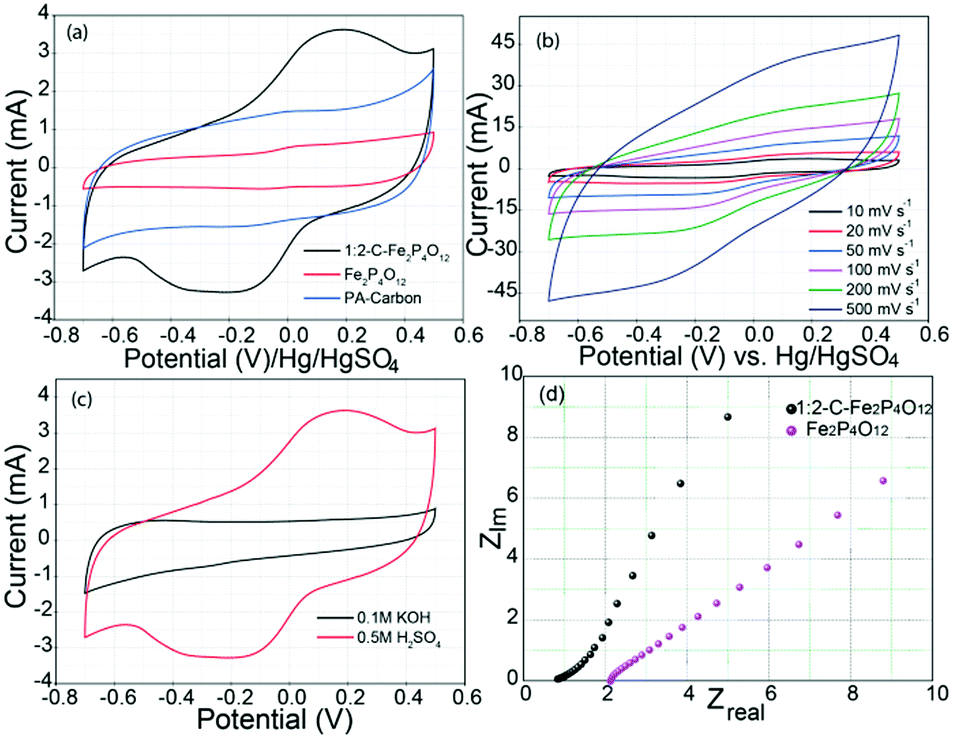 | ||
| Fig. 5 (a) Cyclic voltammograms recorded at a scan rate of 10 mV s−1 comparing electrochemical behaviour of 1:2-C–Fe2P4O12, Fe2P4O12 and phytic acid-derived carbon, (b) CVs recorded at progressively increasing scan rates for 1:2-C–Fe2P4O12 depicting its rate behaviour, (c) CV profiles of 1:2-C–Fe2P4O12 recorded at a scan rate of 10 mV s−1 in acidic and basic media and (d) Nyquist plots showing the ESR and frequency response of 1:2-C–Fe2P4O12 and Fe2P4O12. | ||
To assess the role of H+ ion in the charge storage behaviour of Fe2P4O12, cyclic voltammograms were recorded in acidic and basic solutions. CV profiles recorded in 0.5 M H2SO4 (pH 0.301) and 0.1 M KOH (pH 13) are given in Fig. 5c. The H2SO4 used here is highly acidic, containing 1 M H+ ions while the KOH used contains only 10−13 M H+ ions. As can be seen, capacitance is very high in the acidic medium compared to the basic medium. Such a huge difference in the capacitance at the two extreme values of pH gives an indication of the involvement of H+ ions in the redox reaction of C–Fe2P4O12. The probable mechanism for charge storage in this case is the surface adsorption of H+ ions, as with ruthenium oxide (RuO2).38 Furthermore, the amount of phytic acid was varied in the composite to assess the effect of carbon content. A C–Fe2P4O12 composite was prepared in 1:4 composition, which contains 60% Fe2P4O12 compared to 82% in the 1:2 composite. 1:4-C–Fe2P4O12 shows less capacitance (100 F g−1) than its 1:2 counterpart, as can be seen from the corresponding CV profile shown in Fig. S5 (ESI†). The decreased capacitance can be explained by the low amount of Fe2P4O12 in the composite, which results in a low degree of redox reaction. Also, the carbon obtained from the phytic acid has a low conductivity due to its reduced graphitization caused by the angular structure of the phytic acid that restricts the condensation into sheets associated with an aromatic structure, as observed in the XPS analysis.39
The equivalent series resistance (ESR) and the frequency response of 1:2-C–Fe2P4O12 were also studied through electrochemical impedance spectroscopy. Nyquist plots of 1:2-C–Fe2P4O12 and Fe2P4O12 are given in Fig. 5d. The ESR measured for 1:2-C–Fe2P4O12 is 0.82 Ω; however, after the removal of carbon from the system, the ESR increases to 2.14 Ω and also the frequency behaviour deteriorated compared to that of 1:2-C–Fe2P4O12, as evidenced by the leaning of the capacitive line towards the abscissa.40 Also, a semicircle representative of the charge-transfer resistance is not observed in either case. This can be explained by the fast redox reaction, which is not limited by the kinetics of the Fe2+/Fe3+ redox couple. For the capacitance measurement and to study the charge discharge behaviour, galvanostatic charge–discharge (GCD) curves were recorded for 1:2-C–Fe2P4O12 and Fe2P4O12 at a current density of 1 A g−1, as shown in Fig. 6a. The GCD curves presented here show typical pseudocapacitive behaviour; the curves show a small plateau in the potential region of 0 to −0.2 V. The plateau represents the redox reaction of Fe2P4O12. The capacitance measured at a current density of 1 A g−1 for 1:2-C–Fe2P4O12 is 251 F g−1, which is quite a significant value that is higher than many of the reported capacitance values of Fe2O3 composites.41’42 GCD at different current densities was also carried out to understand the rate behaviour and capacitance retention at a high power output, with the corresponding plots shown in Fig. 6b. The change in capacitance observed at increasing current densities was also calculated, and it is plotted as a function of current density in Fig. 6c. At a current density of 5 A g−1, a capacitance retention of 39% could be obtained when taking the capacitance at 1 A g−1 as a reference. This modest rate capability can be attributed to the restricted diffusion of the electrolyte ions at higher current densities and to the low conductivity of 1:2-C–Fe2P4O12. However, the capacitance retention is superior for 1:4-C–Fe2P4O12 compared to its 1:2 counterpart, as evident from the data shown in Fig. S6 (ESI†). At a current density of 5 A g−1, it could retain 70% of the capacitance obtained at 0.5 A g−1. This superior retention can be related to the higher carbon content, which provides better conductivity and facilitates easier ion diffusion compared to the 1:2 composite, which has a low carbon content.
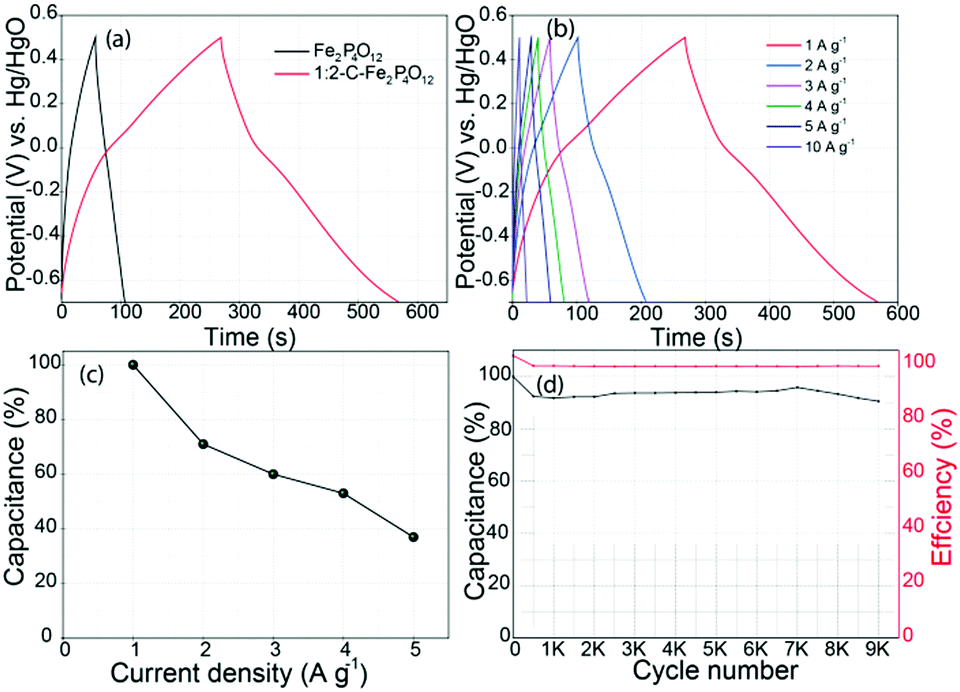 | ||
| Fig. 6 (a) The GCD curves recorded at a current density of A g−1, (b) the GCD curves recorded at different current densities for 1:2-C–Fe2P4O12, (c) the plot depicting variation in the capacitance of 1:2-C–Fe2P4O12 at different current densities and (d) plot showing the durability and coulombic efficiency of 1:2-C–Fe2P4O12. | ||
To assess the cycling performance of the composite, a durability test was conducted at a current density of 5 A g−1, which is shown in Fig. 6d; it shows an initial 10% loss of capacitance after 500 cycles. However, after the first 500 cycles, the capacitance does not change appreciably even after 9000 cycles. Such a high stability for a pseudocapacitive material is highly significant and can be attributed to the highly reversible redox reaction. It also maintains 100% coulombic efficiency throughout the durability testing, signifying high efficiency and minimum energy loss over long periods of application. The cycling stability of 1:2-C–Fe2P4O12 is much better than that of iron oxide composites whose reduced stability during cycling is a pertinent issue that inhibits their application in supercapacitors.
A supercapacitor test was also conducted using a symmetrical full cell configuration with 1 mg loading of 1:2-C–Fe2P4O12 on the negative and positive electrodes in 0.5 M H2SO4 as electrolyte. Fig. 7a represents the CV recorded at a scan rate of 10 mV s−1 in which the curve shows a near-rectangular shape with slight deviation at the inception of the discharge cycle due to the internal resistance of the device.43 However, the CV feature is highly symmetrical, exhibiting high reversibility, which will help it to achieve high durability. Furthermore, the CVs recorded at increasing scan rates of 5, 10, 20, 50, 100, 200, and 500 mV s−1 are shown in Fig. 7b. At a scan rate of 200 mV s−1, the CV maintains its rectangular shape, which demonstrated good rate behaviour up to a scan rate of 200 mV s−1. However, at 500 mV s−1, it shows considerable deviation from the rectangular behaviour. Such behaviour can be attributed to the limited redox reaction and reduced electrolyte accessibility to the active surface at high scan rates.43 The GCD curves are shown in Fig. 7c; the GCD curves show a triangular waveform typical of a symmetric supercapacitor device. Capacitance calculated from the charge–discharge curve at a current density of 0.5 A g−1 is 123 F g−1. Ideally, the value should be half of the capacitance value obtained for the single electrode.44 However, the low capacitance obtained here is caused by the high equivalent series resistance of 7 Ω, as shown in the Nyquist plot in Fig. 7d. The device exhibits an energy density of 6.2 W h kg−1 at a power output of 151 W kg−1 whereas the maximum power delivered is 25 kW kg−1 (V2/4R).
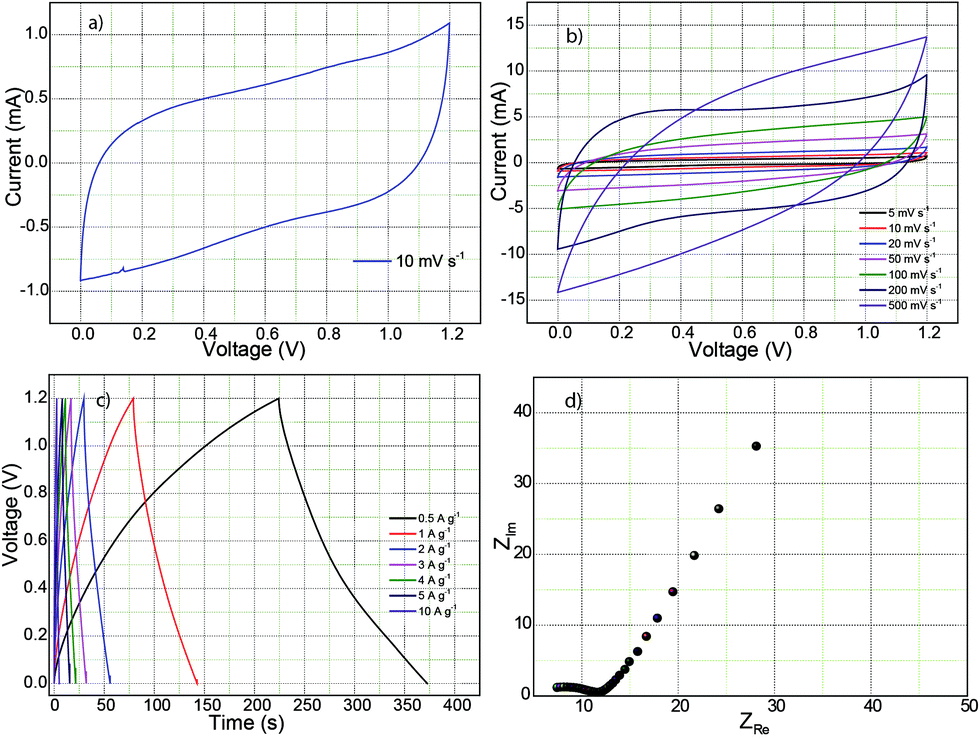 | ||
| Fig. 7 Electrochemical characterization carried out for full cell in 0.5 M H2SO4: (a) cyclic voltammogram recorded at a scan rate of 10 mV s−1; (b) CVs measured at different scan rates; (c) the GCD curves recorded at different current densities; (d) Nyquist plot depicting the frequency behavior. | ||
The performance of 1:2-C–Fe2P4O12 was also tested in a solid-state device and compared to the performance in liquid electrolyte. Full cell testing was done by fabricating symmetric cells containing 1:2-C–Fe2P4O12 composite on both negative and positive electrodes with 1 mg loading on the respective electrodes. Solid supercapacitor prototypes were fabricated using 10 wt% PVA–H2SO4 gel electrolyte. Fig. S7a (ESI†) shows the CV behaviour of 1:2-C–Fe2P4O12 in the liquid- and solid-state devices. The CV for the solid-state device shows a steep slope at the beginning of the discharge cycle, caused by the resistance experienced by the device due to restricted and slow ion movement in the solid-state configuration. Also, the area under the CV is substantially decreased compared to its liquid counterpart due to the low impregnation of the available active surface. The capacitance was measured by recording the GCD curves at a current density of 0.5 A g−1 and compared with its liquid counterpart, as shown in Fig. S7b (ESI†). The capacitance obtained in the solid-state configuration is 72 F g−1, which is 58% of the capacitance in the liquid system. The capacitance behaviour in the solid-state configuration can be improved by enhancing the electrolyte penetration and impregnation so that it can form a large electrode–electrolyte interface.
Conclusions
This report documents the application of an iron cyclotetraphosphate–carbon composite as a highly stable electrode material for an electrochemical capacitor. The 1:2-C–Fe2P4O12 composite is thoroughly characterized through X-ray photoelectron spectroscopy, Raman spectroscopy, electron microscopy, etc. Fe2P4O12 crystallized in the monoclinic crystal system, as confirmed by the XRD pattern. Raman spectra and XPS measurements indicated the phenomenon of phosphorus doping in the carbon present in the composite, which also contributes to the pseudocapacitance of the composite. In addition, the electrochemical measurements in acidic and basic media pointed to the role of H+ ions in the charge storage mechanism of Fe2P4O12. 1:2-C–Fe2P4O12 shows a high capacitance of 251 F g−1 with very high cycling stability that does not exhibit any significant degradation after 9000 cycles at a current density of 5 A g−1. The material also possesses a high energy density of 6.2 W h kg−1 with a power output of 151 W kg−1. The performance of Fe2P4O12 can be further optimized by manipulating the shape and size, and by making composites with carbon or other materials. The material needs significant improvement in terms of rate capability, which can be realized by utilizing different carbon morphologies that have high conductivity. Owing to its eco-friendly and non-toxic nature coupled with the very high stability, C–Fe2P4O12 has noteworthy prospects as a green material for energy storage applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their sincere gratitude to the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for the funding to carry out this project (TLP003526). RS thanks the CSIR for the research fellowship.References
- H. Ji, X. Zhao, Z. Qiao, J. Jung, Y. Zhu, Y. Lu, L. L. Zhang, A. H. MacDonald and R. S. Ruoff, Nat. Commun., 2014, 5, 3317 CrossRef PubMed.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CrossRef CAS PubMed.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- C. Kim and K. S. Yang, Appl. Phys. Lett., 2003, 83, 1216–1218 CrossRef CAS.
- R. Soni, A. Raveendran and S. Kurungot, Nanoscale, 2017, 9, 3593–3600 RSC.
- Y. Zhu, X. Ji, C. Pan, Q. Sun, W. Song, L. Fang, Q. Chen and C. E. Banks, Energy Environ. Sci., 2013, 6, 3665 RSC.
- H. Xia, C. Hong, B. Li, B. Zhao, Z. Lin, M. Zheng, S. V. Savilov and S. M. Aldoshin, Adv. Funct. Mater., 2015, 25, 627–635 CrossRef CAS.
- K. Lota, V. Khomenko and E. Frackowiak, J. Phys. Chem. Solids, 2004, 65, 295–301 CrossRef CAS.
- B. Anothumakkool, A. T. A. Torris, S. N. Bhange, S. M. Unni, M. V. Badiger and S. Kurungot, ACS Appl. Mater. Interfaces, 2013, 5, 13397–13404 CrossRef CAS PubMed.
- P. A. Basnayaka, M. K. Ram, L. Stefanakos and A. Kumar, Graphene, 2013, 02, 81–87 CrossRef CAS.
- Y.-H. Dai, L.-B. Kong, K. Yan, M. Shi, Y.-C. Luo and L. Kang, Ionics, 2016, 22, 1461–1469 CrossRef CAS.
- Y. Lin, X. Wang, G. Qian and J. J. Watkins, Chem. Mater., 2014, 26, 2128–2137 CrossRef CAS.
- Y. Zeng, M. Yu, Y. Meng, P. Fang, X. Lu and Y. Tong, Adv. Energy Mater., 2016, 6, 1601053 CrossRef.
- A. Maitra, A. K. Das, S. K. Karan, S. Paria, R. Bera and B. B. Khatua, Ind. Eng. Chem. Res., 2017, 56, 2444–2457 CrossRef CAS.
- Y. Li, Q. Li, L. Cao, X. Cui, Y. Yang, P. Xiao and Y. Zhang, Electrochim. Acta, 2015, 178, 171–178 CrossRef CAS.
- F. Yan, J. Ding, Y. Liu, Z. Wang, Q. Cai and J. Zhang, Synth. Met., 2015, 209, 473–479 CrossRef CAS.
- Y.-H. Kim and S.-J. Park, Curr. Appl. Phys., 2011, 11, 462–466 CrossRef.
- P. Yang, Y. Ding, Z. Lin, Z. Chen, Y. Li, P. Qiang, M. Ebrahimi, W. Mai, C. P. Wong and Z. L. Wang, Nano Lett., 2014, 14, 731–736 CrossRef CAS PubMed.
- W. Wang, P. Gao, S. Zhang and J. Zhang, J. Alloys Compd., 2017, 692, 908–914 CrossRef CAS.
- P. Rerksompus, K. Sarasamak, B. Boonchom and P. Thanomngam, Ferroelectrics, 2015, 482, 113–120 CrossRef CAS.
- Y. Wen, B. Wang, C. Huang, L. Wang and D. Hulicova-Jurcakova, Chemistry, 2015, 21, 80–85 CrossRef CAS PubMed.
- T. E. Anders, G. Nord and P.-E. Werner, Z. Kristallogr., 1990, 192, 83–90 Search PubMed.
- Y. Li, S. Li, Y. Wang, J. Wang, H. Liu, X. Liu, L. Wang, X. Liu, W. Xue and N. Ma, Phys. Chem. Chem. Phys., 2017, 19, 11631–11638 RSC.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- V. Thirumal, A. Pandurangan, R. Jayavel, K. S. Venkatesh, N. S. Palani, R. Ragavan and R. Ilangovan, J. Mater. Sci.: Mater. Electron., 2015, 26, 6319–6328 CrossRef CAS.
- D.-S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J.-S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S. K. Saha, U. V. Waghmare, K. S. Novoselov, H. R. Krishnamurthy, A. K. Geim, A. C. Ferrari and A. K. Sood, Nat. Nanotechnol., 2008, 3, 210–215 CrossRef CAS PubMed.
- V. U. N. a. G. A. K. Vishwanathan, Proc. - Indian Acad. Sci., Chem. Sci., 1985, 95, 463–469 Search PubMed.
- H. W. Chang, Y. R. Lu, J. L. Chen, C. L. Chen, J. F. Lee, J. M. Chen, Y. C. Tsai, C. M. Chang, P. H. Yeh, W. C. Chou, Y. H. Liou and C. L. Dong, Nanoscale, 2015, 7, 1725–1735 RSC.
- W. C. Y. He, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7, 174–182 CrossRef PubMed.
- M. B. D. Pech, H. Durou1, P. Huang, V. Mochalin, Y. Gogotsi and P.-L. T. a. P. Simon, Nat. Nanotechnol., 2010, 5, 651–654 CrossRef PubMed.
- J. Zhi, W. Zhao, X. Liu, A. Chen, Z. Liu and F. Huang, Adv. Funct. Mater., 2014, 24, 2013–2019 CrossRef CAS.
- W. Zhou and H. F. Greer, Eur. J. Inorg. Chem., 2016, 941–950 CrossRef CAS.
- L. Guan, L. Yu and G. Z. Chen, Electrochim. Acta, 2016, 206, 464–478 CrossRef CAS.
- L. Liu, Q. Li, Z. Wang and Y. Chen, Mater. Technol., 2018, 33, 748–753 CrossRef CAS.
- M. A. Patel, F. Luo, K. Savaram, P. Kucheryavy, Q. Xie, C. Flach, R. Mendelsohn, E. Garfunkel, J. V. Lockard and H. He, Carbon, 2017, 114, 383–392 CrossRef CAS.
- J. Patino, N. Lopez-Salas, M. C. Gutierrez, D. Carriazo, M. L. Ferrer and F. d. Monte, J. Mater. Chem. A, 2016, 4, 1251–1263 RSC.
- Y. Liu, F. Zhou and V. Ozolins, J. Phys. Chem. C, 2012, 116, 1450–1457 CrossRef CAS.
- P. L. Walker and A. Weinstein, Carbon, 1967, 5, 13–17 CrossRef CAS.
- P. L. Taberna, P. Simon and J. F. Fauvarque, J. Electrochem. Soc., 2003, 150, A292 CrossRef CAS.
- D. Wang, Y. Li, Q. Wang and T. Wang, J. Solid State Electrochem., 2012, 16, 2095–2102 CrossRef CAS.
- S. Shivakumara, T. R. Penki and N. Munichandraiah, J. Solid State Electrochem., 2014, 18, 1057–1066 CrossRef CAS.
- W. G. P. B. E. Conway, J. Power Sources, 2002, 105, 169–181 CrossRef.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj04671a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |