
High-temperature phase transitions, switchable dielectric behaviors and barocaloric effects in three new organic molecule-based crystals†
Rui-Xia
Li
,
Lin
Zhou
,
Ping-Ping
Shi
,
Xuan
Zheng
,
Ji-Xing
Gao
,
Qiong
Ye
 * and
Da-Wei
Fu
*
* and
Da-Wei
Fu
*
Ordered Matter Science Research Center, Jiangsu Key Laboratory for Science and Applications of Molecular Ferroelectrics, Southeast University, Nanjing 211189, People's Republic of China. E-mail: yeqiong@seu.edu.cn; dawei@seu.edu.cn
First published on 12th November 2018
Abstract
Three new organic molecule-based compounds, ethyl-trimethyl-phosphonium picrate (1, [ETtmp][picrate]), hydroxymethyl-trimethyl-phosphonium picrate (2, [HMtmp][picrate]), and cyclopentyl-trimethyl-phosphonium picrate (3, [CPtmp][picrate]), have been corroborated as high-temperature phase transition materials possessing switchable dielectric behaviors. Compounds 1, 2 and 3 undergo dielectric anomalies which could be tuned in two pronounced dielectric states and switched by reversible phase transitions at 320.8 K, 393.9 K and 398.1 K, respectively. For compounds 1, 2 and 3, not only the phase transition temperatures but also the magnitudes of the dielectric anomalies presented variations by regulating the guest cations. The respective dielectric constants in the high dielectric states are 1.6, 2.7 and 3.6 times those in the low dielectric states for compounds 1, 2 and 3. And considering the big entropy changes (ΔS) in the three title compounds, we predicted the sensitivities of the phase transition temperatures (Tc) to the applied pressure. And the estimated barocaloric coefficients (δTc/δP) in the three title compounds indicate that they potentially perform barocaloric effects of interest for cooling applications under adequate pressure.
Introduction
Solid-state phase transition compounds have provoked renowned attention because of their potential applications in data communication, signal processing, switchable dielectric devices, etc.1–7 Phase transitions are usually accompanied by interesting optical, dielectric, and magnetic responses and even ferroelectric properties under external stimuli.8–20 Generally speaking, dielectric responses are relevant to the internal motion and structural changes of materials. Hence, building molecular compounds with motional and/or disordered moieties is a promising strategy to acquire new phase transition materials bearing functional dielectric properties.21–30 Particularly, as one class of striking dielectric materials, switchable molecular dielectrics with convertible dielectric behaviors have shown their foreseeable applications in electronic technology fields.31–36,62 Such typical dielectric behaviors can be reversibly altered between the distinct high and low dielectric states. And much progress has been accomplished in the development of switchable molecular dielectrics.37–44 However, constructing them with high phase transition temperatures is still presently challenging. Recently, organic molecule-based switchable dielectrics have aroused great attention because they possess advantages, such as being environmentally friendly, easy to dispose, processed at low cost, etc.45–49 Until now, some organic molecule-based switchable dielectrics have been discovered, but they mostly have low phase transition temperatures, Tc, which severely hamper their practical applications.45–52 Therefore, it is vital to unearth new high-temperature organic molecule-based switchable dielectrics.The picrate anion easily forms π–π stacking and hydrogen bonding interactions with a cation to construct an interesting three-dimensional supramolecular structure. Meanwhile, the rotation of the nitro group and the sliding of the π–π stacking trigger a phase transition to obtain new organic switchable dielectrics. In recent years, picric acid chosen to assemble organic switchable dielectrics has drawn much attention.49–52 However, compounds which possess high-temperature phase transitions are sporadic. Inspired by this stage, considering that quaternary phosphonium picrates are still unexplored, we introduced different cations and have successfully synthesized three new organic molecule-based compounds, ethyl-trimethyl-phosphonium picrate (1, [ETtmp][picrate]), hydroxymethyl-trimethyl-phosphonium picrate (2, [HMtmp][picrate]) and cyclopentyl-trimethyl-phosphonium picrate (3, [CPtmp][picrate]). As expected, compounds 1, 2 and 3 all depict interesting reversible high-temperature phase transitions at 320.8 K, 393.9 K and 398.1 K, respectively, accompanied by step-like dielectric responses coinciding fairly well with switchable molecular dielectrics. The phase transition temperatures and magnitudes of the dielectric anomalies all get regulated by tuning the guest cations. Nowadays, barocaloric materials play an important role in the development of a new generation of eco-friendly solid-state refrigeration technologies.53–58 They usually experience large entropy changes in the first-order solid-state phase transitions and display interesting pressure dependence of the phase transition temperature (δTc/δP). Therefore, considering the large entropy changes (ΔS) in the three title compounds, we also estimated the pressure dependence of the phase transition temperature. And the calculated barocaloric coefficients (δTc/δP = 20.2 K kbar−1, 5.95 K kbar−1 and 11.9 K kbar−1) in compounds 1, 2 and 3 reveal that the three title compounds show large pressure responses and suggest that they are likely to display barocaloric effects of interest for cooling applications under adequate pressure. Systematic measurements afford three title compounds as promising candidates for application in switchable molecular dielectrics.
Experimental section
Synthesis
All of the chemical reagents were used without any further purification.Preparation of ethyl-trimethyl-phosphonium bromide. Dehydrated dichloromethane (50 ml) was added into a dry flask under nitrogen at room temperature. Then, trimethylphosphine (5.20 ml, 50 mmol) and ethyl bromide (4.51 ml, 60 mmol) were added to this solution using a syringe. The mixed solution was stirred and refluxed for three days at 37 °C. A white solid was obtained after filtering and drying (yield: 7.97 g, 86.1%).
Preparation of hydroxymethyl-trimethyl-phosphonium chlorine was performed in accordance with literature procedure.43
Preparation of cyclopentyl-trimethyl-phosphonium bromide. Dehydrated acetonitrile (50 ml) was added into a dry flask under nitrogen at room temperature. Then, trimethylphosphine (6.21 ml, 60 mmol) and cyclopentyl bromide (5.36 ml, 50 mmol) were added to this solution using a syringe. The mixed solution was heated to 70 °C and stirred for 40 h. The solvent was evaporated under vacuum to yield cyclopentyl-trimethyl-phosphonium bromide as a white solid (yield: 6.52 g, 58%).
Single-crystal X-ray diffraction
The variable temperature single-crystal structural analyses of 1, 2 and 3 were performed using a Rigaku Saturn 924 diffractometer with Mo Kα radiation (λ = 0.71073 Å). Data processing including empirical absorption corrections was performed by using the CrystalClear software package (Rigaku, 2005). The crystal structures were solved by direct methods and refined by a full-matrix method based on F2 by means of the SHELXLTL-97 software package. All non-hydrogen atoms were refined anisotropically and the positions of all hydrogen atoms were generated geometrically. The crystal structure and packing views were drawn with DIAMOND. For compounds 1, 2 and 3, the crystallographic data and structure refinements are listed in Table S1 (ESI†).Powder X-ray diffraction
For compounds 1, 2 and 3, variable-temperature powder X-ray diffraction (PXRD) measurements were carried out using a Rigaku D/MAX 2000 PC X-ray diffractometer. The PXRD patterns were obtained in the 2θ range of 5°–50° with a step size of 0.02°. As shown in Fig. S2 (ESI†), the PXRD patterns obtained at 298 K accord fairly well with the simulated patterns, indicating the purity of the as-grown crystals of the three title compounds.DSC and TGA measurements
Differential scanning calorimetry (DSC) experiments were performed using a Netzsch Model DSC 200 F3 instrument by heating and cooling the crystalline samples of compounds 1, 2 and 3 which were placed in an aluminum crucible under nitrogen at atmospheric pressure with a rate of 10 K min−1 over the temperature ranges of 280–360 K, 330–420 K and 340–430 K, respectively. The crystalline samples of three compounds were placed in an aluminum crucible under nitrogen at atmospheric pressure. Thermogravimetric analysis (TGA) was performed using a Netzsch Model TG 209 F3 instrument. The measurements were performed in a nitrogen flow at a rate of 10 K min−1 over temperature ranges of 380–880 K, 302–850 K and 345–880 K, respectively. And the results show that compounds 1, 2 and 3 begin to decompose at the respective temperatures of 576.7 K, 474.6 K and 586.5 K (Fig. S3, ESI†).Dielectric constant measurements
The powder-pressed pellets of compounds 1, 2 and 3 pasted with carbon conductive glue were used to discuss dielectric properties. The real parts (ε′) of the complex dielectric constants (ε = ε′ − iε′′, where ε′′ represents the imaginary part) and the dielectric losses (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ = ε′′/ε′) were measured using a Tonghui TH2828A impedance analyzer at different frequencies with a heating and cooling rate of about 10 K min−1 within temperature ranges of 285–342 K, 338–418 K and 355–425 K, respectively, with the applied AC field fixed at 1 V.
δ = ε′′/ε′) were measured using a Tonghui TH2828A impedance analyzer at different frequencies with a heating and cooling rate of about 10 K min−1 within temperature ranges of 285–342 K, 338–418 K and 355–425 K, respectively, with the applied AC field fixed at 1 V.
Results and discussion
Compounds 1, 2 and 3 can be easily obtained from the acetonitrile or ethanol solution. The crystal structures of the three title compounds are listed in Table S1 (ESI†), and at 223 K, [ETtmp][picrate] (1) crystallizes in monoclinic centrosymmetric space group P21/n, with cell parameters a = 11.325(5) Å, b = 19.765(9) Å, c = 6.921(3) Å, β = 100.830(8)°, and V = 1521.6(12) Å3. By changing guest cations, compound 2 belongs to the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) at 298 K, with cell parameters a = 7.4008(15) Å, b = 10.413(2) Å, c = 10.972(2) Å, α = 109.76(3)°, β = 99.84(3)°, γ = 104.20(3)°, and V = 741.0(4) Å3. And for compound 3, at 298 K, it has the same space group and similar unit cell parameters from those of compound 1, with a = 12.300(3) Å, b =19.943(4) Å, c = 7.3103(15) Å, β = 98.55(3)°, and V = 1773.3(7) Å3. As illustrated in Fig. 1, the asymmetric units of compounds 1, 2 and 3 all contain a common picrate anion and one [ETtmp]+, [HMtmp]+ or [CPtmp]+ cation. As listed in Table S2 (ESI†), for cations of three title compounds, the bond parameters all fall within normal ranges. In order to show more clearly about the conformational differences in cations, the overlapped view of cations in the three title compounds is depicted in Fig. S4, ESI† and the partial cationic torsion angles are listed in Table 1. Therefore, in the cations of compounds 1, 2 and 3, the ethyl, hydroxymethyl and cyclopentyl can be regarded as occupying the pseudo-axial positions, respectively. And the distances of the P atom from the C3 plane (C7, C8 and C9) in compounds 1, 2 and 3 are 0.5898 Å, 0.5756 Å and 0.6029 Å, respectively. The torsion angles of C7–P1–C10–C11, C8–P1–C10–C11 and C9–P1–C10–C11 in compound 3 are very similar to those of compound 1 when the ethyl is replaced by the cyclopentyl. Peculiarly, the C8–P1–C10–C11 torsion angle (178.8(3)°) in compound 3 is nearly the same as the corresponding value (178.3(2)°) in compound 1. However, the torsion angles of C7–P1–C10–C14, C8–P1–C10–C14 and C9–P1–C10–C14 generated accompanying the change from ethyl to cyclopentyl present tremendous changes compared with the torsion angles of C7–P1–C10–C11, C8–P1–C10–C11 and C9–P1–C10–C11 in compound 1. Similarly, the corresponding torsion angles also change dramatically upon replacing the ethyl with hydroxymethyl, with the values of C7–P1–C10–O8, C8–P1–C10–O8 and C9–P1–C10–O8 torsion angles in compound 2 being −174.5(3)°, 66.0(4)° and −53.0(4)°, respectively. Additionally, for compound 3, accompanied by the mild distortion of the C5 plane, the corresponding torsion angles of C10–C11–C12–C13, C11–C12–C13–C14, C12–C13–C14–C10, C13–C14–C10–C11, and C14–C10–C11–C12 are −10.0(8)°, −8.2(11)°, 22.7(10)°, −27.6(7)° and 23.3(6)°, respectively, which make the C5 plane adopt the irregular envelope conformation. Therefore, by tuning the cations, the cationic conformations display different degrees of variations.
at 298 K, with cell parameters a = 7.4008(15) Å, b = 10.413(2) Å, c = 10.972(2) Å, α = 109.76(3)°, β = 99.84(3)°, γ = 104.20(3)°, and V = 741.0(4) Å3. And for compound 3, at 298 K, it has the same space group and similar unit cell parameters from those of compound 1, with a = 12.300(3) Å, b =19.943(4) Å, c = 7.3103(15) Å, β = 98.55(3)°, and V = 1773.3(7) Å3. As illustrated in Fig. 1, the asymmetric units of compounds 1, 2 and 3 all contain a common picrate anion and one [ETtmp]+, [HMtmp]+ or [CPtmp]+ cation. As listed in Table S2 (ESI†), for cations of three title compounds, the bond parameters all fall within normal ranges. In order to show more clearly about the conformational differences in cations, the overlapped view of cations in the three title compounds is depicted in Fig. S4, ESI† and the partial cationic torsion angles are listed in Table 1. Therefore, in the cations of compounds 1, 2 and 3, the ethyl, hydroxymethyl and cyclopentyl can be regarded as occupying the pseudo-axial positions, respectively. And the distances of the P atom from the C3 plane (C7, C8 and C9) in compounds 1, 2 and 3 are 0.5898 Å, 0.5756 Å and 0.6029 Å, respectively. The torsion angles of C7–P1–C10–C11, C8–P1–C10–C11 and C9–P1–C10–C11 in compound 3 are very similar to those of compound 1 when the ethyl is replaced by the cyclopentyl. Peculiarly, the C8–P1–C10–C11 torsion angle (178.8(3)°) in compound 3 is nearly the same as the corresponding value (178.3(2)°) in compound 1. However, the torsion angles of C7–P1–C10–C14, C8–P1–C10–C14 and C9–P1–C10–C14 generated accompanying the change from ethyl to cyclopentyl present tremendous changes compared with the torsion angles of C7–P1–C10–C11, C8–P1–C10–C11 and C9–P1–C10–C11 in compound 1. Similarly, the corresponding torsion angles also change dramatically upon replacing the ethyl with hydroxymethyl, with the values of C7–P1–C10–O8, C8–P1–C10–O8 and C9–P1–C10–O8 torsion angles in compound 2 being −174.5(3)°, 66.0(4)° and −53.0(4)°, respectively. Additionally, for compound 3, accompanied by the mild distortion of the C5 plane, the corresponding torsion angles of C10–C11–C12–C13, C11–C12–C13–C14, C12–C13–C14–C10, C13–C14–C10–C11, and C14–C10–C11–C12 are −10.0(8)°, −8.2(11)°, 22.7(10)°, −27.6(7)° and 23.3(6)°, respectively, which make the C5 plane adopt the irregular envelope conformation. Therefore, by tuning the cations, the cationic conformations display different degrees of variations.
 | ||
| Fig. 1 Molecular structures of compounds 1 (a), 2 (b), and 3 (c), showing an atom labelling scheme with 30% probability thermal ellipsoids. The hydrogen atoms are omitted for clarity. | ||
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| C7–P1–C10–C11 | −62.1(3) | −60.7(4) | |
| C8–P1–C10–C11 | 178.3(2) | 178.8(3) | |
| C9–P1–C10–C11 | 58.2(3) | 61.1(4) | |
| C7–P1–C10–O8 | −174.5(3) | ||
| C8–P1–C10–O8 | 66.0(4) | ||
| C9–P1–C10–O8 | −53.0(4) | ||
| C7–P1–C10–C14 | 60.0(4) | ||
| C8–P1–C10–C14 | −60.6(4) | ||
| C9–P1–C10–C14 | −178.2(4) |
Furthermore, differences can also be found in the anions of the three title compounds. As depicted in Fig. 2a and c, for [ETtmp][picrate] (1) and [CPtmp][picrate] (3), π–π stacking interactions were observed and the respective Ci–Cj (distance of centers in aromatic rings) are 3.422 Å and 3.537 Å, 3.674 Å and 3.678 Å. And both picrate anions are stacked in opposite orientations alternately along the possible π–π stacking interactions producing one-dimensional zigzag chains along the a-axis. And as shown in Fig. 2b, for [HMtmp][picrate] (2), only π–π interactions can be found between partial adjacent picrate anions and the distance is 3.581 Å. It can be observed that the O–H⋯O hydrogen bonds impede the formation of the π–π stacking interactions in compound 2. Therefore, no one-dimensional chain was observed which was formed by π–π stacking interactions.
 | ||
| Fig. 2 The diagrams of π–π stacking interactions of picrate anions in compounds 1 (a), 2 (b) and 3 (c). And the O–H⋯O hydrogen bonding interactions in compound 2 (b). | ||
In many cases, hydrogen bond conformations also present differences in the three title compounds by altering the guest cations. As shown in Fig. S5 and Table S3 (ESI†), for [ETtmp][picrate] (1), ten hydrogen bonds are discovered between one [ETtmp]+ cation and the surrounding picrate anions. Four weak hydrogen bonds involving the carbon bound hydrogen atoms of ethyl are within the range of 3.354(4) Å to 3.569(5) Å. The hydrogen bond formation displays noticeable changes when the ethyl is replaced by hydroxymethyl. Therefore, in the case of [HMtmp][picrate] (2), each [HMtmp]+ cation forms five weak C–H⋯O hydrogen bonds with four picrate anions and one [HMtmp]+ cation. The weak hydrogen bonds vary from 3.278(8) Å to 3.565(8) Å. Moreover, the substitution of cyclopentyl for ethyl also makes more complicated hydrogen bond architectures. Therefore, in [CPtmp][picrate] (3), there are nine weak hydrogen bonds connecting methyl groups with picrate anions and the hydrogen bond lengths differ from 3.142(5) Å to 3.581(6) Å. Additionally, the methylene hydrogen atoms of the cyclopentyl ring take part in three weak C12–H12A⋯O3B, C12–H12B⋯O2A and C13–H13B⋯O3A interactions with adjoining picrate anions. Unlike compounds 1 and 3, there still exist two strong O8–H8⋯O1 and O8–H8⋯O2 hydrogen bonds in compound 2 (as indicated in red dashed line). It can be inferred that the discrepancies in cations lead to different three dimensional hydrogen-bonding networks in compounds 1, 2 and 3 (Fig. S6, ESI†). Consequently, at LTP, compounds 1 and 3 possess more similar packing structures, in comparison with a quite distinct packing structure in compound 2 (Fig. S7, ESI†).
In general, the differences in crystal structures of compounds 1, 2 and 3 should be attributed to the variations of space groups and unit cell dimensions caused by the changes of guest cations.
To investigate whether phase transitions occur under the external temperature stimulus in compounds 1, 2 and 3, DSC measurements were performed on the three title compounds over different temperature ranges. As shown in Fig. 3a, b and c, in the DSC curves of compounds 1, 2 and 3, the reversible heat anomalies at 320.8/303.5 K, 393.9/370.5 K and 398.1/380.8 K (heating/cooling) were obviously observed, respectively. Under the circumstance of changing guest cations, the discrepancies in the phase transition temperatures (Tc) may be correlated with the distinct structures of the three title compounds. For compound 1, the large thermal hysteresis of 17.3 K indicates a reversible first-order phase transition. Based on the DSC curves, the average enthalpy change ΔH(1) and entropy change ΔS(1) are obtained as 17.82 kJ mol−1 and 44.03 J mol−1 K−1, respectively. Compared to compound 1, the phase transition temperature of compound 2 increases by 73.1 K when ethyl is replaced by the hydroxymethyl group. And considering the shape of the pronounced endothermic and exothermic peaks with a relatively large thermal hysteresis of about 23.4 K, the phase transition is also attributed to the first-order type. In view of the DSC curves, the average ΔH(2) and ΔS(2) are estimated as 20.84 kJ mol−1 and 54.56 J mol−1 K−1, respectively. For compound 2, the O–H⋯O hydrogen bonds contribute to the formation of more stable structure networks than compound 1 leading to the increase of the phase transition temperature. Moreover, the change from ethyl to cyclopentyl makes the phase transition temperature increase to 398.1 K. And for compound 3, the calculated average values of ΔH(3) and ΔS(3) are 21.32 kJ mol−1 and 54.78 J mol−1 K−1, respectively. The large thermal hysteresis of 17.3 K also manifests the typical feature of the first-order phase transition. In compound 3, the large cyclopentyl causes a closer packing structure (Fig. S7, ESI†), which makes the movement of [CPtmp]+ cations difficult. As a result, compared to [ETtmp]+ cations, compound 3 exhibits a higher phase transition temperature. In conclusion, compounds 1, 2 and 3 all undergo reversible first-order phase transitions and the phase transition temperatures could be tuned by changing the guest cations. Moreover, it is obvious that the substitution of cyclopentyl for ethyl has more influence on thermal properties. And for convenience, the phases below and above Tc(1), Tc(2) and Tc(3) are designated as low temperature phases (LTPs) and high temperature phases (HTPs), respectively.
 | ||
| Fig. 3 DSC curves of compounds 1 (a), 2 (b), and 3 (c) obtained in the heating and cooling cycles. | ||
To better understand the reversible high-temperature phase transitions firstly confirmed by the DSC measurements, we tried obtaining the high-temperature crystal structures of the three title compounds. Unfortunately, only the high temperature structure of compound 1 was obtained. At 333 K, compound 1 crystallizes in the monoclinic space group C2/c with cell parameters a = 11.412(2) Å, b = 19.766(4) Å, c = 7.048(3) Å, β = 113.10(4)°, and V = 1462.4(9) Å3 (Table S1, ESI†). Particularly, in comparison with their cell parameters, the notable discrepancy in the crystal graphical data for the HTP (333 K) and LTP (223 K) is the β angle. With the increase in temperature, the β angle changes from 100.830(8)° to 113.10(4)°. And as shown in Fig. 4, it is clear that the ellipsoidal degree of each atom is greatly increased because thermally induced atomic vibrations become severe with the increase of temperature. And this point can be well explained from the changes of atomic thermal vibration parameters from LTP to HTP (Table S4, ESI†). As a result, the cationic part is highly disordered in the HTP. To meet the crystal symmetry requirements, each carbon atom of two methyl groups and one ethyl group is orientationally disordered over two equivalent positions related to two-fold axes. And as shown in Fig. S8 (ESI†), the two-fold axis is parallel to the b-axis and along the P1–C6 bond. For picrate anions, accompanied by the phase transition, the π–π stacking interactions also display changes. As presented in Fig. S9 (ESI†), only π–π stacking interactions (Ci–Cj = 3.532 Å) were observed in the HTP compared with those in the LTP. The adjoining picrate anions are also stacked in opposite orientations alternately together with the π–π stacking interactions forming one-dimensional zigzag chains along the a-axis. At the HTP, the respective planes that the picrate anions locate on are all nearly parallel to the (001) plane. In addition, the two-fold axis can also be observed in picrate anions which passes through the O1, C1, C4, and N2 atoms of aromatic rings and is parallel to the b-axis as well (Fig. S8, ESI†). From this detailed structural analysis, conspicuously, the most evident discrepancy between the LTP and HTP is the order–disorder transformation of the structural component. Thus, it is deduced that the high-temperature phase transition of compound 1 is mainly dominated by the order–disorder changes of the cations.
 | ||
| Fig. 4 Molecular structures of 1 (a) at 223 K and 1 (b) at 333 K. Thermal ellipsoids for all atoms are shown at the 10% probability level. Hydrogen atoms are omitted for clarity. Symmetry codes: (I) (−x, y, −2.5 − z); (II) (−1 − x, y, −2.5 − z). | ||
In consideration of the difficulties in obtaining single-crystal structures of compounds 2 and 3 at the HTP, variable-temperature PXRD patterns were measured to validate the phase transitions further. As illustrated in Fig. 5a, for compound [HMtmp][picrate] (2), at 413 K, a relatively large number of peaks disappeared and eight new diffraction peaks were observed at 7.98°, 14.61°, 16.97°, 21.06°, 22.86°, 26.17°, 38.56°, and 45.04°. And the diffraction peaks at 30.37° and 35.39° displayed some shifts. Additionally, the change of the diffraction peak at 24.47° suggests a swelling process accompanied by heating. In summary, the changes of diffraction peaks indicate the occurrence of phase transition, which is consistent with the DSC result. In the case of compound [CPtmp][picrate] (3), as depicted in Fig. 5b, on further heating, a large number of diffraction peaks disappeared and five new diffraction peaks were discovered at 20.66°, 22.90°, 24.52°, 39.63° and 46.10°. The diffraction peaks at 14.00° and 14.54° display shifts and swell to visible peaks. Generally speaking, the apparent changes of the PXRD patterns also provide strong evidence of the existence of phase transition in compound 3, which is compatible with the thermal measurement of DSC. In addition, simulating the cell parameters of compounds 2 and 3 by using the Material Studio software suggests that compounds 2 and 3 crystallize in monoclinic space group P21/n and monoclinic space group C2/c, respectively. And the corresponding cell parameters are listed in Table S5 (ESI†).
 | ||
| Fig. 5 Variable-temperature PXRD patterns of compounds 2 (a) and 3 (b) measured in the heating modes. | ||
Generally speaking, the temperature-dependent dielectric constants always present obvious changes in the vicinity of phase transitions, and dielectric constants are usually detected by way of dielectric measurements. On the basis of the reversible phase transitions ascertained by DSC and PXRD results, compounds 1, 2 and 3 tend to display notable dielectric responses triggered by temperature. The dielectric measurements were performed on polycrystalline samples of the three title compounds. Consequently, the real parts (ε′) of the dielectric constants taken at 1 MHz are presented in Fig. 6. For [ETtmp][picrate] (1), as depicted in Fig. 6a, during the heating process, in the vicinity of the Tc(1) of 320.8 K, the temperature-dependent dielectric constant ε′ changed abruptly from 5.5 at LTP to 8.7 at HTP. The ε′ in the high dielectric state (HTP) is approximately 1.6 times that in the low dielectric state (LTP), denoting the obvious step-like feature of dielectric anomaly. For [HMtmp][picrate] (2), as illustrated in Fig. 6b, when ethyl is replaced by the hydroxymethyl, the temperature-dependent ε′ exhibited a remarkable change increasing sharply from 5.9 to 15.8 around the Tc(2) of 393.9 K. The value of ε′ in the high dielectric state (HTP) is almost 2.7 times that in the low dielectric state (LTP), also demonstrating an evident step-like dielectric anomaly. And as shown in Fig. 6c, upon heating, compared to compound 1, a similar notable step-like dielectric anomaly was also observed in [CPtmp][picrate] (3). The substitution of cyclopentyl for ethyl makes ε′ change from 7.4 at LTP to 26.4 at HTP around the Tc(3) of 398.1 K. And ε′ in the high dielectric state is estimated to be about 3.6 times that in the low dielectric state. In general, for compounds 1, 2 and 3, the curves recorded in cooling processes are similar to those obtained in the heating codes, indicating the phenomenon of reversible phase transitions. And the large temperature hystereses further verify the first order characteristics of the phase transitions of compounds 1, 2 and 3, in very good accordance with the DSC measurements. As presented in Fig. S10 (ESI†), upon heating and cooling, the temperature-dependent dielectric losses (tanδ) measured at 1 MHz also show apparent anomalies, further confirming the reversible phase transitions in the three title compounds. And as plotted in Fig. S11 and S12 (ESI†), in compounds 1, 2 and 3, it is noteworthy that the ε′ and dielectric losses measured at frequencies of 100 kHz and 1 MHz all exhibit frequency-dependent properties. In general, for compounds 1, 2 and 3, the ε′ could be switched by the reversible phase transitions and be tuned in two pronounced dielectric states. And the corresponding dielectric constants in the high dielectric states are 1.6, 2.7 and 3.6 times those in the low dielectric states for compounds 1, 2 and 3. By altering the guest cations, not only the phase transition temperatures but also the magnitudes of dielectric anomalies displayed different degrees of variations. And this means that the modification on cations can affect the dielectric properties in the three title compounds.
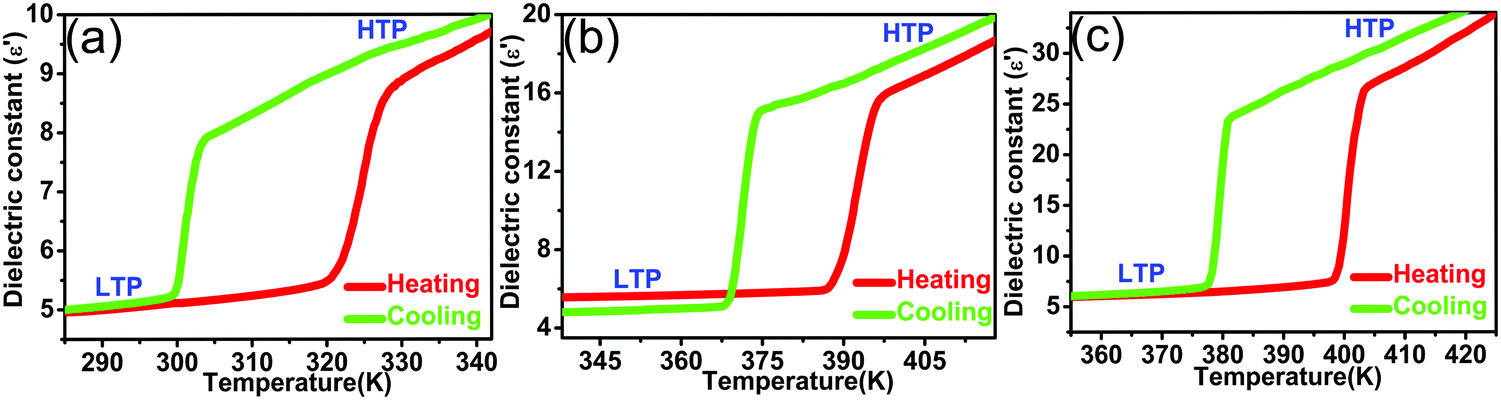 | ||
| Fig. 6 Temperature-variable dielectric constants measured at 1 MHz of compounds 1 (a), 2 (b) and 3 (c). | ||
Finally, considering the large entropy change (ΔS) in the three title compounds, we predicted how sensitive the phase transition temperatures are to the applied pressure. And the pressure dependence of phase transition temperatures of three compounds, the barocaloric coefficients (δTc/δP), are calculated by using the indirect Clausius–Clapeyron equation:53,59–61

| Compound | T c (K) exp. | ΔS (J mol−1 K−1) exp. | M.W. (g mol−1) | ΔS (J kg−1 K−1) exp. | |ΔV| × 10−5 (m3 kg−1) exp. | |δTc/δP| (K kbar−1) calc. |
|---|---|---|---|---|---|---|
| 1 | 320.8 | 44.03 | 333.24 | 132.13 | 2.6736 | 20.2 |
| 2 | 393.9 | 54.56 | 335.21 | 162.76 | 0.9680 | 5.95 |
| 3 | 398.1 | 54.78 | 373.30 | 146.75 | 1.7404 | 11.9 |
Conclusion
In summary, three new organic molecule-based compounds, [ETtmp][picrate] (1), [HMtmp][picrate] (2) and [CPtmp][picrate] (3), have been successfully synthesized and characterized as high-temperature phase transition materials, which undergo reversible first-order phase transitions at different temperatures of 320.8 K, 393.9 K and 398.1 K, with large thermal hystereses of 17.3 K, 23.4 K and 17.3 K, respectively. Compounds 1, 2 and 3 all present striking step-like dielectric anomalies in the process of phase transitions, which are characteristics of switchable dielectrics. The phase transition temperatures and the magnitudes of the dielectric anomalies all get adjusted by changing the guest cations. And the corresponding dielectric constants in the high dielectric states are 1.6, 2.7 and 3.6 times those in the low dielectric states for compounds 1, 2 and 3. And to a limited extent, the computed barocaloric coefficients (δTc/δP) disclose that the three title compounds present large pressure responses and imply that they possibly exhibit barocaloric effects of interest for cooling applications. In terms of potential barocaloric effects, specific investigations of the three compounds remain to carry out further. What is expected is that the present findings would open up a new realm to tap and devise organic high-temperature switchable dielectrics.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21471032, 21771037 and 21805033) and the Natural Science Foundation of Jiangsu Province (JSNSF) (BK20170659).References
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- Y. Tian, S. Shen, J. Cong, L. Yan, S. Wang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 782–785 CrossRef CAS PubMed.
- B. Champagne, A. Plaquet, J. L. Pozzo, V. Rodriguez and F. Castet, J. Am. Chem. Soc., 2012, 134, 8101–8103 CrossRef CAS PubMed.
- D. Lencer, M. Salinga and M. Wuttig, Adv. Mater., 2011, 23, 2030–2058 CrossRef CAS PubMed.
- W. Zhang and R. G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS PubMed.
- P. P. Shi, Y. Y. Tang, P. F. Li, W. Q. Liao, Z. X. Wang, Q. Ye and R. G. Xiong, Chem. Soc. Rev., 2016, 45, 3811–3827 RSC.
- Y. L. Liu and W. Zhang, Dalton Trans., 2017, 47, 45–48 RSC.
- D. W. Fu, H. L. Cai, Y. Liu, Q. Ye, W. Zhang, Y. Zhang, X. Y. Chen, G. Giovannetti, M. Capone, J. Li and R. G. Xiong, Science, 2013, 339, 425–428 CrossRef CAS PubMed.
- D. W. Fu, W. Zhang, H. L. Cai, J. Z. Ge, Y. Zhang and R. G. Xiong, Adv. Mater., 2011, 23, 5658–5662 CrossRef CAS PubMed.
- H.-L. Cai, W. Zhang, J.-Z. Ge, Y. Zhang, K. Awaga, T. Nakamura and R.-G. Xiong, Appl. Phys. Lett., 2011, 107, 147601 CrossRef PubMed.
- T. Akutagawa, H. Koshinaka, D. Sato, S. Takeda, S. Noro, H. Takahashi, R. Kumai, Y. Tokura and T. Nakamura, Nat. Mater., 2009, 8, 342–347 CrossRef CAS PubMed.
- Q. Ye, T. Akutagawa, N. Hoshino, T. Kikuchi, S.-i. Noro, R.-G. Xiong and T. Nakamura, Cryst. Growth Des., 2011, 11, 4175–4182 CrossRef CAS.
- Q. Ye, T. Akutagawa, T. Endo, S. Noro, T. Nakamura and R. G. Xiong, Inorg. Chem., 2010, 49, 8591–8600 CrossRef CAS PubMed.
- H. L. Cai, Y. Zhang, D. W. Fu, W. Zhang, T. Liu, H. Yoshikawa, K. Awaga and R. G. Xiong, J. Am. Chem. Soc., 2012, 134, 18487–18490 CrossRef CAS PubMed.
- H. Y. Ye, Y. Zhang, D. W. Fu and R. G. Xiong, Angew. Chem., Int. Ed., 2014, 53, 6724–6729 CrossRef CAS PubMed.
- P.-P. Shi, Q. Ye, Q. Li, H.-T. Wang, D.-W. Fu, Y. Zhang and R.-G. Xiong, Chem. Mater., 2014, 26, 6042–6049 CrossRef CAS.
- S.-Y. Zhang, J.-S. Liao, W.-J. Xu, F.-Y. Liang, G.-H. Peng, S.-J. Liu, H.-R. Wen and Z.-Y. Du, New J. Chem., 2017, 41, 9963–9968 RSC.
- D. W. Fu, W. Zhang, H. L. Cai, Y. Zhang, J. Z. Ge, R. G. Xiong, S. D. Huang and T. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 11947–11951 CrossRef CAS PubMed.
- Z. Sun, J. Luo, T. Chen, L. Li, R.-G. Xiong, M.-L. Tong and M. Hong, Adv. Funct. Mater., 2012, 22, 4855–4861 CrossRef CAS.
- Y.-L. Sun, B.-B. Zheng and W. Zhang, New J. Chem., 2017, 41, 5142–5150 RSC.
- T. Hang, W. Zhang, H. Y. Ye and R. G. Xiong, Chem. Soc. Rev., 2011, 40, 3577–3598 RSC.
- D. W. Fu, W. Zhang, H. L. Cai, Y. Zhang, J. Z. Ge, R. G. Xiong and S. D. Huang, J. Am. Chem. Soc., 2011, 133, 12780–12786 CrossRef CAS PubMed.
- W. Zhang, Y. Cai, R. G. Xiong, H. Yoshikawa and K. Awaga, Angew. Chem., Int. Ed., 2010, 49, 6608–6610 CrossRef CAS PubMed.
- W.-Y. Zhang, Q. Ye, D.-W. Fu and R.-G. Xiong, Adv. Funct. Mater., 2017, 27, 1603945 CrossRef.
- H. Y. Ye, J. Z. Ge, Y. Y. Tang, P. F. Li, Y. Zhang, Y. M. You and R. G. Xiong, J. Am. Chem. Soc., 2016, 138, 13175–13178 CrossRef CAS PubMed.
- Z. Sun, T. Chen, C. Ji, S. Zhang, S. Zhao, M. Hong and J. Luo, Chem. Mater., 2015, 27, 4493–4498 CrossRef CAS.
- T. Akutagawa, S. Takeda, T. Hasegawa and T. Nakamura, J. Am. Chem. Soc., 2004, 126, 291–294 CrossRef CAS PubMed.
- Z.-Y. Du, Y.-P. Zhao, C.-T. He, B.-Y. Wang, W. Xue, H.-L. Zhou, J. Bai, B. Huang, W.-X. Zhang and X.-M. Chen, Cryst. Growth Des., 2014, 14, 3903–3909 CrossRef CAS.
- K. Awaga and R. G. Xiong, J. Am. Chem. Soc., 2012, 134, 18487–18490 CrossRef PubMed.
- Z. Sun, Y. Tang, S. Zhang, C. Ji, T. Chen and J. Luo, Adv. Mater., 2015, 27, 4795–4801 CrossRef CAS PubMed.
- H. Zheng, J. B. Rivest, T. A. Miller, B. Sadtler, A. Lindenberg, M. F. Toney, L.-W. Wang, C. Kisielowski and A. P. Alivisatos, Science, 2011, 333, 206–209 CrossRef CAS PubMed.
- K. Gao, M. Gu, X. Qiu, X. N. Ying, H.-Y. Ye, Y. Zhang, J. Sun, X. Meng, F. M. Zhang, D. Wu, H.-L. Cai and X. S. Wu, J. Mater. Chem. C, 2014, 2, 9957–9963 RSC.
- Z. Sun, X. Wang, J. Luo, S. Zhang, D. Yuan and M. Hong, J. Mater. Chem. C, 2013, 1, 2561 RSC.
- C. Ji, Z. Sun, S.-Q. Zhang, T. Chen, P. Zhou and J. Luo, J. Mater. Chem. C, 2014, 2, 567–572 RSC.
- C. Ji, Z. Sun, S.-Q. Zhang, T. Chen, P. Zhou, Y. Tang, S. Zhao and J. Luo, J. Mater. Chem. C, 2014, 2, 6134–6139 RSC.
- P. Zhou, Z. Sun, S. Zhang, C. Ji, S. Zhao, R.-G. Xiong and J. Luo, J. Mater. Chem. C, 2014, 2, 2341–2345 RSC.
- Y. Z. Tang, Y. M. Yu, J. B. Xiong, Y. H. Tan and H. R. Wen, J. Am. Chem. Soc., 2015, 137, 13345–13351 CrossRef CAS PubMed.
- W.-Q. Liao, J.-X. Gao, X.-N. Hua, X.-G. Chen and Y. Lu, J. Mater. Chem. C, 2017, 5, 11873–11878 RSC.
- P. P. Shi, Q. Ye, Q. Li, H. T. Wang, D. W. Fu, Y. Zhang and R. G. Xiong, Dalton Trans., 2015, 44, 8221–8231 RSC.
- L. Zhou, X. Zheng, P. P. Shi, Z. Zafar, H. Y. Ye, D. W. Fu and Q. Ye, Inorg. Chem., 2017, 56, 3238–3244 CrossRef CAS PubMed.
- Y.-L. Liu, D.-H. Wu, Z. Wang and Y. Zhang, New J. Chem., 2017, 41, 3211–3216 RSC.
- Y. Lu, X. N. Hua, W. Q. Liao, J. X. Gao and Z. Yin, Dalton Trans., 2017, 46, 12760–12765 RSC.
- X. Zheng, P.-P. Shi, Y. Lu, L. Zhou, J.-X. Gao, F.-J. Geng, D.-H. Wu, D.-W. Fu and Q. Ye, Inorg. Chem. Front., 2017, 4, 1445–1450 RSC.
- F. J. Geng, D. H. Wu, L. Zhou, P. P. Shi, P. F. Li, J. X. Gao, X. Zheng, D. W. Fu and Q. Ye, Dalton Trans., 2017, 46, 9528–9534 RSC.
- Y. Tang, C. Ji, Z. Sun, S. Zhang, T. Chen and J. Luo, Chem. – Asian J., 2014, 9, 1771–1776 CrossRef CAS PubMed.
- J.-Z. Ge, X.-Q. Fu, T. Hang, Q. Ye and R.-G. Xiong, Cryst. Growth Des., 2010, 10, 3632–3637 CrossRef CAS.
- Y.-Z. Tang, Z.-F. Gu, J.-B. Xiong, J.-X. Gao, Y. Liu, B. Wang, Y.-H. Tan and Q. Xu, Chem. Mater., 2016, 28, 4476–4482 CrossRef CAS.
- D.-H. Wu, J.-Z. Ge, H.-L. Cai, W. Zhang and R.-G. Xiong, CrystEngComm, 2011, 13, 319–324 RSC.
- M. A. Asghar, J. Zhang, S. Han, Z. Sun, C. Ji, A. Zeb and J. Luo, Chin. Chem. Lett., 2018, 29, 285–288 CrossRef CAS.
- Y. Zhou, T. Chen, Z. Sun, S. Zhang, C. Ji, C. Song and J. Luo, Chem. – Asian J., 2015, 10, 247–251 CrossRef CAS PubMed.
- K. Tao, Z. Wu, S. Han, J. Zhang, C. Ji, Y. Wang, W. Zhang, J. Luo and Z. Sun, J. Am. Chem. Soc., 2018, 6, 4150–4155 CAS.
- T. Khan, M. A. Asghar, Z. Sun, C. Ji, L. Li, S. Zhao and J. Luo, RSC Adv., 2016, 6, 69546–69550 RSC.
- J. M. Bermudez-Garcia, M. Sanchez-Andujar and M. A. Senaris-Rodriguez, J. Phys. Chem. Lett., 2017, 8, 4419–4423 CrossRef CAS PubMed.
- M. Sánchez-Andújar, L. C. Gómez-Aguirre, B. Pato Doldán, S. Yáñez-Vilar, R. Artiaga, A. L. Llamas-Saiz, R. S. Manna, F. Schnelle, M. Lang, F. Ritter, A. A. Haghighirad and M. A. Señarís-Rodríguez, CrystEngComm, 2014, 16, 3558 RSC.
- J. M. Bermudez-Garcia, M. Sanchez-Andujar, S. Castro-Garcia, J. Lopez-Beceiro, R. Artiaga and M. A. Senaris-Rodriguez, Nat. Commun., 2017, 8, 15715 CrossRef CAS PubMed.
- P. Lloveras, E. Stern-Taulats, M. Barrio, J. L. Tamarit, S. Crossley, W. Li, V. Pomjakushin, A. Planes, L. Manosa, N. D. Mathur and X. Moya, Nat. Commun., 2015, 6, 8801 CrossRef CAS PubMed.
- X. Moya, S. Kar-Narayan and N. D. Mathur, Nat. Mater., 2014, 13, 439–450 CrossRef CAS PubMed.
- Y. Liu, J. Wei, P.-E. Janolin, I. C. Infante, X. Lou and B. Dkhil, Appl. Phys. Lett., 2014, 104, 162904 CrossRef.
- J. M. Bermudez-Garcia, M. Sanchez-Andujar, S. Yanez-Vilar, S. Castro-Garcia, R. Artiaga, J. Lopez-Beceiro, L. Botana, A. Alegria and M. A. Senaris-Rodriguez, Inorg. Chem., 2015, 54, 11680–11687 CrossRef CAS PubMed.
- D. Matsunami, A. Fujita, K. Takenaka and M. Kano, Nat. Mater., 2015, 14, 73–78 CrossRef CAS PubMed.
- M. V. Gorev, E. V. Bogdanov and I. N. Flerov, J. Phys. D: Appl. Phys., 2017, 50, 384002 CrossRef.
- L. Zhou, P. Shi, X. Zheng, F. Geng, Q. Ye and D. Fu, Chem. Commun., 2018, 54, 13111–13114 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra, PXRD patterns, dielectric losses, packing diagrams, hydrogen-bond geometries and parameters of the crystal structures in compounds 1, 2 and 3. CCDC 1858051 (1, 223 K), 1858056 (1, 333 K), 1858057 (2, 298 K) and 1858060 (3, 298 K). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8nj03845g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |