
In silico design, synthesis, characterization and pharmacological evaluation of captopril conjugates in the treatment of renal fibrosis†
Swapnil Dashrath
Jadhav
 *,
Prafulla Balkrishna
Choudhari
and
Manish Sudesh
Bhatia
*,
Prafulla Balkrishna
Choudhari
and
Manish Sudesh
Bhatia
Department of Pharmaceutical Chemistry, Bharati Vidyapeeth College of Pharmacy, Near Chitranagari, Kolhapur, Maharashtra 416013, India. E-mail: swapnil.mpharm@gmail.com; bhatiamanish13@gmail.com; Fax: +91-0231-2638833; Tel: +91-0231-2637286
First published on 14th November 2018
Abstract
Renal fibrosis is a renal disorder whereby production of excess fibrous connective tissue in the glomerulus and proximal convoluted tubules will occur in a reparative or reactive process leading to severe conditions like surgery, replacement, etc. Such a condition needs pharmacotherapy with drugs reducing renal overload (captopril) and inflammation (taurine). In this research project, two chemical conjugates of captopril with taurine and glutamic acid were developed using in silico analysis for an improvement in bioavailability with a reduction in inflammation. The stability of these conjugates in sheep kidney cells and in human plasma along with transport across renal cells was investigated using in vitro protocols. The results of these studies have revealed that conjugates have retained desired interactions for transportability across renal epithelial cells and their bioactivity against ACE and TGF-β. Both conjugates A and B were found to be stable over a period of 14 h in plasma and transported nearly 2 times more than captopril across renal epithelial cells. These conjugates were almost entirely hydrolyzed in renal lysosomes over a period of 14 h (87.39 ± 2.59%). Thus a combination of these two conjugates would be an effective chemotherapy to prevent progression of renal fibrosis to renal failure after in vivo studies of these conjugates.
1. Introduction
New drug development requires more than 15 years with thousand millions’ worth of costs incurred and most of them may fail in clinical trials or may produce adverse or side effects at both desired as well as undesired sites of action. Hence it is urgent to design selective and specific drugs, acting at desired sites.1 Fibrosis is the formation of excess fibrous connective tissue in lungs, skin, liver, kidney, bones, heart, shoulders, etc. in a reparative or reactive process.2 Most of these will lead to severe conditions like surgery, organ replacement, etc. Several cellular pathways, including mesangial and fibroblast activation as well as tubular epithelial–mesenchymal transitions, have been identified as the major avenues for the generation of matrix-producing cells in diseased conditions.3 Amongst the many fibrogenic factors that regulate renal fibrotic processes, transforming growth factor-beta (TGF-β) is one that plays a central role. Activated signalling cascades in the proximal tubular cells of the kidneys play a crucial role in the development of tubule-interstitial fibrosis. Inhibition of these signalling cascades with locally delivered therapeutics is an attractive approach to minimize the risk of unwanted side effects and to enhance their efficacy within the renal tissue.4Anti-hypertensive agents, anti-fibrotic agents, anti-inflammatory agents or steroids are used to prevent progression of fibrosis to chronic kidney failure. The treatment of fibrosis with ACE inhibitors not only reduces proteinuria but also shows long term renoprotective effects that appear to be independent, at least partially, of the reduction of blood pressure.5,6 Thus, selective inhibition of renal ACE by captopril (Fig. 1) will reduce systemic side effects and thereby prevent the progression of renal fibrosis to chronic renal failure.7
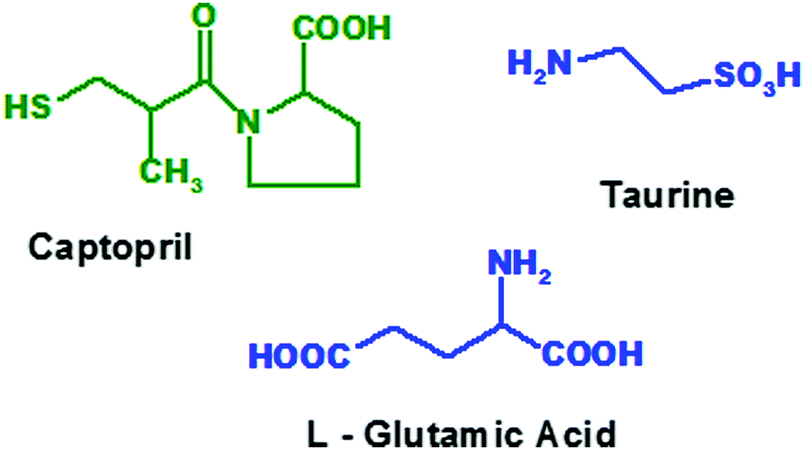 | ||
| Fig. 1 Structures of captopril, glutamic acid and taurine. | ||
Among the many fibrogenic factors that regulate renal fibrotic processes, TGF-β is one that plays a central role. Activated signalling cascades in the proximal tubular cells of the kidneys play a crucial role in the development of tubule-interstitial fibrosis. Inhibition of these signalling cascades with locally delivered therapeutics is an attractive approach to minimize the risk of unwanted side effects and to enhance their efficacy within the renal tissue.4 Moreover, taurine (Fig. 1), an antioxidant amino acid, is also found to reduce the TGF-β induced fibrosis and thus it is useful in preventing progressive renal fibrosis.8
Earlier, captopril has been conjugated with low molecular weight proteins leading to a six fold increase in its concentration as reported by Kok.9 But due to its macromolecular size, it needed to be administered subcutaneously as an infusion.10 As per the findings of Peng et al.,11 it has been observed that in most kidney targeted drug delivery systems the delivery of drugs is based on improving targeting efficiency and seeking new carriers. As the conjugation of prednisolone with glutamic acid will increase its bio-distribution in kidneys with lower toxicity,12 glutamic acid has been shortlisted for renal targeting.
It has been observed that a significant decrease in extracellular matrix protein expression was obtained using ACE inhibitors and taurine.8 Hence in the proposed project, we focused on the chemical conjugation of captopril with glutamic acid to form conjugate A i.e. acylated-captopril-glutamic acid (ACE-CAP-GLU) and with taurine to form conjugate B i.e. acylated-captopril-taurine (ACE-CAP-TAU) (Fig. 2). The screening of these conjugates computationally and using in vitro protocols for transportability and biological assessment could lead to effective therapeutic agents for the treatment of renal fibrosis.
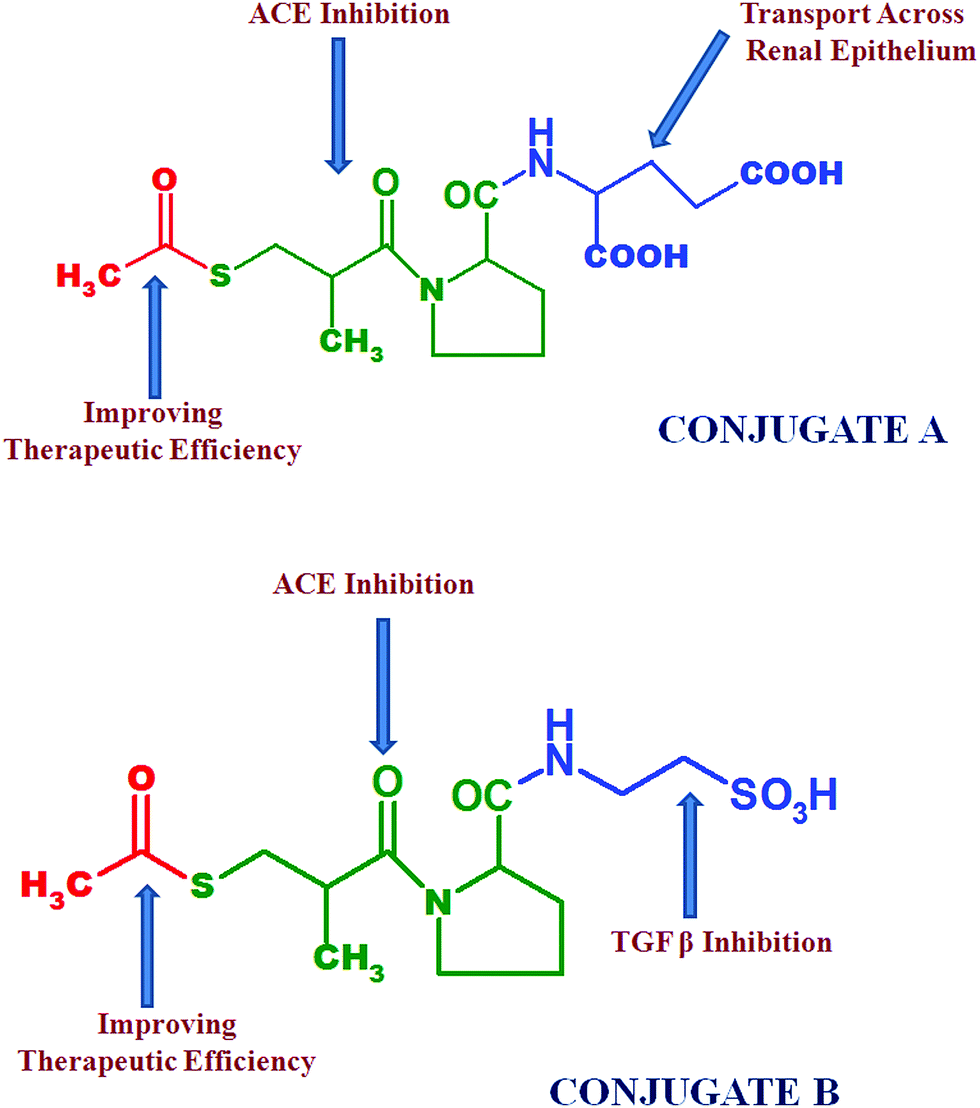 | ||
| Fig. 2 Structures of conjugate A and conjugate B; emphasizing the specific structural parts responsible for targeted purposes. | ||
2. Materials and methods
2.1 Materials
2.2 Methods
Homology modelling of EAAT3. The excitatory amino acid transporter 3 (EAAT3) primary amino acid sequence was downloaded from the UniProtKB database with accession number P43005. Protein blast was performed to identify suitable homologue sequences for development of the protein structure with known protein databases. 5mju.1.A was utilised as a template for homology modelling based on the percentage similarity and method of identification. Using the selected template, a homology model of EAAT3 was generated with the SWISS-MODEL protein server.13,14 The Vlife MDS 4.4 software was utilised to visualize the 3D structure and to generate the Ramachandran plot (Fig. 3) to ascertain the structure validity.
 | ||
| Fig. 3 Ramachandran plot for the homology model of EAAT3 generated for its structural validity using Vlife MDS 4.4 software. | ||
Docking analysis using the homology model of EAAT3.
Ligand preparation. The structures of the glutamic acid and conjugate A were built in the 2D drawing mode of Vlife MDS 4.4. These 2D conformations were converted to 3D conformations using the molecule design suite. For docking studies, the ligand geometries were optimized by energy minimization using force field MMFF and modified Qeq charges for the atoms, until a gradient of 0.001 kcal mol−1 Å−1 was reached. A conformation search was carried out to identify the lowest energy conformation, using the systematic search of the MDS engine.
Docking studies. Docking analysis was performed to predict the transport mechanism of the synthesised conjugates across the kidney cell. The developed homology model of the glutamate transporter was utilised to study the docking analysis. A grip based docking simulation was performed, keeping the protein structure of the EAAT3 in a rigid conformation, with the synthesized conjugate in a flexible conformation. The L-glutamic acid was utilised as a reference to validate the docking results.
Docking analysis using angiotensin converting enzyme (PDB ID – 1UZF). To explore the interactions of conjugate A, binding simulations have been carried out using the BioPredicta module of Vlife Molecular Design Suite 4.4.15 The crystal structure of angiotensin converting enzyme (PDB ID – 1UZF) was downloaded from http://www.rcsb.org to perform docking simulations. The downloaded protein structure was optimized using Vlife Engine, keeping the native coordinates intact. Water molecules were removed and hydrogens were added to keep the native geometry of the protein structure. Energy minimized ligand structures were selected as micro-molecules for docking analysis. Grip based docking simulations were performed where the protein structure was kept rigid and ligand structures were kept flexible to attain a number of conformations. The best docked conformation of the ligand was selected based on binding energy and interaction profiles.
Pharmacophore identification. Pharmacophore identification of captopril was carried out using the MolSign module of Vlife MDS 4.4, to get insight into the basic pharmacophoric requirements for ACE inhibition. Identified pharmacophoric features were considered in the design of conjugate B, keeping the native pharmacophore intact. The pharmacophore of the designed conjugate was further utilised to validate the results.
Docking analysis using ACE (PDB ID – 1UZF) and TGF-β (PDB ID – 1TGK). To explore the interactions of captopril and conjugate B, binding simulations were carried out using the BioPredicta module of Vlife Molecular Design Suite 4.4. The crystal structures of the angiotensin converting enzymes (PDB ID – 1UZF) and TGF-β (PDB ID – 1TGK) were downloaded from http://www.rcsb.org to perform docking simulations. Each of the downloaded protein structures was optimized using Vlife Engine, keeping the native coordinates intact. Water molecules were removed and hydrogens were added to keep the native geometry of the protein structure. Energy minimized ligand structures were selected as micro-molecules for docking analysis. Grip based docking simulations were performed where the protein structure was kept rigid and ligand structures were kept flexible to attain a number of conformations. The best docked conformation of the ligands was selected based on binding energy and interaction profiles.
Step I: synthesis of acylated captopril. Captopril (6.2 g, 29 mM) was put in a 100 mL glass beaker. Acetyl chloride (1.5 mL, 24 mM) was added to it dropwise using a dropping funnel with continuous stirring. The product was kept aside for 5 min. It was then dried at 60 °C for 15 min to remove unreacted acetyl chloride (b.p. 57 °C) in a hot air oven.
Step II: synthesis of mixed anhydride of acylated captopril. Acylated captopril (5.425 g, 21 mM) from the above step I was dissolved in 15 mL of dry chloroform containing 3.4 mL of dried and redistilled triethylamine. It was cooled in ice. During cooling, 2.4 mL (25 mM) of ethyl chloroformate was added steadily with continuous shaking. The product was set aside at 0 °C for 15 min.
Synthesis of conjugate A (Fig. 4).
Step III: synthesis of ACE-CAP-GLU. L-Glutamic acid (3.7 g, 25 mM) was dissolved in 20 mL of dry chloroform containing 3.4 mL of triethylamine. The product from step II was added to this reaction mixture with vigorous shaking. This reaction mixture was stirred until evolution of carbon dioxide was completed and the solid was converted into solution. This mixture was set aside for 30 min and thereafter warmed at 50 °C for 10 min. The product was washed with 15 mL of water and then twice (10 mL each) with 1 M hydrochloric acid. The product was dried over anhydrous sodium sulfate and if present, chloroform was removed using a vacuum pump. The crystals of the product were recrystallized from dilute ethanol.
Synthesis of conjugate B (Fig. 4).
Step III: synthesis of ACE-CAP-TAU conjugate. Taurine (3.7 g) was dissolved in 20 mL of dry chloroform containing 3.4 mL of triethylamine. The mixture from step II was added to this reaction mixture with vigorous shaking. This mixture was stirred until evolution of carbon dioxide was completed and the solid was converted into solution. This mixture was set aside for 30 min and thereafter warmed at 50 °C for 10 min. The product was washed with 15 mL of water and then twice (10 mL each) with 1 M hydrochloric acid. The product was dried over anhydrous sodium sulfate and chloroform was removed using a vacuum pump. The crystals of the product were recrystallized from dilute ethanol.
The melting points of captopril, taurine, and both conjugates were determined by the capillary tube method using Thiele tubes. The purity of conjugates was assessed by analysis on the HPLC system using a Luna HILLIC column as the stationary phase and acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (70:30% v/v) with 0.1% acetic acid at a flow rate of 1 mL min−1 and 237 nm as the wavelength of analysis.17 The percentage purities of individual synthesized compounds were determined by considering the areas of peaks of starting materials and synthesized compounds.
:water (70:30% v/v) with 0.1% acetic acid at a flow rate of 1 mL min−1 and 237 nm as the wavelength of analysis.17 The percentage purities of individual synthesized compounds were determined by considering the areas of peaks of starting materials and synthesized compounds.
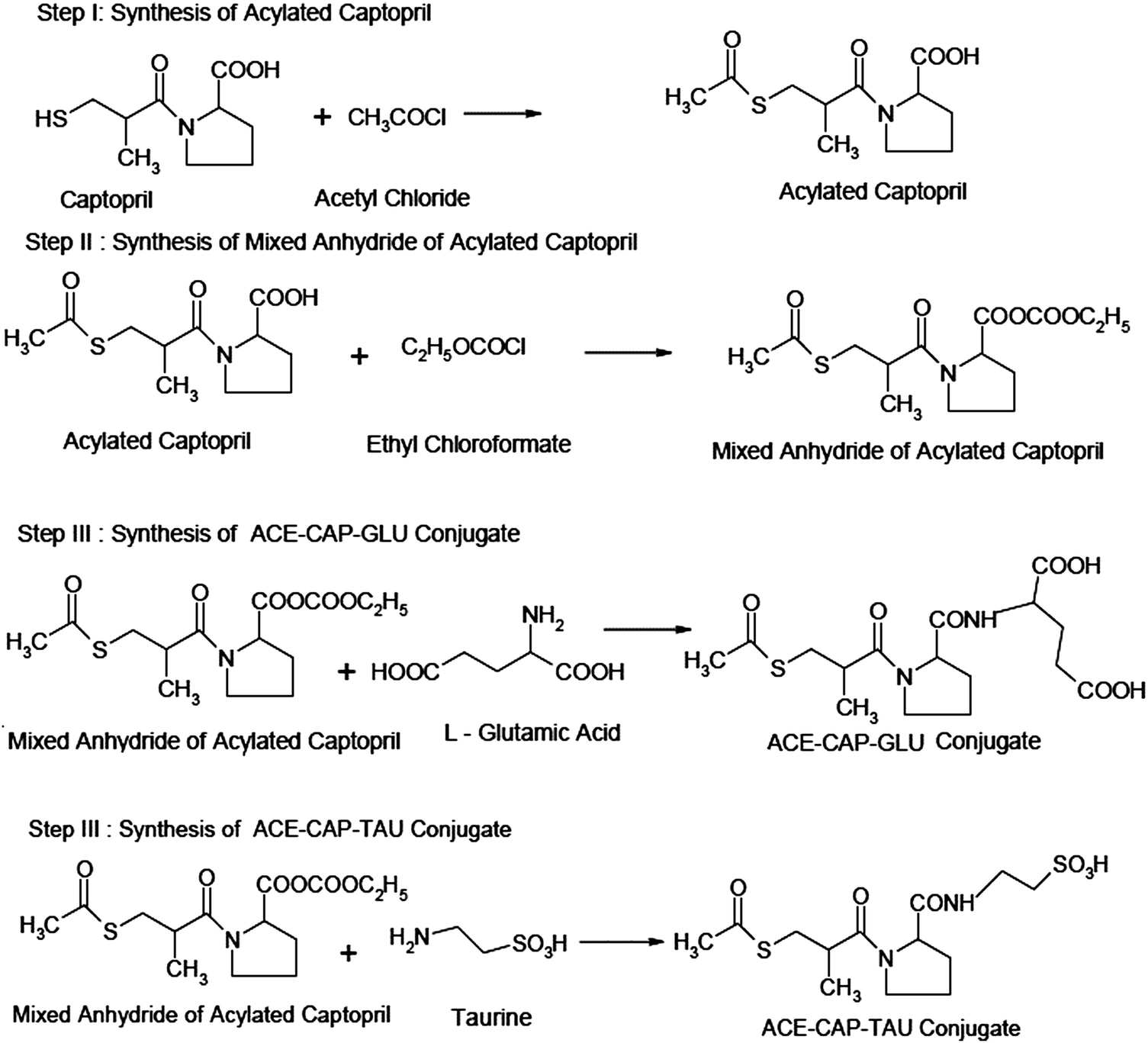 | ||
| Fig. 4 Synthesis of conjugate A and conjugate B: the first two steps are the same for both conjugates with different third steps of the synthesis. | ||
Conjugate A. Color: colorless, yield: 89%, purity: 92.45%, m.p.: 190–194 °C, mass: [M + 1]+m/z 389, IR: C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O as amide (1681 cm−1), NH (3399 cm−1), NMR: 1H NMR (CDCl3-d3, 300 MHz): δ = 1.11 (s, 3H, methyl), 1.82, 2.05, 2.36 (m, 6H, CH2 in pyrrolidine), 4.25 (t, 1H, CH in pyrrolidine), 3.49 (d, 2H, CH2), 3.74 (d, 2H, pyrrolidine), 2.61 (s, 3H, methyl), 2.88 (m, 1H, methane), 4.31 (t, 1H, methine), 8.21 (s, 1H, NH sec. amide), and 11.07 (s, 2H, carboxylic acid).
O as amide (1681 cm−1), NH (3399 cm−1), NMR: 1H NMR (CDCl3-d3, 300 MHz): δ = 1.11 (s, 3H, methyl), 1.82, 2.05, 2.36 (m, 6H, CH2 in pyrrolidine), 4.25 (t, 1H, CH in pyrrolidine), 3.49 (d, 2H, CH2), 3.74 (d, 2H, pyrrolidine), 2.61 (s, 3H, methyl), 2.88 (m, 1H, methane), 4.31 (t, 1H, methine), 8.21 (s, 1H, NH sec. amide), and 11.07 (s, 2H, carboxylic acid).
Conjugate B. Color: pale yellow, yield: 88%, purity: 95.19%, m.p.: 204–206 °C, mass: [M + 1]+m/z 367, IR: C
O as amide (1683 cm−1), OSO (1427, 1147 cm−1), NH (3209 cm−1), OH (3350–3540 cm−1) NMR: 1H NMR (CDCl3-d3, 300 MHz): δ = 1.67, 2.25, 3.19 (m, 6H, CH2 in pyrrolidine), 2.59 (s, 3H, CH3), 1.92 (s, 1H, OH), 2.72 (m, 1H, methine), 4.24 (t, 1H, CH in pyrrolidine), 3.40 (d, 2H, CH2) 1.31 (s, 3H, CH3), 7.72 (s, 1H, NH), 3.61, and 3.84 (t, 4H, CH2).
Stability in plasma. The blood required for stability studies of the conjugates was obtained in our laboratory by our campus doctor. The investigators’ own blood samples were obtained by medial cubital vein puncture. Approximately 4 IU of heparin was added to each μL of blood to prevent coagulation. Immediately after the collection of plasma, one of the conjugates was added to produce blood concentrations of 40 μg mL−1 (n = 3), and the samples were incubated at 37 °C in a shaking water bath for 24 h. At predetermined time points, one mL of sample was taken for the concentration assay of free captopril or taurine or acylated captopril by the above developed HPLC method. For a control, samples were also prepared in an isotonic phosphate buffer of pH = 7.4, instead of blood, and subjected to identical procedures as those for the blood samples. The results of these studies are reported in Fig. 5.
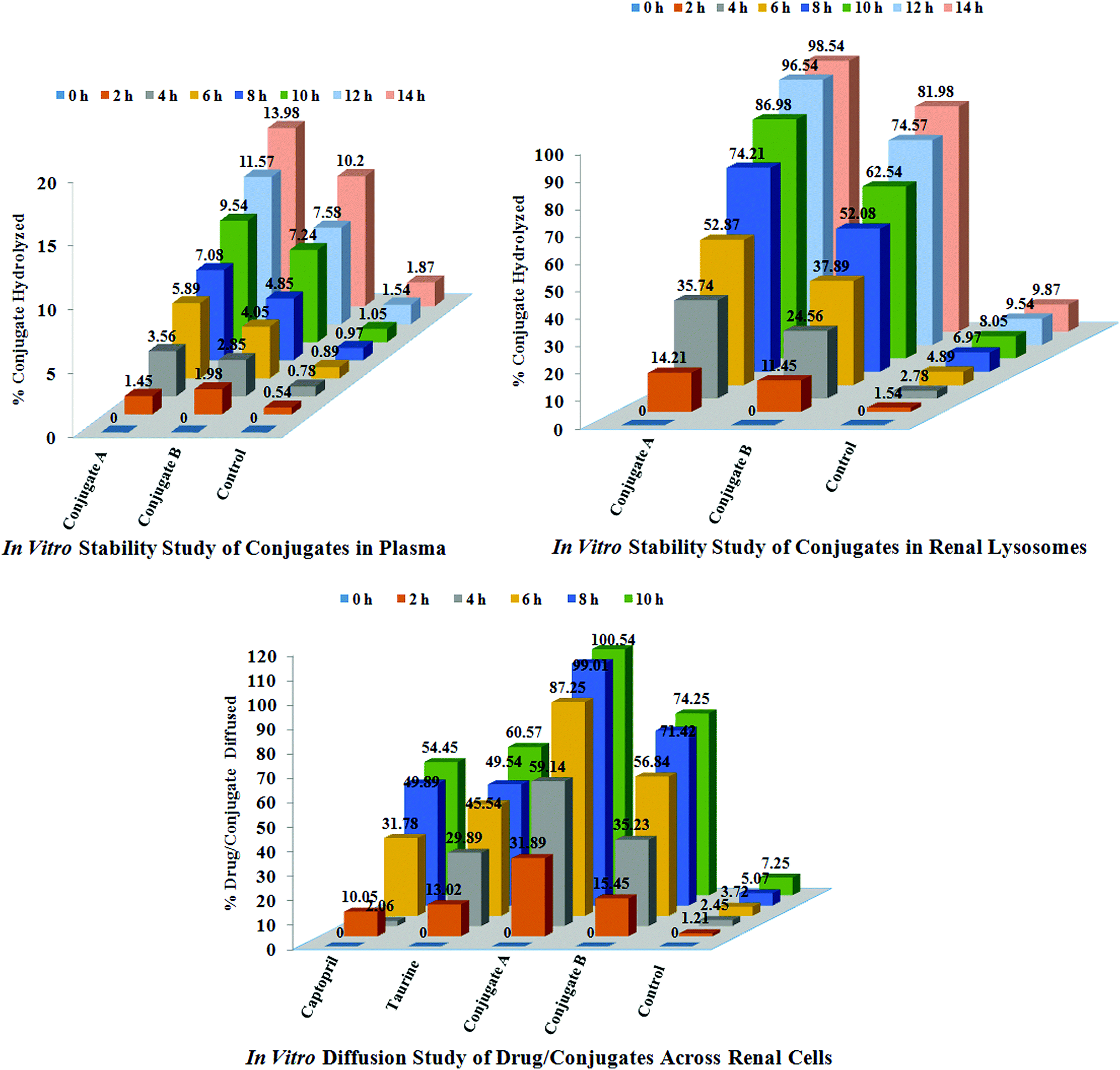 | ||
| Fig. 5 In vitro studies of conjugates and drugs; stability studies of conjugates A and B in plasma and renal lysosomes and diffusion studies of conjugates and drugs across renal cells of sheep. | ||
Diffusion across renal cells. The intact kidneys of sheep were obtained from a slaughter house. These were repeatedly washed with hot water and sterile phosphate buffered saline, pH 7.6. After washings, these kidneys were stored in fresh 50 mL sterile phosphate buffered saline, pH 7.6 (n = 3). The standard solutions (0.5 mM) of captopril, glutamic acid, taurine, ACE-CAP-GLU and ACE-CAP-TAU were prepared and 1 mL of this solution was incubated with the kidneys at 37 °C separately. After 60 min, 5 mL of sterile buffer solution was taken and diluted with the mobile phase. These solutions were further diluted if required and analysed using HPLC and quantification of individual analytes was carried out. These processes were repeated up to 10 h. Based on these results, concentrations of these analytes diffused across kidneys were calculated using HPLC and expressed as percentages of analytes diffused as shown in Fig. 5.
Stability in renal lysosomes. Lysosomes were isolated from sheep kidneys according to a reported method with minor modifications.19 Kidneys were removed and then homogenized in 0.25 mol L−1 sucrose and subjected to differential centrifugation which resulted in the collection of a lysosomal fraction. The potency of the lysosomal fraction was tested by the activity of acid phosphatase which is the marker for the lysosomes. The specific activity of the enzyme was calculated, in both the kidney homogenate and the lysosomal fraction, as the ratio of the enzyme activity per mg of protein. The activity of acid phosphatase was measured using a colorimetric assay. In agreement with previous studies, the specific enzyme activity in the lysosomal fraction was 30-fold higher than that in the homogenate.
The lysosomal fraction was stored at −40 °C and used within one week of preparation. For lysosomal hydrolysis studies (n = 3), a solution of conjugate was added to the lysosomal fraction to produce final concentrations of 40 μg mL−1, and samples were incubated at 37 °C in a shaking water bath for 24 h. At a predetermined time interval, one mL of solution was removed, and the concentration of captopril or taurine was analysed using HPLC. For the control, similar samples were prepared by addition of drug solutions to 0.25 M sucrose (instead of the lysosomal fraction), and subjected to procedures identical to those for the lysosome samples. The results of these studies are reported in Fig. 5.
Cytotoxicity assay. The cytotoxicity was studied using the brine-shrimp lethality assay method.20,21 Brine-shrimp (Artemia salina) eggs were hatched in artificial sea water (40 g sea salts per L) at room temperature (22–29 °C). After two days, these shrimps were transferred to vials (10 shrimps per vial) containing artificial sea water (5 mL) with 500, 50 and 10 μg mL−1 final concentrations of each compound taken from their stock solutions of 20 mg mL−1 in DMSO. After 24 h, the number of surviving shrimps was counted. Data was analyzed with a Finney computer programme (Probit analysis) to determine LC50 values. The experimental model chosen for toxicity studies does not require the approval of the institutional animal ethics committee.
3. Results and discussion
The project is mainly focused on the development of active or passive targeting drug delivery systems for the treatment of renal fibrosis. This focus was proposed to be achieved by developing drug conjugates with maximum bio-distribution in the glomerulus and proximal tubular cells. The developed targeted drug delivery systems contain therapeutic agents, targeting molecules (biomolecules) and carrier systems.Captopril selected as a therapeutic agent is known for being useful in inhibiting ACE in kidneys, and in the developed product is intended to reduce the preload over kidneys and thereby contribute to the prevention of chronic renal disorders.6,9,10,22 Like alacepril, thiol group acylated captopril is also likely to have advantages of improved potency, gradual onset and long lasting action after oral administration.23 The hypothesis supporting the selection of glutamic acid for targeted delivery is based on its relatively higher biodistribution in the kidney coupled with relatively higher expression of glutamic acid metabolising enzymes like glutamic acid decarboxylase,24,25 gamma-glutamyl transpeptidase and aromatic L-amino acid decarboxylase,26 glutamine synthetase and gluconeogenesis enzymes27 in the kidneys. Most conjugates of drugs with glutamic acid monomers or polymers would be effectively hydrolysed in kidneys before the metabolism of glutamic acid.28 Similarly, taurine has been found to inhibit production of reactive oxygen species (anti-oxidant) and it inhibits TGF-β induced age related progressive renal fibrosis.8 Thus, the developed products which are conjugates of thiol group acylated captopril with taurine and glutamic acid are likely to be promising tools for therapy to prevent the progression of renal fibrosis to chronic kidney failure.
3.1 Designing of conjugate A for improving renal transport
Reports in the literature prompted the choice of glutamic acid for conjugation with captopril to improve delivery and selective biodistribution to the targeted renal tissues. This hypothesis was supported by results of our in silico docking experiments with glutamic acid transporters that are responsible for transportability across renal epithelial cells. EAAC1 (EAAT3), GLT1 (EAAT2), GLAST (EAAT1), EAAT4, and EAAT5 are five isoforms of glutamate transporters in humans.29 Bailey's30 and Robert's31 findings have proven that amongst these five isoforms, EAAT3, also known as SLC1A1, is the major renal transporter of glutamate and aspartate in humans in human kidneys across the renal epithelium. But protein database records for EAAT3 were not available for computational studies, hence it became necessary to develop a homology model for this transporter protein. The homology model for EAAT3 was developed using the SWISS-MODEL protein server using 5mju.1.A as the template. The homologous model was validated using a Ramachandran plot (Fig. 3) to ascertain the protein structure. The developed model showed similar structural features with the model reported by Heinzelmann.32,33The in silico transport study of the synthesised conjugate was performed using the homology modelled protein structure of EAAT3 using glutamic acid as the reference molecule in Vlife MDS 4.4 software. Glutamic acid was found to be interacting with EAAT3 via formation of hydrogen bond interactions with R447, T448, and N451, charge interactions with D444, R447, and T448 and hydrophobic interactions with R447 and T448. The modelled conjugate A has shown hydrogen bond interactions with R447, T448, and N451, charge interactions with R447 and hydrophobic interactions with T370, D444, R447, T448, and N451. The hydrogen bond and charge interactions of conjugate A have more than 80 percent similarities with interactions of the native ligand, glutamic acid. These results (Fig. 6) indicate that the transport across renal epithelia of conjugate A and glutamic acid would follow the same mechanism and conjugation of glutamic acid with captopril would not affect transport significantly. Thus, conjugation of captopril with glutamic acid will definitely lead to improved transport of captopril to the target site.
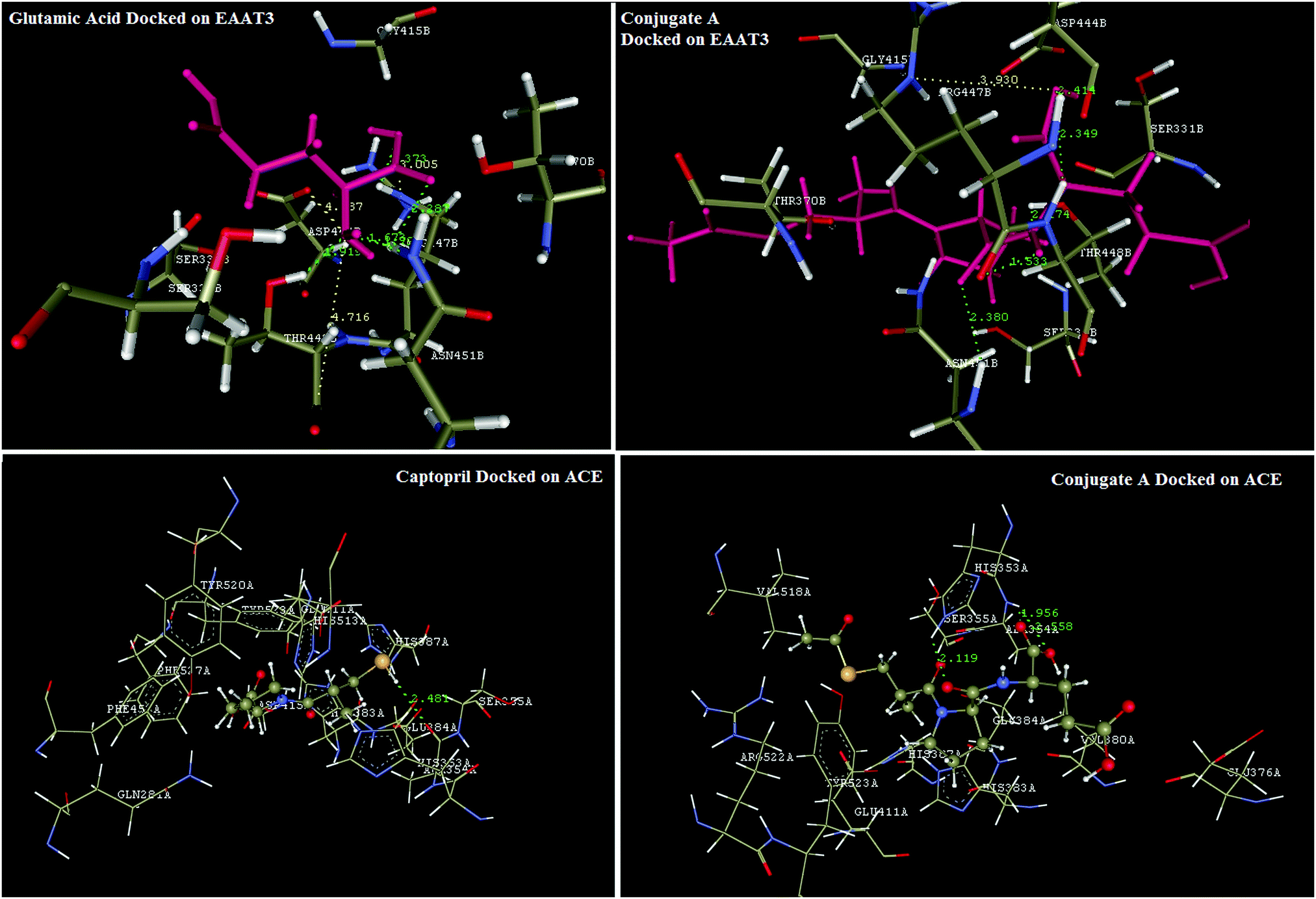 | ||
| Fig. 6 In silico designing of conjugate A for renal transport across kidneys; docking studies of glutamic acid and conjugate A on the EAAT3-glutamic acid transporter in human kidney and docking studies of captopril and conjugate A on ACE using Vlife MDS 4.4 software. | ||
Docking simulations were also performed to validate the binding mode of captopril and its synthesized glutamyl conjugate on ACE. Captopril has shown characteristic interactions with key amino acid residues like Q281, H353, E384, K511, H513, and Y520 via formation of hydrogen bond and charge interactions. Synthesized conjugate A has also shown binding interactions similar to those of captopril. These results confirm that the conjugation would not significantly affect bonding of the conjugate A to ACE and thus the bioactivity of captopril will not be affected. Results of the docking simulation are reflected in Fig. 6.
3.2 Designing of conjugate B for reducing inflammation
Pharmacophore identification indicated that a four point pharmacophore was an essential feature of captopril for ACE inhibition. In captopril the hydrogen bond donor, aliphatic carbon and negative ionisables are the key features. Design of the conjugate B has not affected any of the critical features of captopril essential for interaction with ACE. In conjugate B, the sulfhydryl group of the captopril was acylated for increasing therapeutic efficacy as in that of alacepril23 and the free carboxylic group of captopril was modified too by forming a neutral amide bond with taurine. These modifications have led to a novel structure with predictable pharmacokinetic improvements as in alacepril along with possibilities of TGF-β mediated benefits in renal fibrosis. Docking interaction analysis of this designed conjugate B was done using both ACE and TGF-β (Fig. 7).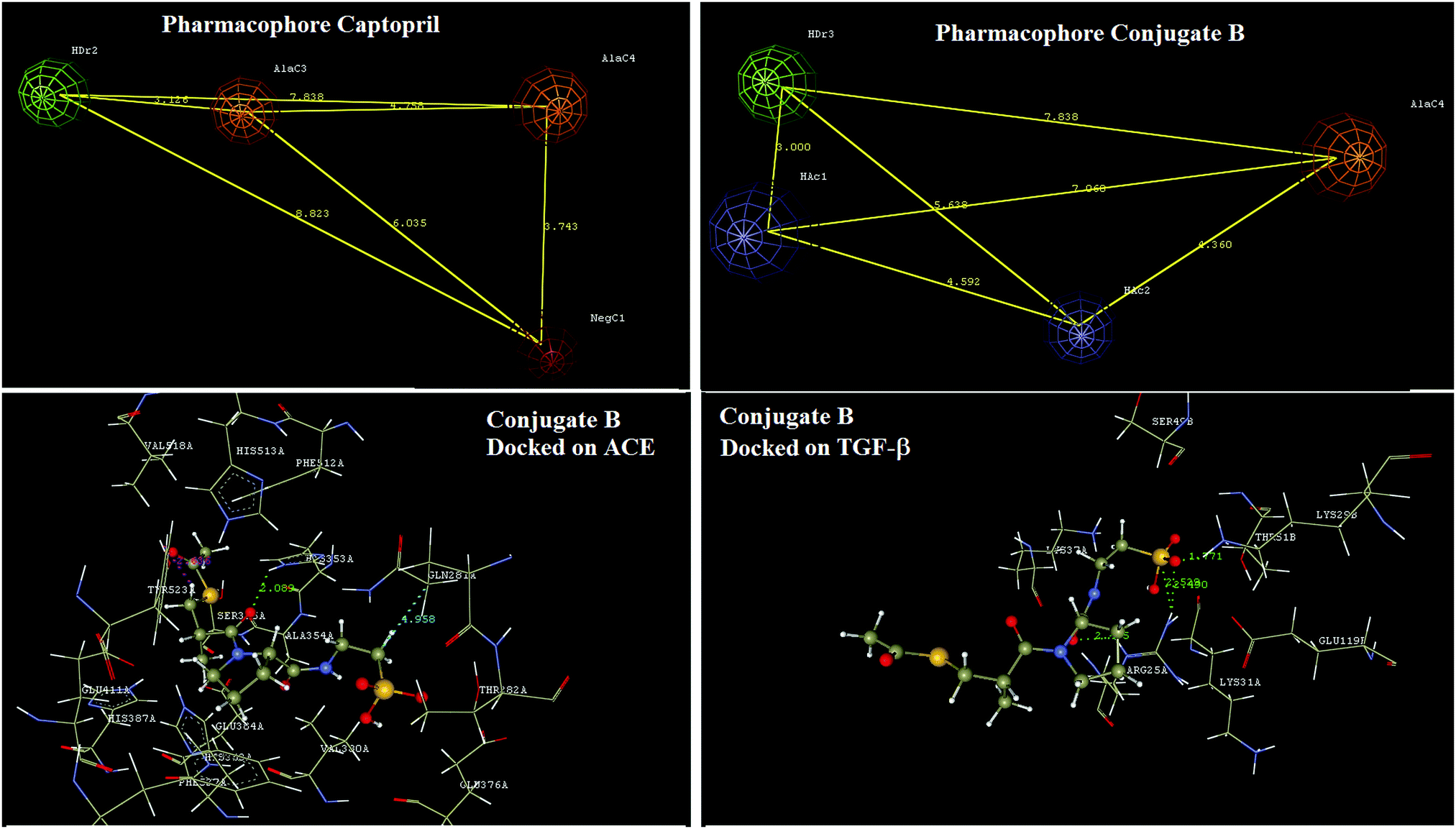 | ||
| Fig. 7 In silico designing of conjugate B for reducing inflammation; pharmacophore development for captopril, conjugate B and docking studies of conjugate B on ACE and TGF-β using Vlife MDS 4.4 software. | ||
Docking interaction analysis has shown conjugate B interacting with ACE via formation of hydrogen bond interactions with H353 and a charge interaction with Y523. Similarly, conjugate B has shown docking interactions with TGF-β via formation of hydrogen bonds with R25 and K29. Docking analysis results have also shown that two exclusively different structural domains of conjugate B interact with the above two proteins and thus will have no structural interference in the binding at either protein.
3.3 Synthesis of conjugates
The first step in synthesizing both conjugates involved acylation of captopril at the terminal thiol group, as Kumar23 has reported an increase in efficacy due to acylation of the thiol which is exemplified by alacepril. Acylation was carried out using acetyl chloride which offers the advantage of easily removable by-products like hydrochloric acid and excess acetyl chloride (b.p. 57 °C) which are removed by heating at 60 °C. Initially acylation was done to protect the thiol group which would have interfered in subsequent reactions but the deacylation step was omitted keeping the above cited advantage in view. In the next step, the free carboxyl group of acylated captopril could have been activated either by chlorination or by mixed anhydride formation.16 But, mixed anhydride formation method was adopted because it led to high purity products with least and easily separable by-products. In the last step, acylated and carboxyl activated captopril was shaken vigorously with glutamic acid or taurine in dry chloroform to form conjugate A or B, respectively. The reaction progressed with evolution of carbon dioxide and subsequent complete solubilisation of the entire solid mass. On heating at about 50 °C, the solid separated out in about 10 min. The product was then washed once with water and twice with hydrochloric acid. The product was dried over anhydrous sodium sulfate. Thus these conjugates were produced with the highest purity as unreacted reagents, by-products, and the reaction medium were removed.3.4 Characterization of conjugates
The melting points confirmed formation of these conjugates as the melting points were different from those of captopril, taurine and glutamic acid, determined under similar conditions. Melting points of both conjugates were higher than that of captopril which is in agreement with the increased polarity of the conjugates and thus indicates formation of the designed product. Chromatographic analysis of these conjugates was carried out using reverse phase HPLC for which the chromatographic conditions were optimized after several trials. The results have confirmed the identity and purity of the conjugates synthesized.The structures of synthesized conjugates were confirmed by spectroscopic studies i.e. FT-IR, NMR and mass spectrometry. In the cases of conjugates A and B, the absence of an IR peak corresponding to free OH and SH as in captopril, and the presence of two characteristic peaks for both amide linkages has confirmed conjugation of the carboxyl carbonyl of captopril with the amino group of the respective amino acids. The number of protons indicated by P-NMR data of each conjugate and the presence of molecular ion peaks at m/z 389 and m/z 368 confirmed structures of both the conjugates A and B, respectively. The presence of two protons at δ 10.80 in the P-NMR data confirmed the presence of two free carboxyl groups of glutamic acid in conjugate A. The presence of a proton at δ 2.05 in P-NMR data has confirmed the presence of free SO2OH i.e. SO3H of taurine in conjugate B. Thus, spectral data has confirmed the structures of both conjugates.
3.5 In vitro studies of conjugates
Both the designed and developed conjugates of captopril involved conjugation of the drug with an amide linkage to amino acids which would aid the desired biodistribution of the conjugates. As amide prodrugs are reported to have insignificant hydrolytic release in the upper GIT,34–36 the primary objective of this study was focussed on generating data supporting the claim of targeting renal tissues. On the basis of reported work on amide prodrugs, we have presumed that the developed conjugates would have insignificant hydrolytic release of captopril in the upper GIT.| Compound | Concentration (μg mL−1) | Shrimp survived | Total number of shrimp alive | % inhibition | LC50 (μg mL−1) | ||
|---|---|---|---|---|---|---|---|
| Conjugate A | 1000 | 7 | 6 | 8 | 21 | 18.85 | 2652.51 |
| 100 | 8 | 7 | 9 | 24 | 14.58 | ||
| 10 | 9 | 8 | 8 | 25 | 12.28 | ||
| Conjugate B | 1000 | 7 | 8 | 7 | 22 | 17.21 | 2905.28 |
| 100 | 8 | 8 | 8 | 24 | 14.58 | ||
| 10 | 9 | 8 | 9 | 26 | 10.74 | ||
4. Conclusions
The synthesized conjugates are stable in systemic circulation and thus are likely to reach the target site of action (kidney) in considerable amounts. Conjugate A would ensure greater biodistribution of the conjugate in the renal cells and hence larger concentrations of captopril generated on hydrolysis by enzymes in renal lysosomes. Conjugate B would ensure biodistribution to renal cells and on subsequent hydrolysis in renal lysosomes, generate two bioactive compounds, captopril and taurine. Coupled to the benefits of captopril, taurine would aid in reducing inflammation and oxidative degeneration of renal cells. After generation of necessary in vivo data, an appropriate combination of the two developed conjugates could be a potential therapy for the prevention and treatment of renal fibrosis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are thankful to Principal, Bharati Vidyapeeth College of Pharmacy, Kolhapur for providing facilities for research work and Wockhardt Limited, Waluj, Maharashtra for providing gift sample of drug captopril for this research work.References
- R. Langer, Nature, 1998, 392, 5–10 CAS.
- B. F. Palmer, J. Invest. Med., 1997, 45, 346–361 CAS.
- A. A. Eddy, Pediatr. Nephrol., 2000, 15, 290–301 CrossRef CAS.
- M. E. M. Dolman, S. Harmsen, G. Storm, W. E. Hennink and R. J. Kok, Adv. Drug Delivery Rev., 2010, 62, 1344–1357 CrossRef CAS.
- M. Ruizortega, O. Lorenzo, M. Ruperez and J. Egido, Nephrol., Dial., Transplant., 2000, 15, 561–565 CrossRef CAS.
- M. R. Weir, Curr. Hypertens. Rep., 2000, 2, 497–499 CrossRef CAS.
- W. A. Windt, J. Prakash, R. J. Kok, F. Moolenaar, C. A. Kluppel, Z. D. De, D. R. P. Van and R. H. Henning, J. Renin-Angiotensin-Aldosterone Syst., 2004, 5(4), 197–202 CrossRef CAS.
- I. D. L. C. Carmen, R. T. Piedad, G. A. D. M. Raimundo, R. G. P. Manuel and R. G. P. Diego, Am. J. Physiol. Renal. Physiol., 2000, 278, F122–F129 CrossRef.
- R. J. Kok, R. F. G. Haverdings, F. Grijpstra, J. Koiter, D. Moolenaar, D. E. Zeeuw and D. K. F. Meijer, J. Pharmacol. Exp. Ther., 2002, 301(3), 1139–1143 CrossRef CAS.
- J. Prakash, L. W. Van, M. Annemiek, M. Haas, J. H. Proost, K. F. Dirk, M. F. Meijer, K. Poelstra and R. J. Kok, Drug Metab. Dispos., 2005, 33(5), 683–688 CrossRef CAS.
- Z. Peng, S. Xun and Z. Zhirong, Acta Pharm. Sin. B, 2014, 4(1), 37–42 CrossRef.
- M. Su, Q. He, Z. R. Zhang, B. Hu and S. W. Liu, Acta Pharmacol. Sin., 2003, 38(8), 627–630 CAS.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46(W1), W296–W303 CrossRef.
- M. Bertoni, F. Kiefer, M. Biasini, L. Bordoli and T. Schwede, Sci. Rep., 2017, 7(1), 10480 CrossRef.
- A. A. Patravale, A. H. Gore, G. B. Kolekar, M. B. Deshmukh, P. B. Choudhari, M. S. Bhatia, S. Prabhu, M. D. Jamdhade, M. S. Patole and P. V. Anbhule, J. Taiwan Inst. Chem. Eng., 2016, 68, 105–118 CrossRef CAS.
- B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, Longman Group, UK, 1989 Search PubMed.
- S. D. Jadhav and M. S. Bhatia, Indian Drugs Search PubMed , in press.
- R. Govindarajan, S. Manoharan, V. Purushothaman, N. D. J. Chandran and T. G. Prabhakar, Tamilnadu Journal of Veterinary Animal Sciences, 2008, 4(6), 215–218 Search PubMed.
- I. Yuji, Y. Hidenori, N. Miyuki, Y. Ayako, U. Tadashi and I. Taiji, Eur. J. Biochem., 1993, 213, 649–658 CrossRef.
- M. A. Berghot, E. M. Kandeel, A. H. Abdel-Rahman and M. Abdel-Motaal, Med. Chem., 2014, 4, 381–388 Search PubMed.
- M. J. Rieser, Z. M. Gu, X.-P. Fang, L. Zeng, K. V. Wood and J. L. McLaughlin, J. Nat. Prod., 1996, 59, 100–108 CrossRef CAS.
- Y. Sun and F. A. O. Mendelsohn, J. Cardiovasc. Pharmacol., 1991, 18(4), 478–486 CrossRef CAS.
- R. Kumar, R. Sharma, K. Bairwa, R. K. Roy, A. Kumar and A. Baruwa, Pharm. Lett., 2010, 2(3), 388–419 CAS.
- D. T. Whelan, C. R. Scriver and F. Mohyuddin, Nature, 1969, 224, 916–917 CrossRef CAS.
- Z. H. Liu, L. J. Striker, M. Hattori, C. W. Yang and G. E. Striker, Nephron, 1996, 72(4), 662–666 CrossRef CAS.
- M. Barthelmebs, A. Caillette, J. D. Ehrhardt, J. Velly and J. L. Imbs, Kidney Int., 1990, 37(6), 1414–1422 CrossRef CAS.
- D. A. Hems, Biochem. J., 1972, 130(3), 671–680 CrossRef CAS.
- M. C. G. van de Poll, P. B. Soeters, N. E. P. Deutz, C. H. F. Kenneth and H. C. D. Cornelis, Am. Soc. Clin. Nutri., 2004, 79, 185–197 CrossRef CAS.
- M. A. Hediger, Am. J. Physiol., 1999, 277(46), F487–F492 CAS.
- C. G. Bailey, R. M. Ryan, A. D. Thoeng, N. Cynthia, K. Kara, M. Jessica, V. C. Auray-Blais, R. J. Vandenberg, S. Bröer and E. J. R. John, J. Clin. Invest., 2011, 121(1), 446–453 CrossRef CAS PubMed.
- A. Robert, C. Michael and W. M. Orson, Seldin and Giebisch's The Kidney Physiology and Pathophysiology, Academic Press, USA, 2012 Search PubMed.
- G. Heinzelmann and S. Kuyucak, Biophys. J., 2014, 106, 2675–2683 CrossRef CAS.
- G. Heinzelmann and S. Kuyucak, Molecular Dynamics Simulations of the Mammalian Glutamate Transporter EAAT3, PLoS One, 2014, 9(3), e92089 CrossRef.
- T. Elsaman, O. A. A. Aldeeb, T. Aboul-Fadl and E. I. Hamedelneil, Bioorg. Chem., 2016, 70, 144–152 CrossRef.
- A. Rasheed, C. K. A. Kumar and A. Mishra, J. Enzyme Inhib. Med. Chem., 2011, 26(5), 688–695 CrossRef CAS PubMed.
- Y. Sun, W. Gan, M. Lei, W. Jiang, M. Cheng, J. He, Q. Sun, W. Liu, L. Hu and Y. Jin, Asian J. Pharm. Sci., 2018, 1–11 Search PubMed.
- S. Rajabi, A. Ramazani, M. Hamidi and T. Naji, Daru, J. Pharm. Sci., 2015, 23(1), 20 CrossRef PubMed.
- G. Libralato, E. Prato, L. Migliore, A. M. Cicero and L. Manfra, Ecol. Indic., 2016, 69, 35–49 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj03836h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |