
C2/C3 alkynylation of L-ascorbic acid by Sonogashira coupling and efficient access to some potent and highly selective novel anticancer agents†
Santosh Rangnath
Deshmukh
and
Shankar Ramchandra
Thopate
 *
*
Department of Chemistry, Prof. John Barnabas Post Graduate School for Biological Studies, Ahmednagar College, Ahmednagar, Station Road, Ahmednagar, Maharashtra 414001, India. E-mail: srthopate@gmail.com
First published on 14th November 2018
Abstract
Natural products are a valuable source of bioactive compounds for the development of new drugs. Herein, we report an undemanding synthesis of novel C2/C3 alkynyl derivatives of L-ascorbic acid (AsA) via the Sonogashira cross-coupling reaction with moderate to good yield. All new derivatives exhibited superior potency against human MCF7, A549 and HT29 cancer cell lines. Compound 7k showed 2.7, 16.6 and 6.2 fold more potency against MCF7, A549 and HT29 cell lines, respectively, compared to doxorubicin and 643 fold more potency compared to AsA against the MCF7 cancer cell line. Notably, it showed 25 fold less cytotoxicity towards the normal human umbilical vein endothelial cell (HUVEC) line as compared to the doxorubicin anticancer drug. Pleasantly, it showed 333 times more cytotoxicity to human cancer cells compared to human normal cells. The flow cytometry study indicated that compound 4h and 7k arrest A549 cells in the G0/G1 phase via apoptosis. Furthermore, these compounds have high potential to be developed as promising anticancer agents.
Introduction
Cancer is one of the most common causes of death worldwide.1,2 In 2015, an estimated 8.8 million deaths occurred due to cancer, accounting for 15.7% of the overall deaths.3 The number of cancer related deaths is estimated to reach 13.1 million by 2030. Among the different type of cancers, lung cancer is by far the leading cause of cancer related death in both men and women. Each year, more people die due to lung cancer than colon, breast and prostate cancers combined.4 With a low response rate to current chemotherapeutic treatments, there is an immediate need for new anticancer agents that particularly provide more options to lung cancer patients.In drug development, currently, natural products and their analogues play a vital role, especially for treatment of infections and cancer. Structural derivatisation of a range of natural products aims to increase activity and decrease toxicity. Preclinical in vitro screening of these derivatives in a panel of selected human cell lines is a major tool to identify the most promising target for anticancer drug development. In several cases these analogues proved to display superior biological activity than their parent natural products.5,6
L-Ascorbic acid (vitamin C, AsA) is an essential nutrient as well as an important biomolecule widely distributed in aerobic organisms. It is a biological cofactor which plays a vital role in numerous biological pathways, fundamental to cellular function.7 At physiological pH (7.35–7.45) it acts as a strong reducing agent. The ascorbate anion acts as a radical scavenger directly or by recycling other antioxidants such as α-tocopherol or glutathione. In the presence of trace metals, it reacts with O2 to form H2O2 and dehydroascorbic acid via the formation of O2˙− and hence exhibits prooxidant effects.8 For this reason, high doses of intravenous ascorbate have been recently proven to deliver hydrogen peroxide to tissue fluids and to retard tumor growth in numerous animal models.9,10
Although AsA is suitable for prevention and treatment of cancer and other diseases, its susceptibility to thermal and oxidative degradation limits its use in many cases. Considerable efforts have been made in recent years to address the above problem. Several reports have appeared in the literature for synthesis of various AsA derivatives with enhanced stability and activity. For example, 2-O and 3-O-alkylated derivatives of AsA protect lipids of bio-membranes from peroxidation.11–13 5,6-O-modified AsA derivatives exhibit effective anti-tumor activity against various human cancers by inducing apoptosis in tumor cells,14–16 while 2-C-alkyl derivatives show immuno-stimulant activity.17–19
The conjugated electron-rich ene-diol unit in AsA is a rather inimitable, reactive and fascinating functionality. Such an unusual olefin is expected to demonstrate interesting reactivity via various noteworthy reactions. In fact it has been proved true, as reported in the literature on Claisen rearrangement20 and intramolecular [2+2] photocycloaddition21 reactions of AsA derivatives. We have shown that the above ene-diol motif of AsA smoothly undergoes the Paternò-Büchi22 reaction with various aromatic aldehydes and ketones to produce chiral oxetane bicycles. As part of our continued efforts to discover various derivatives of AsA with improved anti-cancer activity, we have demonstrated that 2,3-di-O-alkyl derivatives of 5,6-O-isopropylidene-L-ascorbic acid show enhanced antitumor activity against various human cancer cell lines.23 The inherent ene-diol functionality at C2 and C3 in AsA is well-suited for diverse cross-coupling reactions. Being very much focused on this aspect, our research group has recently reported the Suzuki cross-coupling reaction of 5,6-O-isopropylidene-L-ascorbic acid.24 A series of C2 aryl derivatives of AsA have been synthesized with special emphasis given to the anticancer activity with establishment of the mechanism of action. The resulting C2-aryl derivatives have displayed potent anti-cancer activity in the lower micro molar range as compared to AsA. As a continued approach to screening for new cancer chemotherapeutic agents using a small molecule library that contains AsA structures, herein we report the Sonogashira cross-coupling reaction of AsA to access novel C2/C3 alkynyl derivatives of AsA. These new derivatives of AsA are expected to exhibit an array of biological activity, particularly antiproliferative activity against various human cancers. Herein, we report initial results of our investigations in this area.
Results and discussion
Chemistry
Our strategy was to first establish a synthetic tactic applicable to access a set of structure-based analogues. The succinct synthetic endeavour began with discovering the optimum reaction conditions for the Sonogashira cross-coupling reaction of 5,6-O-isopropylidene-3-O-methyl-2-O-triflyl-L-ascorbic acid 2, which was obtained from 1,24,25 with phenyl acetylene using PdCl2(dppf) as a catalyst (10 mol%), CuI as a co-catalyst (10 mol%) and Et3N as both a base and solvent. Stirring the above reaction mixture at room temperature (RT) for 24 h did not lead to the desired product 4a (Table 1, entry 1), while at reflux temperature (4 h) the desired product 4a was obtained only in 5% yield (Table 1, entry 2). Use of THF as a solvent at room temperature (24 h) also gave none of the product 4a (Table 1, entry 3), while heating the reaction mixture under reflux for 4 h gave the product 4a in 15% yield (Table 1, entry 4). Replacement of base Et3N with K3PO4 in THF improved the yield a little bit (Table 1, entry 5). Changing the ligand of the Pd catalyst from dppf to PPh3 did not alter the yield much (Table 1, entries 6 and 7). When Pd(PPh3)4 was used as a catalyst and K2CO3 as a base in THF at RT (24 h), the coupling reaction provided desired product 4a in 35% yield (Table 1, entry 8). When the same reaction was performed at reflux temperature for 2 h it gave 4a in 85% yield (Table 1, entry 9). Decreasing the catalyst load to 5 mol% resulted in a lowering of the yield to 50% (Table 1, entry 10). Thus the use of 10 mol% Pd(PPh3)4 as a catalyst, CuI as a co-catalyst (10 mol%) and K2CO3 as a base in THF at reflux temperature, was identified as the optimum condition for the above Sonogashira cross-coupling reaction (Table 1).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Temp. (°C) | Time (h) | Yieldb (%) |
| a The reactions were carried out under nitrogen atmosphere on a 0.5 mmol scale using 10 mol% catalyst according to the conditions indicated in the above equation, unless otherwise specified; 1.1 equivalents of phenyl acetylene and 1.5 equivalents of base were employed. b Isolated yields. c Starting material was recovered. d Along with the desired product a substantial amount of starting material was isolated. e 5 mol% catalyst was employed. f No reaction. | ||||||
| 1c | PdCl2(dppf) | Et3N | Et3N | RT | 24 | NRf |
| 2d | PdCl2(dppf) | Et3N | Et3N | Reflux | 4 | 5 |
| 3c | PdCl2(dppf) | Et3N | THF | RT | 24 | NRf |
| 4d | PdCl2(dppf) | Et3N | THF | reflux | 4 | 15 |
| 5d | PdCl2(dppf) | K3PO4 | THF | reflux | 4 | 19 |
| 6d | Pd(PPh3)2Cl2 | K3PO4 | THF | RT | 24 | 10 |
| 7d | Pd(PPh3)2Cl2 | K3PO4 | THF | Reflux | 4 | 20 |
| 8d | Pd(PPh3)4 | K2CO3 | THF | RT | 24 | 35 |
| 9 | Pd(PPh 3 ) 4 | K 2 CO 3 | THF | Reflux | 2 | 85 |
| 10e | Pd(PPh3)4 | K2CO3 | THF | Reflux | 2 | 50 |
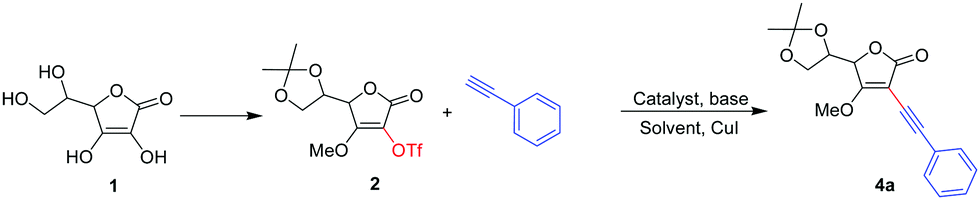
Table 2 shows the scope of the Sonogashira cross-coupling reaction of 2 with various terminal alkynes under the developed conditions. In general, aromatic alkynes bearing groups like –OMe, –F, –NH2, and –CH3 were efficiently coupled with 2 to afford the corresponding products 4a–f in moderate to good yield. Whereas, linear aliphatic alkynes, such as 1-pentyne, 1-heptyne, 1-octyne and 1-decyne gave desired products 4g–j in comparatively low yield, maybe due to the steric effect of the long chain part of alkynes. When 5,6-O-isopropylidene-2-O-methyl-3-O-triflyl-L-ascorbic acid 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24,25 was subjected to the Sonogashira cross-coupling reaction under optimized reaction conditions, various terminal alkynes offered the corresponding C3 alkynyl derivatives 5a–d with moderate yield (Table 2).
24,25 was subjected to the Sonogashira cross-coupling reaction under optimized reaction conditions, various terminal alkynes offered the corresponding C3 alkynyl derivatives 5a–d with moderate yield (Table 2).
|
To enable further diversification, we adopted a “deprotection” strategy. Thus removal of the 5,6-O-isopropylidene group from 4a, 4c–g, 5b and 5d derivatives with 10% H2SO4 in THF at RT smoothly gave C2 alkynyl derivatives 6a, 6c, 6d–g and C3 alkynyl derivatives 7b, 7d and 7k of AsA with good to excellent yield, respectively.
Biology
Initially the in vitro antiproliferative efficacy of the C2/C3 alkynyl derivatives of AsA 4a–h, 4j, 5b–d, 6a, 6c, 6e–g, 7b, 7d, and 7k was evaluated against human breast adenocarcinoma MCF7 using MTT assay; doxorubicin, the standard antitumor drug, was used as a positive control. All the new derivatives showed enhanced antiproliferative activity as compared to AsA, while compounds 4h and 7k even exhibited superior activity to doxorubicin (Fig. 1 and Table 3). In the C2 alkynyl series compound 4h having an n-pentyl group showed potent antiproliferative activity in the nano-molar range (IC50 = 300 ± 15 nM) and the carcinogenic potency also appears to decrease abruptly with the increase in (4j) or decrease in (4g) the length of hydrocarbon side chain indicating that lipophilicity of the pentyl tail is suitable to enhance the anticancer activity. Furthermore, compound 4e (4-methyl phenyl) and 4f (4-methoxy phenyl) showed IC50 of 2 ± 0.017 and 5 ± 0.027 μM respectively. While the rest of the compounds 4a–d, 4g, 4j, 6a, 6c and 6e–g showed IC50 values of more than 10 μM (Table 2 and Fig. 1). In the C3 alkynyl series compound 7k bearing a 3-amino phenyl group showed the most potent antiproliferative activity (IC50 = 200 ± 10 nM) while compound 7b (3-fluoro phenyl) showed an IC50 value of 1 ± 0.02 μM, and the other compounds 5a–d and 7d showed IC50 values of more than 10 μM (Table 3 and Fig. 1). | ||
| Fig. 1 Anticancer activity of novel compounds against the MCF7 cancer cell line. | ||
| Entry | Ar/R | Compound | IC50a (μM) MCF7 |
|---|---|---|---|
| a Data represent mean values for three independent determinations. | |||
| 1 | Phenyl | 4a | >10 |
| 2 | 3-Methyl phenyl | 4b | >10 |
| 3 | 3-Fluoro phenyl | 4c | >10 |
| 4 | 3-Amino phenyl | 4d | >10 |
| 5 | 4-Methyl phenyl | 4e | 2 ± 0.017 |
| 6 | 4-Methoxy phenyl | 4f | 5 ± 0.027 |
| 7 | n-Propyl | 4g | >10 |
| 8 | n-Pentyl | 4h | 0.3 ± 0.015 |
| 9 | n-Hexyl | 4i | — |
| 10 | n-Octyl | 4j | >10 |
| 11 | Phenyl | 5a | — |
| 12 | 3-Fluoro phenyl | 5b | >10 |
| 13 | 4-Methoxy phenyl | 5c | >10 |
| 14 | n-Propyl | 5d | >10 |
| 15 | Phenyl | 6a | >10 |
| 16 | 3-Fluoro phenyl | 6c | >10 |
| 17 | 3-Amino phenyl | 6d | — |
| 18 | 4-Methyl phenyl | 6e | >10 |
| 19 | 4-Methoxy phenyl | 6f | >10 |
| 20 | n-Propyl | 6g | >10 |
| 21 | 3-Fluoro phenyl | 7b | 1 ± 0.02 |
| 22 | n-Propyl | 7d | >10 |
| 23 | 3-Amino phenyl | 7k | 0.2 ± 0.01 |
| 24 | Doxorubicin | — | 0.55 ± 0.014 |
| 25 | AsA | — | 128.54 |
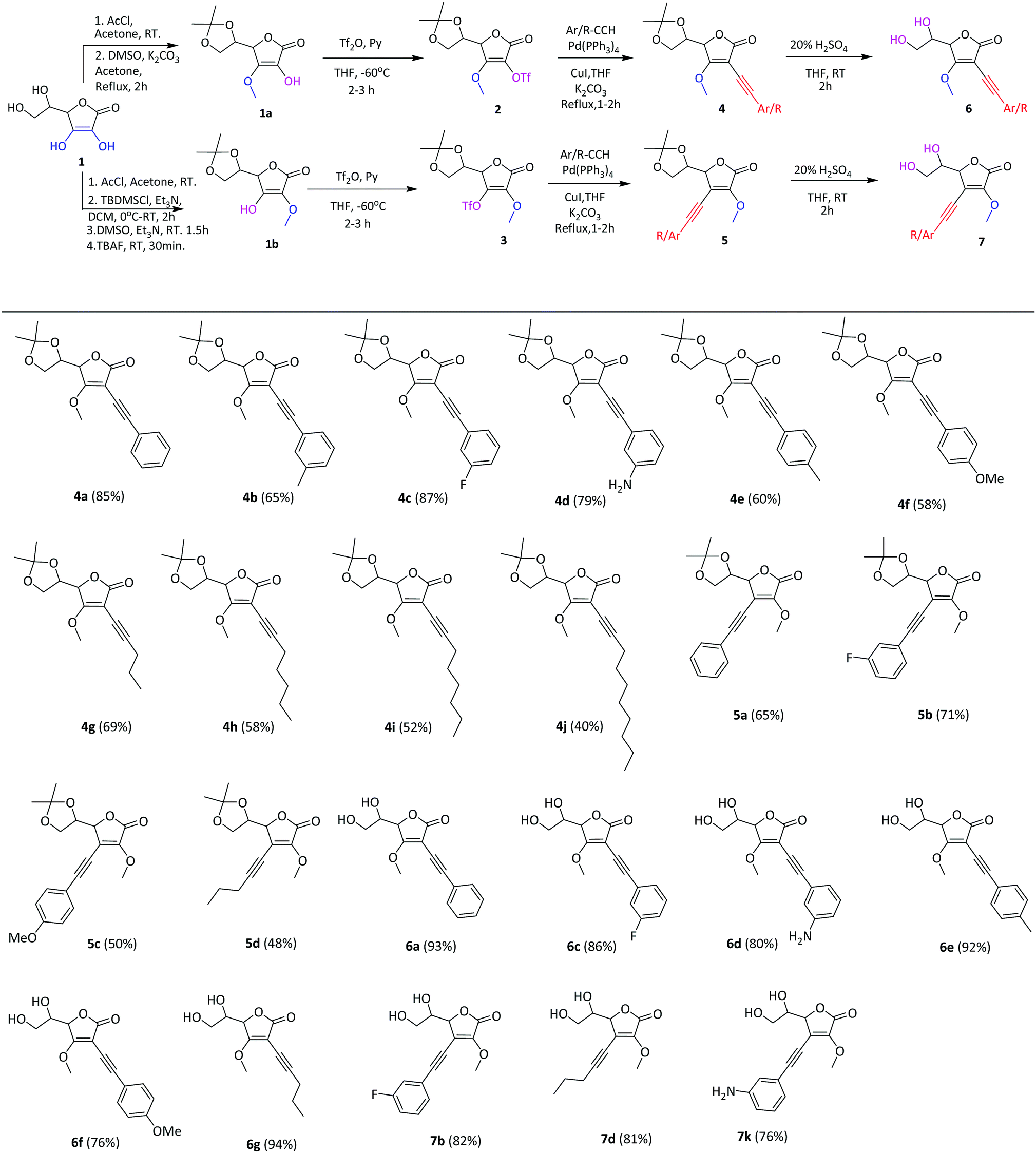
Next, we tested the two most potent compounds 4h and 7k against the A549 human lung cancer cell line. Compound 4h showed a slight decrease in antiproliferative activity with IC50 = 400 ± 18 nM as compared to the MCF7 cell line (IC50 = 300 ± 15 nM), while 7k exhibited 6.6 fold enhanced activity against the A549 cancer cell line (IC50 = 30 ± 04 nM) compared to the MCF7 cell line (IC50 = 200 ± 10 nM). Further, compound 7k when tested against human colorectal adenocarcinoma (HT 29) cell lines showed an IC50 value of 80 ± 07 nM (Fig. 2).
 | ||
| Fig. 2 Cytotoxic activity of compounds 4h, 7b, 7k and doxorubicin against MCF7, A549, HT29 and HUVEC cell lines. | ||
In order to check the selectivity of these compounds towards normal cells, compounds 4h, 7b and 7k were screened against normal human umbilical vein endothelial cells (HUVEC) and were found to exhibit an IC50 value of 1 ± 0.03, 4 ± 0.025 and 10 ± 0.014 μM respectively against the HUVEC cell line (Fig. 2). These results showed that compound 4h is 1.8 and 1.2 fold more potent than doxorubicin against MCF7 and A549 cancer cell lines while it is 2.5 fold less cytotoxic towards the normal HUVEC cell line than doxorubicin. Even the compound 7b is 1.8 times less potent than doxorubicin against the MCF7 cancer cell line, but it is 10 times more selective towards the normal HUVEC cell line than doxorubicin. Compound 7k showed the most promising results i.e. 2.7, 16.6 and 6.2 fold potency against MCF7, A549 and HT29 cancer cell lines, respectively, compared to doxorubicin. More importantly, 7k showed 25 fold less cytotoxicity against the normal HUVEC cell line than doxorubicin (Fig. 2). Despite the high potency, it also exhibited 50, 125 and 333 fold more cytotoxic selectivity to MCF-7, HT-29 and A549 cancer cell lines than the normal human cell line (HUVEC). 7k has prominently surpassed the potency against cancer cells as well as selectivity towards normal cells compared to doxorubicin.
In order to shed light on the underlying antitumor mechanism of compounds 4h and 7k, their effects on cell cycle progression were evaluated in the A549 cell line by employing flow cytometry. The results showed that compounds 4h and 7k arrest A549 cells in the G0/G1 phase by inducing apoptosis (Fig. 3).
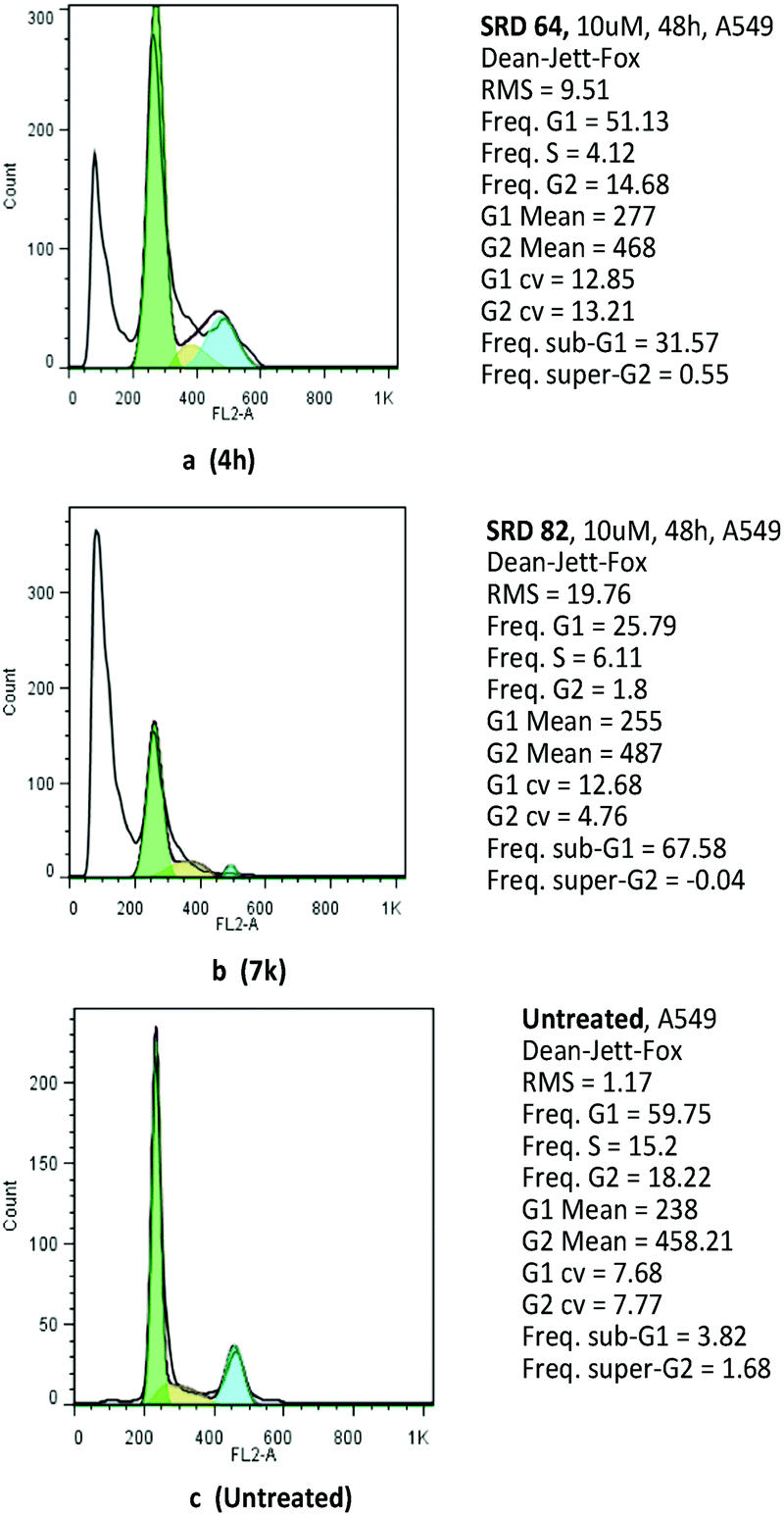 | ||
| Fig. 3 Compound 4h and 7k were treated with A549 cells for the given time period and concentration. The cells were trypsinized and washed with PBS twice. The single cell suspension was fixed with methanol and stained with propidium iodide. The sample was analysed on a BD FACS calibre flow cytometer. | ||
Conclusions
In conclusion, we have developed a simple and straightforward protocol for the synthesis of various C2/C3 alkynyl derivatives of AsA via the Sonogashira cross-coupling reaction with various terminal alkynes. To the best of our knowledge this is the first report of the Sonogashira cross coupling reaction of AsA. The screening of all these derivatives against the MCF7 cancer cell line showed potent anticancer activity as compared to AsA, while on the other hand compounds 4h and 7k even revealed superior antitumor activity to the doxorubicin drug. Flow cytometry data reveal that the mode of action of compounds 4h and 7k is by apoptosis. Due to their high potency against cancer cells, superior selectivity towards normal cells and easy accessibility, compounds 4h and 7k could be promising candidates for anticancer drug development.Materials and methods
Chemistry
All the reactions were performed in oven dried glassware and under a N2 atmosphere. Solvents were dried and degassed by standard methods before use. Thin-layer chromatography (TLC) was performed on silica gel Kieselgel (Merck) 60 F254 (0.25 mm thickness) plates and they were visualized under short (254 nm) and long (365 nm) UV light. Column chromatography was performed using silica gel 200–400 mesh (Acme). Melting points (m.p.) were determined in open capillary tubes using a paraffin oil bath and are uncorrected. Optical rotations were recorded on JASCO DIP Digital-181 polarimeter. 1H and 13C NMR spectra were recorded on a 500 and 125 MHz NMR spectrometer respectively using CDCl3 or DMSO-d6 as a solvent. Chemical shifts δ are reported in ppm relative to a Me4Si internal standard. The multiplicity of signals is designated by the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). FT-IR was recorded on an IR-Affinity1 Shimadzu DRS-8000A instrument. High-resolution mass spectra (HRMS) were obtained using a micromass-QTOF machine operating in electrospray ionization (ESI) mode. The purity (≥99%) of the compounds was verified by HPLC (Shimadzu Pvt. Ltd Japan CHT-2010 with auto sampler) study performed on a Shimadzu Luna C18 phenomenix column (250 mm × 4.6 mm, 5 μm maintained at 30 °C) using a mixture of solvent acetonitrile and water at a flow rate of 1.0 mL min−1 and peak detection at 257–325 nm under UV.Biological evaluations (cytotoxic activity)
Cytotoxic evaluation was done using MTT assay.26,27 Briefly the compounds were dissolved in DMSO and serially diluted with complete medium to get a range of test concentrations. The DMSO concentration was kept <0.1% in all the samples. Cells maintained in appropriate conditions were seeded in 96 well plates and treated with different concentrations of the test samples and incubated at 37 °C, 5% CO2 for 96 hours. MTT reagent was added to the wells and incubated for 4 hours; the dark blue formazan product formed by the cells was dissolved in DMSO under a safety cabinet and visualized at 550 nm. Percentage inhibitions were calculated and plotted with the concentrations used to calculate the IC50 values.Experimental
General procedure for synthesis of 2/3
To a dry dichloromethane solution (10 mL) of 2-O-methyl or 3-O-methyl-5,6-O-isopropylidene-L-ascorbic acid24,25 (1a/1b) (5 g, 21.718 mmol) and pyridine (2.624 mL, 32.578 mmol), triflic anhydride (Tf2O) (4.202 mL, 24.976 mmol) was added at −60 °C; the resulting mixture was stirred at −60 °C for 2–3 hours. The reaction mixture was filtered off to remove salt which was washed with dichloromethane. The filtrate was washed with 5% dil. HCl (2 × 50 mL) followed by water (2 × 50 mL). The organic extract was dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by silica column chromatography using n-hexane and ethyl acetate (3:1) as an eluent to afford compound 2/3 as colorless oil.
General procedure for synthesis of 4a–j & 5a–d
Compound 2/3 (0.5 g, 1.37 mmol), Pd(PPh3)4 (10 mol%), terminal alkynes (1.51 mmol), anhydrous potassium carbonate (K2CO3) (2.76 mmol) and copper iodide (5 mol%) were dissolved in 5 mL of THF and refluxed for 1–2 h under an inert atmosphere. After completion of the reaction (TLC check) the mixture was cooled to room temperature and solvent was removed under vacuo. Water (30 mL) was added to the residue which was extracted using ethyl acetate (3 × 25 mL). The organic layer was dried over anhydrous Na2SO4, and concentrated under reduced pressure to give the crude product which was purified by column chromatography using ethyl acetate:n-hexane (10–30%) as the eluent to yield the pure product as a white solid.General procedure for synthesis of 6 & 7
A mixture of compound 4/5 (1 mmol), THF (5 mL) and 10% H2SO4 (2 mL) was stirred for 2–3 h at room temperature. After completion of the reaction (TLC check) solid NaHCO3 was added to the reaction mixture in small portions to ensure the complete neutralization of acid. Then this solution was extracted using ethyl acetate (3 × 25 mL). The combined organic layer was dried over anhydrous Na2SO4, and concentrated under reduced pressure to give the crude product which was purified by column chromatography using ethyl acetate:n-hexane (50–70%) as the eluent to yield the pure product as a white solid.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from Council of Scientific and Industrial Research (CSIR), New Delhi India (File No. 02(0288)/17/EMR-II) and Board of Colleges and University Development (BCUD), (Proposal No. 13SCI000286) Savitribai Phule Pune University, Pune-411 007, MS, India. We thank Dr R. J. Barnabas (Ahmednagar College, Ahmednagar) for his helpful discussion and suggestions.Notes and references
- World Health Organization, Cancer, http://www.who.int/cancer/en/, accessed Sept 16, 2014.
- F. Bray, J. S. Ren, E. Masuyer and J. Ferlay, Int. J. Cancer, 2012, 132, 1133 CrossRef PubMed.
- World health statistics 2017: monitoring health for the SDGs, Sustainable Development Goals, Geneva, World Health Organization, 2017, Licence: CC BY-NC-SA3.0 IGO.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, CA Cancer J. Clin., 2011, 61, 69 CrossRef PubMed.
- S. N. A. Bukhari, G. B. Kumar, H. M. Revankar and H. L. Qin, Bioorg. Chem., 2017, 72, 130 CrossRef CAS PubMed.
- L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem., Int. Ed., 2003, 42, 3996 CrossRef CAS PubMed.
- B. Uttara, A. V. Singh, P. Zamboni and R. T. Mahajan, Curr. Neuropharmacol., 2009, 7, 65 CrossRef CAS PubMed.
- G. W. Burton and K. U. Ingold, in Mechanisms of antioxidant action: preventive and chain-breaking antioxidants, ed. A. Miguel, A. T. Quintanilha and H. Weber, CRC Handbook of Free Radicals and Antioxidants in Biomedicine, Press Inc, Boca Raton, FL, 1990, p. 29 Search PubMed.
- Q. Chen, M. G. Espey, A. Y. Sun, C. Pooput, K. L. Kirk, M. C. Krishna, D. B. Khosh, J. Drisko and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11105 CrossRef CAS PubMed.
- J. Verrax and P. B. Calderon, Free Radical Biol. Med., 2009, 47, 32 CrossRef CAS PubMed.
- Y. Nihro, H. Miyataka, T. Sudo, H. Matsumoto and T. J. Satoh, J. Med. Chem., 1991, 34, 2152 CrossRef CAS PubMed.
- K. Kato, S. Terao, N. Shimamoto and M. Hirata, J. Med. Chem., 1988, 31, 793 CrossRef CAS PubMed.
- F. M. El-Demerdash, M. I. Yousef and M. A. Zoheir, Food Chem. Toxicol., 2005, 43, 1743 CrossRef CAS PubMed.
- S. Tanuma, D. Shiokawa, Y. Tanimoto, M. Ikekita, H. Sakagami, M. Takeda, S. Fukuda and M. Kochi, Biochem. Biophys. Res. Commun., 1993, 194, 29 CrossRef CAS PubMed.
- T. Gazivoda, S. Raić-Malić, M. Marjanović, M. Kralj, K. Pavelić, J. Balzarini, E. D. Clercq and M. Mintas, Bioorg. Med. Chem., 2007, 15, 749 CrossRef CAS PubMed.
- K. Wittine, M. S. Babić, D. Makuc, J. Plavec, S. K. Pavelić, M. Sedić, K. Pavelić, P. Leyssen, J. Neyts, J. Balzarini and M. Mintas, Bioorg. Med. Chem., 2012, 20, 3675 CrossRef CAS PubMed.
- R. W. Veltri, G. Fodor, C. M. Liu, C. J. Woolverton and M. W. Baseler, J. Biol. Response Modif., 1986, 5, 444 CAS.
- C. J. Woolverton, R. W. Veltri and I. S. Snyder, J. Biol. Response Modif., 1986, 5, 527 CAS.
- H. Sakagami, M. Takeda, A. Utsumi, S. Fujinaga, A. Tsunoda, N. Yasuda, M. Shibusawa, T. Koike, H. Ota, K. Kazama, D. Shiokawa, S. Tanuma and M. Kochi, Anticancer Res., 1993, 13, 65 CAS.
- K. Wimalasena and M. P. D. Mahindaratne, J. Org. Chem., 1994, 59, 3427 CrossRef CAS.
- S. Redon and O. Piva, Tetrahedron Lett., 2006, 47, 733 CrossRef CAS.
- S. R. Thopate, M. G. Kulkarni and V. G. Puranik, Angew. Chem., 1998, 110, 1144 ( Angew. Chem., Int. Ed. , 1998 , 37 , 1110 ) CrossRef.
- S. R. Kote, R. Mishra, A. A. Khan and S. R. Thopate, Med. Chem. Res., 2014, 23, 1257 CrossRef CAS.
- S. R. Kote, G. R. Dhage, S. R. Thopate, R. Mishra and A. A. Khan, Synthesis, 2015, 2603 CAS.
- S. R. Thopate, R. A. Dengale and M. G. Kulkarni, Synlett, 2013, 1555 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
- MTT assay was performed at Deshpande Laboratories, Bhopal using the standard operating procedures.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj04477e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |